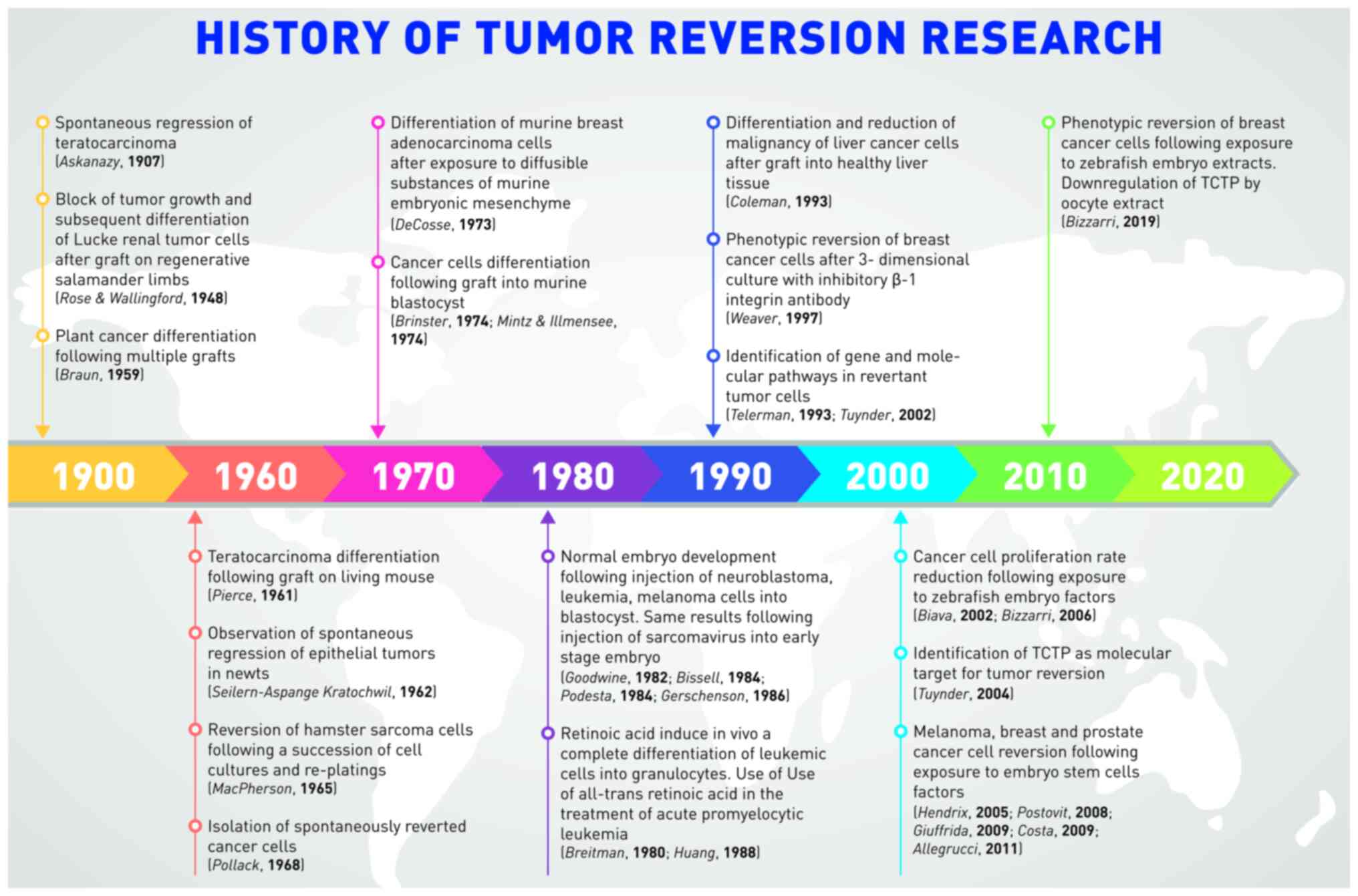
Introduction
Cancer is commonly observed as an irreversible
process. Consistent with this interpretation, the current therapies
are focused on the elimination of cancer cells by the means of
surgery, radiotherapy, chemotherapy/immunotherapy. However, these
approaches are not yet decisive for the oncological pathology, thus
highlighting either the need for improved eradication treatment or
the lack of a general understanding of the cancer process.
In this scenario, the current implicit notion of
irreversibility related to cancer should be questioned: Is cancer
really irreversible? A significant amount of experimental data has
revealed that cancer, under specific conditions, can revert into a
benign phenotype. This fact represents a clear paradox under the
current gene-based model of cancer according to which the primary
cause of cancer is a genetic mutation. Being a genetic mutation
irreversible and being cancer considered essentially caused by
genetic mutations, so this notion has been directly transferred to
cancer. Even though the most updated cancer models take into
consideration numerous other factors such as epigenetic, genetic
mutations are still implicitly considered hierarchically as the
primary cause of cancer.
To improve exploring this issue the literature
related to experimental evidence regarding cancer reversibility was
selected and analyzed. The literature was reviewed in chronological
order, and this work was organized by classifying the different
experimental models in order to improve grasping of the relevant
issues coming from experimental data. The most relevant studies
were selected in consideration of the number of citations received
or considering the fact that they were the first experiment of such
kind ever performed. For the current review, PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Google
scholar (https://scholar.google.com/) were
mainly used. The process of tumor reversion has been differently
named and described, thus different key words were used including
‘tumor regression’, ‘tumor reprogramming’ and ‘tumor
differentiation’. Only studies related to the epigenetic-induced
tumor reversion and not gene editing were considered. In fact, it
was considered that the concept of tumor reversion is related to
physiological complex processes that override genetic mutations. In
fact, one of the issues related to tumor reversion processes is
that it represents a ‘paradox’ within the somatic mutation theory
(SMT) and therefore pushes theoretical biologists to reconsider the
role of the genes in cancer research. A subsequent analysis of the
literature allowed the authors to discern studies that describe
real tumor reversion processes. Furthermore, the bibliography of
each study was analyzed in order to collect the most relevant
literature on tumor reversion. Once new authors that work on this
subject were identified through bibliographic analysis, further
research was performed using the name of the author.
At the end of this work, the implication of these
data were analyzed in consideration of the current model of cancer
and of the current structure of the ‘cancer research systems’
understood as the intertwining of science, medicine, industry,
finance and law.
Embryonal rest theory of cancer
Originally, the very first concept of ‘metastasis’
implicitly considered the possibility that cancer might
spontaneously regress. The word ‘metastasis’ was introduced in 1829
by the French gynecologist, Joseph Claude Anhelme Récamier, after a
clinical observation on a patient with breast cancer. After the
patient's death, the autopsy revealed the complete disappearance of
the breast cancer mass and the presence of a tumor mass localized
in the right lobe of the brain (1).
Commenting on this clinical case, Récamier introduced the term
metastasis for the first time using these words: ‘La résolution
spontanée d'un engorgement carcinomateux, suivie d'un autre
engorgement de même nature, peut conduire à admettre des métastases
cancéreuses’ [the spontaneous resolution of a carcinomatous
engorgement, followed by another engorgement of the same nature,
can lead to admit that cancerous metastases had occurred] (1).
Moreover, Recamier noticed-together with his
pathologist colleague, Jean Lobstein-several histological
similarities between the samples of tumor and embryonic tissue.
They thus presented the hypothesis that cancer could originate from
a residual of embryonic cells still present within the adult
organism (2,3). The German anatomist and physiologist,
Johannes Müller, proposed to associate embryogenesis and
carcinogenesis. He described tumors as the uncontrolled
continuation of embryonic developmental processes (4).
A further endorsement of this hypothesis arised in
1855 from the German pathologist, Rudolph Virchow. He confirmed the
observations of Récamier and Lobstein at the cellular level and
specified that tumor and embryonic cells also share several
functional and structural characteristics (5). This led him to hypothesize that cancer
could originate directly from embryonic-like cells (6). Such a thesis was later developed and
structured by his student, Julius Cohnheim, together with Francesco
Durante, who introduced the ‘embryonal rest theory of cancer’. This
theory states that adult tissues contain residues of embryonic
cells which, under certain conditions, can reactivate and give rise
to tumor masses (7,8). Such a model considered cancer as
reversible, at least in theory. In fact, embryonic cells can
differentiate into normal somatic cells. Therefore, cancer cells
deriving from embryonic stem cells should also be able to transform
themselves into benign differentiated tissue.
In addition, Max Wilms, a German pathologist and
surgeon, indirectly supported the association between embryogenesis
and carcinogenesis when, in 1899, he observed a kidney tumor
populated by embryonic cells in an 8-year-old boy (9). An attempt to explain the activation
mechanisms of residual embryonic cells in adults was made by Hugo
Ribbert between the late 1800s and the early 1900s. He advanced the
hypothesis that the tissue microenvironment exerts a sort of
‘tension’ on embryonic cells, which is capable of keeping them
dormant. When this tension is lost, then the uncontrolled processes
of carcinogenesis start (10,11).
These clinical and histological observations
converge toward the idea that tumor cells and embryonic cells share
some fundamental characteristics. Later on, and following this
hypothesis, it has been theoretically possible to consider cancer
cells as ‘developmental processes gone awry’ (12). This clearly implies a new strategy
for cancer treatments, that is, a re-differentiation approach that
implies a modulation of phenotypic expression.
The embryonal theory of cancer allowed scientists to
consider the hypothesis of the reversibility of tumors. However,
cancer models began to structure according to the SMT after the
1950s. SMT considers cancer an ‘irreversible’ process because it
considers gene mutations that are irreversible, as the primary
cause of cancer.
Teratoma as a model for tumor reversion
research
The cells of teratomas, that is, a type of germ cell
tumor that may contain several different types of tissue, are the
most similar to embryonic cells. Accordingly, they represent an
enlightening link between tumors and embryonic cells. Teratomas are
composed of a heterogeneous series of cells from differentiated
tissues-each of which represent primary germ layers, to which are
added embryonic tumor cells. Ovarian teratomas, for example, in the
early stages of development are composed of a fairly homogeneous
cell population. This turns into a series of differentiated cells
as the teratoma progresses. In some cases, it gives rise to
completely differentiated structures such as teeth and hair. Hence,
the term teratoma, from the Greek ‘τέρας’ (téras), which means
monster. In fact, teratomas appeared monstrous precisely due to the
presence of differentiated structures within shapeless masses,
almost as if a new living being with ‘monstrous’ characteristics
were trying to emerge from these tumor masses. The fact that
portions of differentiated, non-malignant, tissues emerged from
cancer cells suggests that it was possible to transform cancer
cells into normal cells.
The first clinical confirmation of this hypothesis
dates back to 1907 when the Swiss pathologist, Max Askanazy,
studied an ovarian teratoma in the initial stage. He observed and
described the spontaneous regression of the tumor mass where the
teratoma cells differentiated and gave rise to normal tissue
(13). Teratomas are rare tumors
and, therefore, difficult to be systematically studied. The
contribution by Leroy Stevens and Clarence Little was therefore
important: They succeeded in creating an innate strain of mice that
was highly prone to develop teratomas (14). This strain, called 129/SvJ, gave
impulse to the study of teratomas and allowed for increasingly
refined experimental designs.
It is precisely from this animal model that, in
1959, Barry Pierce first observed the spontaneous differentiation
of tumor embryonic cells from testicular teratocarcinomas (15). He isolated some embryonic carcinoma
cells from the differentiated tissues of murine teratocarcinomas
and grafted them into adult mice. Following these grafts, he
experimentally observed the partial differentiation of the
malignant cells into benign cells, which subsequently gave rise to
healthy tissues such as muscle. With these studies, Pierce
confirmed that teratomas can potentially transform into healthy
tissues.
Pierce also highlighted the role of the tissue
context in the differentiation processes of embryonic cancer cells:
In vitro teratocarcinoma cells can remain stable for up to
28 days, but they begin to differentiate as soon as they are
subcutaneously implanted in mice. However, the mechanisms
underlying this differentiation remained unclear. Pierce himself
explained: ‘The data do not rule out a mesenchymal induction in the
embryonal carcinoma as a result of some stimulus originating in the
tissue culture environment. (…) We have observed tissue genesis
from embryonal carcinoma, but the inductive stimuli for most of
these morphogenetic events were not apparent from the study. (…)
Differentiation of a loose reticular type of mesenchyme from
embryonal carcinoma occurred when embryonal carcinoma was overlaid
by visceral yolk sac. Whether this effect depends upon direct
contact by embryonal carcinoma cells to those of visceral yolk sac
or whether a diffusible substance is involved is not as yet known’
(16). Pierce concluded that: ‘This
observation, therefore, suggests the development of methods that
would direct the differentiation of embryonal carcinoma cells to
benign forms as a logical means of controlling this type of cancer’
(16).
In 1974, Brinster provided further evidence on the
possibility of inducing a differentiation of tumor cells and on the
role of the cell microenvironment in guiding these processes. In
his experiments, Brinster injected teratocarcinoma cells from
129/SvJ black agouti mouse testes into a murine blastocyst.
Subsequently, he implanted these blastocysts into female albino
mice (the blastocysts also came from albino mice) and followed them
until birth. He later observed the development of a new, healthy
animal and the consequent disappearance of the malignant cells. In
one case, he reported the presence of tufts of dark hair on the
back of the newborn albino mouse. This most likely revealed the
genetic fingerprint of the mice from which the teratocarcinoma
cells had been received. Cancer cells lost their malignant traits
and participated in the development of the embryo. Commenting on
his results, Brinster stated, ‘the embryo environment can bring
under control the autonomous proliferation of the teratocarcinoma
cells’ (17).
One year later, Mintz and Illmensee (18) confirmed the results of Brinster and
were able to analyze the fate of embryonic carcinoma cells in
detail. Their experimental work took place in its entirety over
eight years: They initially induced a teratocarcinoma on a 129/SvJ
black agouti mouse. They then implanted the embryonic carcinoma
cells into a testis of a brown C57-b/b mouse that metastasized to
the kidney shortly after. The primary testicular tumor was then
extracted, and its embryonic tumor cells were transplanted into the
intraperitoneal space of the abdomen of another C57-b/b mouse. This
gave rise to neoplastic ascites. A series of successive transplants
of embryonic tumor cells into other abdomens of syngeneic mice were
carried out for seven years until, in 1975, when embryonic tumor
cells were injected into a mouse blastocyst of the C57-b/b strain
brown that was then implanted in the uterus of an ‘adoptive
mother.’ Mintz and Illmensee (18)
injected embryonic tumor cells into a total of 280 blastocysts.
These were then implanted in the same number of adoptive mothers'
wombs. A total of 97 of these were sacrificed and analyzed between
the 8th and the 18th day of gestation. The remaining 183 were
allowed to reach the term of pregnancy, giving rise to 48 livng
mice.
Both fetal and offspring analysis revealed that all
animals were healthy and showed no evidence of tumors of any kind.
Even more interesting were the results of the analyses on the
composition of hair, the type of circulating red and white blood
cells, and the protein composition of urine, as well as
characteristics of the kidneys, liver and thymus. The
teratocarcinoma cells deriving from black 129/SvJ agouti mice had
participated in the normal formation of the organs by a ‘mosaic’
integration with the cells of the brown C57-b/b mouse strain. One
mouse was then mated and gave rise to healthy offspring,
demonstrating that the sperm cells were also normal. The authors
highlighted how: ‘In the present experiments, orderly expression of
numerous genes (for example, immunoglobulin, hemoglobin, MUP and
agouti genes) has occurred in vivo after they had been
‘silent’ or undetected in the tumors for 8 years, as well as in
cultures of teratocarcinoma cells’ (18).
Mintz and Illmensee (18) thus advanced the hypothesis that the
mechanisms underlying the neoplastic transformation were to be
sought not at the level of genes but of their expression processes:
‘The capacity of embryonal carcinoma cells to form normally
functioning adult tissues demonstrates that conversion to neoplasia
did not involve structural changes in the genome, but rather a
change in gene expression’.
A similar concept had already been proposed in 1954
by Grobstein (19) on the occasion
of the 13th Symposium of the Society for Development and Growth:
‘The differentiation of such tissues may depend on inductive
interactions between embryonic components’. With regard to the
aforementioned study, the double recurrence of the term ‘reversion’
associated with tumors is underscored. Grobstein specifically
referred to the malignancy of tumors: ‘Reversibility of malignancy
of the core cells’ and ‘the results also furnish an unequivocal
example in animals of a non-mutational basis for transformation to
malignancy and of reversal to normalcy’. Previous studies in fact,
used the term ‘differentiation’.
Although this research sounds promising for new
therapeutic cancer strategies, no systematic study to improved
exploring any ‘differentiation mechanisms’ of cancer cells has
followed. Teratocarcinoma is still considered as a curiosity within
the world of oncology. Possibly, for this reason, those results
were accepted yet ignored as deviations with respect to the general
behavior of other types of cancer cells.
At the same time, the ‘gene-centric’ paradigm was
successfully entering cancer studies as the first chemotherapy
approaches gave promising results. Cancer reversibility data from
teratocarcinoma models need to be verified on other tumor cell
lines. This, in order to substantiate the rationale of
‘differentiation treatments’ as a possible cancer treatment
approach.
Virus-induced tumors and reversion
Significant evidence of non-teratocarcinoma tumor
reversion came from the study of Ian Macpherson (20). He focused on virus-induced tumors,
specifically the Rous sarcoma virus (RSV). This virus, which was
discovered by Peyton Rous in 1911, can induce sarcoma in the cells
that it infects.
Macpherson's series of experiments demonstrated that
RSV-infected cells that had become cancerous could undergo
‘reversion’ (using his term) after repeated in vitro
transplantations of cell cultures. Under the best experimental
conditions, Macpherson was able to obtain a reversion of 19% of the
cells after three weeks of culture and 98% after eight weeks of
culture. It is important to note that Macpherson used the term
‘reversion’ when he observed that tumor cells resumed their
orientation in an orderly manner as in normal tissues, i.e.
re-acquired a ‘normal’ phenotype (21).
An important contribution comes from Pollack et
al (22) who in 1968 isolated
for the first time spontaneously reverted cancer cells. They
obtained cancer cells by infecting NIH3T3 cells with SV40 or
Polyoma virus. It was observed that some of these cells underwent a
spontaneous phenotypic reversion. The cells lost their malignant
traits and acquired a flat morphology. Therefore, these cells were
named ‘flat revertant’. Subsequently, the ‘flat revertant’ cells
were selected by eliminating the non-revertant cells with FUdR
(22). This represents a very
interesting model to study the mechanisms underlying reversion of
cancer cells.
Being inspired by previous studies carried out in
the 1940s (23) In the 1980s,
Dolberg and Bissell (24) carried
out a study on chicken sarcomas, which confirmed the
differentiation and protective potential of the cell
microenvironment against tumors. Early chicken embryos were
infected with RSV. This infection, which gave rise to sarcomas in
adult chickens, did not lead to any malignant degeneration between
the embryonic cells, even though the virus was active inside them
(24). These experiments also
highlighted the potential anticancer role of embryo
microenvironment.
Tumor reversion in plants
At the turn of the 1950s and 1960s, Armin Braun, a
researcher in plant genetics at the Rockefeller Center, developed
an experimental method to differentiate plant tumor cells.
Specifically, Braun worked on teratomas of tobacco. The experiment
aimed to understand how different structures and tissues could
emerge from a single tumor cell. Braun highlighted how specific
environmental factors involved in cell growth and division such as
auxins, and in cell division such as cytokines, were able to
determine cellular differentiation (25,26).
Interestingly, these mechanisms, which control growth and
differentiation, play a role in both germ and cancer cells. In
subsequent studies, Braun observed that it was possible to
transform a malignant phenotype into a benign one by cultivating
plant tumor cells in contexts with no auxins or cytokines, which
are the metabolites necessary to support ‘tumor metabolism’
(26).
A series of experiments involving sequential grafts
of teratomas on healthy plants further demonstrated the possibility
of transforming a malignant tumor phenotype into a benign one.
After growing clonal teratomas, Braun grafted them onto the canes
of healthy plants. The teratomas proliferated but diminished their
degree of malignancy. Braun then took cell samples from these
second tumor masses and grafted them onto other healthy plants. He
repeated these steps three times until the teratoma disappeared
completely. The result was normal plant growth of the plants. Once
planted, the seeds gave rise to new, perfectly normal tobacco
plants (26).
Through his experiments, Braun demonstrated that
cancer cells are endowed with plasticity and that it is possible to
grow a healthy plant from a cancer cell: ‘Results of this study
indicate that the capacity of teratoma tissue of single cell origin
to organize is a reflection of the inherent potentialities of
pluripotent tumor cells (…) clones of teratoma tissue of
single-cell origin developed organized structures (…) a controlled
recovery of crown-gall tumor cells could be accomplished’ (26).
Based on these results, Braun advanced the
hypothesis that there may be a hierarchical relationship between
mutations at the level of genes and control by the cytoplasmic and
tissue context: ‘When tumor shoots derived from tumor buds were
forced into rapid growth by a series of graftings to healthy
plants, they gradually recovered and became normal in every
respect. These results suggested that the cellular alteration in
crown gall did not involve a somatic mutation at the nuclear gene
level since heritable changes of that type are not generally
considered to be lost as a result of rapid growth’ (26).
Since plant dynamics differ from those of animals,
it is difficult to translate observations from one area to another.
However, the idea that a new fertile plant with ‘healthy’ seeds can
be originated from a tumor cell remains stimulating. Anyway, this
research highlights two aspects that have already been observed in
animals: a) embryonic tumor cells can differentiate, and b) the
microenvironment plays a fundamental role in guiding the
differentiation processes.
In vivo model of spontaneous cancer
regression
In the 1940s, Rose (27) and Wallingford (28) documented cases of in vivo
renal tumor regression. They took Lucké kidney tumors from frogs
and implanted them on the limbs of some salamanders that were
undergoing a regeneration process. Following these grafts, Rose and
Wallingford observed the arrest of tumor growth and the subsequent
differentiation of cancer cells into cartilage, muscle and
connective tissue cells. However, they could not define whether the
differentiated cells came from the frog and therefore from the
kidney tumor or the salamander. This prevented them from
establishing with certainty whether the observed process was a
differentiation process of tumor cells or simply an arrest of
cancer cell proliferation (27,28).
It should also be noted that Lucké renal tumor has a viral origin.
Therefore, it could not be a valid model for tumors caused by
carcinogenic chemical agents.
In those same years, Gersch (29) compiled a list of the various types
of tumors observed on different animal species and their frequency.
The list demonstrated that animals with high regenerative capacity
have a very low rate of tumor onset. An association between
regenerative processes and protection from tumors thus became
conceivable (29).
A decade later, Seilern-Aspang and Kratochwil
combined the observations of Gersch with the hypotheses advanced by
Waddington (30) and Needham
(31) according to which tumors
might emerge from a loss of control of cell differentiation by the
context. More precisely, Waddington and Needham took into
consideration the so-called ‘morphogenetic field’, that is, a
biological organizational scheme that emerges from the integration
of biological signals, for example, cell-cell interactions, and
biophysical constraints, for example, forces related to the
stiffness of a given tissue or diffusion processes that alter the
cellular behavior. Starting from these premises, Seilern-Aspang and
Kratochwil (32) designed an
experimental model aimed at studying the processes of
carcinogenesis and tumor plasticity in newts. These animals feature
a highly regenerative power of the limb, so they must host strong
morphogenetic processes. The authors worked on newt epithelial
tumors induced through an exposure to carcinogens on different
sites of an animal's body. In this way, they overcame the
experimental limitation of the work of Rose and Wallingford on the
viral origin of cancer.
Following the exposure to carcinogens, the animals
developed tumors that progressively acquired malignant
characteristics. These evolved from an expansive to an infiltrative
and a metastatic phase. This behavior confirmed the tumor nature of
the processes induced with carcinogens.
By monitoring the spontaneous evolution of tumors, a
strong tendency toward spontaneous regressions was observed. The
frequency of these regressions varied according to the anatomical
areas where the tumor had been induced. In order to verify whether
a differentiation of cancer cells had actually occurred with their
consequent reintegration within healthy tissues, histological
sections were made on both partially regressed and completely
regressed tumors. It emerged that the tumor cells had undergone
differentiation and integration into normal tissues. In some cases,
the differentiated tumor cells had abnormal structures-but not of a
cancerous nature. The authors also noted that the spontaneous
resolution of metastases occurred almost simultaneously with the
resolution of the primary tumor (32). These results made it possible to
advance the following hypothesis: The natural processes of tissue
regeneration were also able to induce and guide the differentiation
of cancer cells.
A further interesting result was reported by
McMichael (33) who observed a
partial tumor regression on a rabbit skin papilloma following the
administration of vitamin A. The role of vitamin A as a potential
anticancer agent was further studied by Saffiotti et al
(34) in mice exposed to the
carcinogen benzopyrene. Vitamin A-administered mice exposed to
benzopyrene tended to develop far fewer squamous lung tumors. Tumor
regression following vitamin A administration was also observed by
Davies on murine skin papilloma (35).
In the early 1990s, Coleman et al (36) studied the fate of two different
tumor cell lines resulting from the neoplastic transformation of
the liver epithelial cell line WB-F344. The two types of tumor
cells (GN6TF and GP7TB) were labeled with the retrovirus BAG2 and
the PKH26-GL dye in order to easily identify them in vivo
through histochemical techniques. When injected subcutaneously,
these two tumor cell lines gave rise to aggressive cancer in 100%
of the animals within 18 to 21 days. This confirmed their high
malignant potential. When, on the other hand, they were injected
into the liver tissue of mice, their aggressiveness was
considerably attenuated. The GN6TF line did not produce any type of
tumor. Instead, it morphologically differentiated and integrated
into the liver tissue. The GP7TB line, on the other hand, gave rise
to highly differentiated and not very aggressive intrahepatic
tumors.
Following these results, it was hypothesized that
the liver microenvironment may exert differentiation action on some
types of cancer cells by eliminating or reducing the tumorigenic
potential: ‘The apparent complete morphological differentiation of
BAG2-GN6TF cells suggests that the microenvironment of the liver
not only suppresses the ability of this particular tumor cell line
to form tumors but also stimulates them to integrate into hepatic
plates and differentiate into hepatocytes. By contrast, BAG2-GP7TB
cells form tumors in the liver that are more highly differentiated
morphologically than are tumors that form at subcutaneous
transplantation sites, suggesting that the regulatory influence of
the liver parenchyma can induce partial reversion of the
transformed phenotype without complete suppression of
tumorigenicity’ (36).
Interestingly, Coleman et al (36) used the term ‘reversion’ (in this
case, ‘partial reversion’) to describe the phenotypic
transformation of cancer cells. The shared elements of this
different experimental evidence obtained on animal models require
further investigation. First, the regenerative properties of
tissues (even liver cells are characterized by a high regenerative
frequency) can exert a control action on cancer cells. This action
is very likely to be exerted by the cellular microenvironment as a
result of regenerative and morphogenetic processes. These
observations are in accordance with the results obtained from the
transplantation of tumors within the blastocyst.
Generative/regenerative processes such as embryonic development
appear to have the ability to regulate the development of cancer
cells. It should be noted that these experimental works did not
find a return of cancer cells to their original stage. Rather, the
loss of their malignant characteristics and the integration within
the healthy tissues was observed. Therefore, the term reversion
should be intended only as related to the benign-malignant
phenotype.
Embryo microenvironment and cancer cell
differentiation
As already aforementioned, Barry Pierce was the
first to set up a systematic study on the role of the embryonic
microenvironment in determining the differentiation of cancer
cells. ‘Alternative to cytotoxic therapies are desperately needed
for the treatment of carcinoma with metastases. I would propose the
direction of differentiation of malignant to benign cells as the
most promising alternative’ (37).
Several experimental observations have highlighted
how embryonic tumor cells could undergo differentiation if injected
into a blastocoele. On the contrary, no regulation took place if
the cells were injected into the perivitelline space (38–40).
According to these observations, the embryonic microenvironment
and, more specifically, the injection site of tumor cells, could
have differentiation potential on tumor cells. This has led to the
deduction that only some types of the cellular microenvironment are
suitable for inducing a differentiation of tumor cells.
Pierce et al (39) took a step further when tried to
inject different types of cancer cells such as leukemia, sarcoma
and neuroblastoma into the blastocyst. With leukemia and sarcoma
cells, no control action was observed on the development of tumor
cells. With neuroblastoma cells, only a slight control action was
observed (39). These data led to
the hypothesis that the control action on cancer cells could only
be possible if the embryo had already developed the respective
‘healthy’ phenotype of cancer cells. Since the cells are still
undifferentiated at the blastula level, it was decided to inject
neuroblastoma cells at a more advanced stage of embryonic
development, specifically the neurula stage during which nerve
tissues are formed. The study was carried out by Podesta, a
collaborator of Pierce. The neuroblastoma cells into a mouse embryo
at 8 ½ days of development and observed normal development of the
embryo in 80% of cases. These results indicated that the tumor
cells had been regulated and directed toward physiological
differentiation processes (41,42).
These data appear to demonstrate that, depending on the specific
embryonic developmental stage, it is possible to regulate different
types of cancer cells.
Subsequent studies confirmed the following
hypothesis. Leukemic cells injected in the blastocyst did not
undergo any differentiation. However, if injected into the placenta
of a 10-day murine fetus, they underwent a correct hematopoietic
maturation with consequent normal development of the embryo
(43). The role of the embryo
microenvironment in preventing tumor growth and promoting a
phenotypic differentiation of sarcoma in chicken was also
highlighted by Dolberg and Bissell (24), as previously described.
Further confirmation of this rationale was provided
by Gerschenson et al (44).
It was managed to obtain a renormalization of B16 strain melanoma
cells following implantation within embryos in a uterus. Both the
identification of the correct phase of embryonic development and
the correct implantation site were crucial. In consideration of the
precise moments in which embryogenesis passes through the phase of
formation of melanocytes, the melanoma cells were implanted both in
the skin of the back of a mouse fetus at 10 days of development and
on the tips of the limbs in formation at the 14th day of embryonic
development. These moments correspond to the phases in which
melanocytes differentiate at those specific anatomical sites.
Consistent with the starting hypotheses, high differentiation rates
of melanoma cells were observed with consequent normal development
of the embryos. As a control, melanoma cells were implanted on the
skin of the back of 14-day-old mice and immediately after being
born. In these cases, no significant differentiation rates were
recorded, but cancer developed in between 70 and 80% of cases.
Gerschenson commented: ‘We have long proposed that cancer is a
problem of developmental biology and that an embryonic field
capable of differentiating a stem-cell lineage should be able to
regulate its closely related kind of cancer. If true, understanding
the mechanism of differentiation could lead to non-cytotoxic cures
for cancer’ (44).
Within these experiments, an issue remains unclear:
What drives cancer reversion processes, the diffusible substances
present in the fluids of the cellular microenvironment or the
physical contact between cells? A first attempt to investigate this
question was made by Pierce et al (45). Embryonic carcinoma cells were
exposed exclusively to the fluid extracted from the blastocoele. In
this case, no differentiation of the tumor was observed. Rather, it
occurred with the graft inside the blastocyst. Thus, it was
concluded that it was the cell-cell contact that played the
fundamental role in determining tumor differentiation (45). These conclusions were not entirely
correct.
In fact, ten years earlier, DeCosse et al
(46) had succeeded in obtaining a
differentiation of murine mammary adenocarcinoma cells (of the BW
10232 line) by exposing them to mammary embryonic mesenchyme.
Commenting on their study, it was hypothesized that: ‘An agent or
agents which was inactivated by formalin, probably stable to heat,
and capable of traversing a 0.45-µm Millipore filter initiated
several morphologic and functional changes in the mammary tumor
compatible with differentiation: Namely, development of tubules;
interruption of DNA synthesis; changes in nuclear and cytoplasmic
morphology; and appearance of a matrix tentatively identified as
containing acid mucopolysaccharides’ (46). This way, potential candidates in
diffusible substances were identified as inducing causes of tumor
differentiation.
A confirmation in favor of the hypothesis on
‘diffusible substances of the microenvironment’ was presented in
1988 by Biava et al (47).
The suppression of tumor development on mice lungs primarily
induced by homogenates of pregnant murine uteri was observed
(47). Subsequent in vitro
experiments on different lines of human tumor cells (glioblastoma,
melanoma, renal adenocarcinoma, breast cancer, lymphoblastic
leukemia), treated with extracts of zebrafish embryos taken during
the different stages of cell differentiation and before the
gastrulation processes, have achieved a reduction in tumor
proliferation on all cell lines. No results were obtained when the
embryonic extracts were received in the phases following
gastrulation-phases in which proliferative activity prevails over
differentiation (48,49).
The zebrafish embryo model was also used by Lee
et al (50) in 2005. Human
melanoma cells were implanted inside zebrafish embryos in their
early stages of development. In this case also, the suppression of
the malignant tumor phenotype and the birth of perfectly healthy
fish were observed. The following year, Cucina et al
(51) treated human colon cancer
cells (Caco2) with protein factors extracted from
zebrafish embryos in the pre-gastrulation stage. Their experiments
confirmed the results that had been previously obtained (49,50)
and demonstrated a reduction in tumor proliferation. Cucina et
al (51) were able to describe
the induction of apoptotic processes mediated by embryonic factors
through the activation of mechanisms independent of p53 and linked
to the pRb system/E2F1. The synergistic effect of these specific
embryonic factors was also demonstrated in vitro when, in
the treatment of colon cancer, they were combined with
5-Fluorouracil (5-FU).
In general, the aforementioned study (51) substantiated Pierce's (37) initial hypotheses, i.e. that the
embryonic microenvironment has specific characteristics that have
yet to be identified and make it able to control the proliferation
of cancer cells directing them toward a path of differentiation and
normal phenotypic maturation: ‘(…) It is our hypothesis that there
must be an embryonic field capable of regulating every carcinoma.
Study of how the embryo regulates malignant cells appears promising
as an alternative to cytotoxic therapy for carcinoma’ (37).
Since it was functional in structuring this model,
Pierce took up the concept of morphogenetic field (30,31).
According to Waddington, cancer emerges as a consequence of the
loss of control of the morphogenetic fields on cells (30). These concepts were taken by Pierce
and Johnson (52,53) and were both applied to the
morphogenetic processes of adult tissues and to the morphogenetic
processes during embryo development. From this perspective, it can
be said that the morphogenetic fields that guide the processes of
embryogenesis are also capable of exercising control over tumor
cells. It is therefore possible to advance an interpretation of the
neoplastic process according to the criteria of developmental
biology. In this sense, the tumor can be described as the
consequence of the loss of control over the cells by the
morphogenetic fields (37) and not
just as the result of a progressive accumulation of genetic
mutations. Clearly, the question remains open as to whether this
‘escape’ from the constraints of the morphogenetic fields depends
on changes in the individual cells or in the microenvironment. It
cannot be excluded that the problem should be analyzed from a
systemic point of view, i.e., considering the cell-microenvironment
equilibrium as an integrated system and therefore hypothesizing
that both causes may intervene in the processes of escape from the
control of morphogenetic fields.
A very interesting fact that emerges from these
different studies is that the various types of cancer cells are
selectively sensitive to those specific embryonic microenvironments
present in the stages of development during which the corresponding
types of healthy cells are differentiated and organized. More
simply, neuroblastoma cells are sensitive to the microenvironment
taken during the neurula stage, epithelial tumor cells are
sensitive to the microenvironments present during the corresponding
development stages. This observation made it possible to predict in
advance which microenvironments to select to re-normalize specific
types of cancer cells.
These concepts are in contrast with the gene-centric
explanation of the tumor that were developing and consolidating
during those same years, absorbing most of the public and private
research funds. As Kenny and Bissel (54) explained, this is most likely why the
research on tumor differentiation/reversion, despite convincing and
promising results, did not arouse the interest of the international
scientific community.
The reversion of acute promyelocytic
leukemia (APL)
Despite the lack of interest in differentiation
approaches, it was in those years that the first clinical success
was achieved: The treatment of APL, a disease that occurs with
bleeding and low platelet counts due to the reduced ability of the
bone marrow to produce platelets. Today, the current clinical
protocol envisages inducing a synergistic effect with a combination
of retinoic acid and arsenic, and about 90% of APL patients achieve
complete remission (55).
At the basis of APL is a genetic mutation: The gene
that codes for the nuclear retinoic acid receptor alpha (RAR-α) and
the one that codes for a protein called promyelocytic leukemia
protein (PML) blend together. The result of this fusion is the
formation of a hybrid protein, PML-RAR-α, which inhibits the
functioning of the retinoic acid receptor (56). Following this inhibition, the
hematopoietic differentiation processes stop, and poorly
differentiated leukemic cells accumulate (57). In parallel, the PML-RAR-α hybrid
protein activates genes that maintain the stem cell phenotype and
suppress the DNA repair genes. The resulting mutated phenotype
promotes tumor progression (58).
Retinoic acid was initially studied as an agent
capable of promoting the differentiation of teratocarcinoma cells
in vitro (59). However, the
results were not as valid in vivo (60). Subsequently, it was observed that
retinoic acid could induce a complete differentiation of leukemic
cells into granulocytes in vivo (61). Granulocytes were then digested by
stromal macrophages, allowing for a rapid elimination of malignant
cells and the complete remission in most patients (62).
In fact, treatment with retinoic acid favors the
initiation of the degradation processes of the hybrid protein
PML-RAR-α (63) and stimulates the
differentiation and possible apoptosis of APL cells (64,65).
APL remains the only cancer treatment capable of inducing the
reversion of the disease and represents a valid ‘proof of
principle‘ of the clinical application of reversion.
The aforementioned rationale identifies the
essential element in the embryonic microenvironment. Here, instead,
a radically different mechanism is at work, that is, the
administration of one single substance, trans-retinoic acid.
Indeed, Gootwine et al (43)
had documented the differentiation of leukemic cells following
their grafting into a mouse embryo at the 10th day of development.
However, the clinical translation of this approach is very
problematic, as it would involve administering a mouse embryo
extract to patients.
These results on APL raise some interesting
questions that require further investigation. First, are there
differences in the differentiation/reversion mechanisms between
liquid tumors and solid tumors? Is it possible to hypothesize
different mechanisms, not necessarily superimposable, which lead to
tumor differentiation/reversion and may involve either the
microenvironment or single substances? Both hypotheses, that of the
microenvironment and that of individual substances, are worthy of
research.
Clinical evidences of tumor reversion
Unlike liquid tumors, such as APL, solid tumors
present a greater biological complexity (66). For this reason, the related clinical
results are most likely not comparable to those achieved for the
treatment of APL (67).
However, numerous clinical studies over the decades
have documented cases of the spontaneous regression of tumors.
Apparently, these processes are comparable to those observed on
salamanders by Seilern-Aspang and Kratochwil (32). The first clinical documentation of
spontaneous cancer regression dates back to the 19th century. In
1918, Rohdenburg published an analytical review of 302 cases of
spontaneous regressions, with 70 of them as certainly valid
(68). Cases of spontaneous cancer
regression were reported periodically in medical literature, for
example, one related to neuroblastoma (69) or lung metastases deriving from renal
cancer (70).
The first monograph on spontaneous cancer regression
appeared in 1966. Everson and Cole (71) presented 176 well-documented cases of
spontaneous cancer regressions that had been published between 1900
and 1964. In their monograph, criteria for ascertaining the
diagnosis of both the disease and its regression were also
proposed, which involved histological and radiological
documentation. To be valid, the regressions must have occurred
without specific therapies, except those whose ineffectiveness was
clear (71). This analytical review
highlighted that most of the regressions were recorded on four main
types of tumors, namely renal tumor, choriocarcinoma, neuroblastoma
and malignant melanoma.
In light of this research, the American National
Cancer Institute sponsored a conference on the topic of spontaneous
tumor regression in 1974. A monograph was later drafted (72). Further significant research on the
subject was conducted by Challis and Stam in 1990 (73). All spontaneous regressions that had
been reported between 1900 and 1987 were analyzed, detailing the
progressive increase in regressions of lymphomas and kidney tumors
(73). In 1993, O'Regan and
Hirshberg published a bibliography of all the reported spontaneous
regressions of both malignant and benign tumors (74).
In 1998, Papac (75)
published an investigative work about the possible mechanisms
underlying spontaneous regressions. It was clarified that the
spontaneous regression of cancer means a complete or partial
disappearance of the malignant tumor in the absence of therapies
capable of inducing anti-neoplastic effects. It was also pointed
out how numerous patients had experienced relapses after a first
regression, which meant that spontaneous regressions were not
always stable and could not be always associated with recovery.
The main mechanisms proposed by Papac involved the
immune system, hormonal changes, the necrosis of tumor cells,
trauma and changes in the vascular system. Mechanisms related to
apoptosis and cell differentiation were also proposed. Tumors were
classified based on the reported frequency of regressions and the
four most recurrent were highlighted, which are kidney cancer,
neuroblastoma, breast cancer and melanoma. It was thus partly
confirmed what had emerged from the studies of Everson and Cole. In
addition, liquid tumors, such as leukemias and lymphomas, were
reported.
Another observation that emerged from Papac's study
is that the rarest tumors were those in which spontaneous
regressions have occurred more easily. On the contrary, spontaneous
regressions have occurred more rarely in the most frequent tumors.
Not only the type of tumor but also the site of its onset was
related to the frequency of regressions. For example, spontaneous
regression occurred more easily in the lungs or on the skin. This
suggested that, perhaps, certain microenvironment factors
facilitated the regression and, more generally, that there were
mechanisms for the endogenous regulation of tumor growth processes
(75).
Further data came from a series of molecular biology
studies on neuroblastoma cells that had experienced reversion: In
these cases, a decrease in telomerase activity was observed.
Similar studies on retinoblastoma cells instead highlighted how
regressing masses also demonstrated an increase in DNA
hypomethylation. Neuroblastomas, together with testicular germ cell
tumors and acute leukemias, were those in which regression due to
cell differentiation was most frequently recorded (75).
Finally, the regression of metastases has been
reported after the surgical removal of primary tumors. This fact
suggested a sort of paracrine mechanism by virtue of which the
primary tumor promoted the growth and proliferation of metastases
at a distance (75). These cases of
spontaneous regression confirmed the hypothesis according to which
the tumor is not a completely irreversible disease. However,
identifying a strategy for developing differentiation treatments
have remained difficult. The only structured clinical trials with a
clear protocol based on differentiation therapy were those related
to APL.
A prospective clinical trial on liver cancer
published in 2005 is therefore remarkable. This study reported some
cases of tumor reversion following non-chemotherapy treatments.
This randomized clinical study involved 179 patients with advanced
hepatocellular carcinoma and no chance to undergo any type of
therapy. One of the two groups of patients was administered, in the
form of compassionate care, a zebrafish egg extract. The second
group was given no treatments other than conventional pain
management for dying patients. The results revealed clear clinical
benefits in favor of the group treated with zebrafish egg extract.
The study was therefore interrupted in its randomization and, for
ethical reasons, the extract was then administered to all 179
patients. The monitoring continued for three years during which
19.8% of the patients had tumor regressions (2.5% were total
regressions), and 16% stabilized the progression of the disease. An
increase in performance was observed in 82% of patients (76). Clearly these are only preliminary
data that need further confirmation.
Molecular mechanisms underlying tumor
reversion
Understanding the mechanisms underlying tumor
differentiation/reversion is key in developing differentiative
treatments. In 1968, for the first time, Robert Pollack isolated
spontaneously reverted cancer cells (Pollack, 1968); afterwards
Telerman et al (77) further
developed this experimental model and produced the first systematic
study on the molecular mechanisms underlying tumor suppression by
focusing on tumor suppressor genes. In 2002, Telerman introduced
the term ‘tumor reversion’. It was defined as follows: ‘The process
by which some cancer cells lose their malignant phenotype-and from
a molecular point of view-tumor reversion can be defined at the
molecular level, not just as the reversal of malignant
transformation, but as a biological process in its own right
involving a cellular reprogramming mechanism, overriding genetic
changes in cancer, by triggering an alternative pathway leading to
suppression of tumorigenicity’ (78).
This concept is very similar to that of ‘reversion
of malignancy,’ introduced in 1975 by Mintz and Illmensee (18), who had studied the malignancy of
specific tumor phenotypic characteristics. To obtain ‘revertant’
cells, Telerman et al used a selection technique based on
the infection of various tumor cell lines with parvovirus H-1. This
particular type of virus has the characteristic of selectively
eliminating only malignant tumor cells. In this way,
virus-resistant tumor cells are selected and have lost their
malignant potential. To confirm that they were ‘revertant’ cells,
these cells were subsequently implanted in mice to observe the
stable reduction of tumorigenicity.
The comparison of the different genetic profiles of
various leukemic and breast cancer cell lines let a common group of
genes stand out among all of the different cells that had undergone
a phenotypic reversion of malignancy. Depending on the type of
tumor, this group of genes had different expression ratios.
However, the ‘overall variable’ of their genetic network seemed
constant among the different types of tumors.
Of the nearly 300 genes involved in the reversion
process, Telerman was able to identify the main ones, namely, seven
in absentia gene (SIAH1), presenilin 1 (PS1), tumor
suppressor-activated pathway (6TSAP6) and translationally
controlled tumor protein (TCTP). Two of these genes, SIAH1 and
TSAP6, are upregulated in the reverting tumor cells. The other two,
PS1 and TCTP, are instead repressed (78–80).
More specifically, SIAH1 is a target gene of the p53
protein. This protein plays a crucial role in apoptosis processes.
SIAH1 promotes the degradation of β-catenin, which is involved in
cell adhesion processes and mitigates the activation of the signal
cascade mediated by wingless-related integration site (Wnt). When
activated, these signals contribute to stimulating cell
proliferation. Overactivation of this gene, therefore, favors tumor
suppressor mechanisms. The overexpression of the TSAP6 gene also
favors the activation of mechanisms that promote apoptosis.
Conversely, when downregulated, the PS1 gene leads to tumor
reversion. This is because PS1 in itself exerts an anti-apoptotic
action; therefore, its downregulation silences this inhibitory
action. The TCTP gene also plays an important role in reversion
when downregulated. Of all genes, this is perhaps the most involved
in the stabilization and promotion of tumor growth processes,
therefore a reduction in its expression levels favors tumor
reversion (79).
TCTP was significantly downregulated in all
‘revertant’ cells compared with malignant tumor cells. This protein
is widespread among most eukaryotes, and its functioning is
associated with tumor growth and acute allergic responses (81). At a physiological level, TCTP has a
pro-tumor action because it promotes the growth and stabilization
of the cytoskeleton, thus favoring the spread and invasiveness of
tumor cells. It also blocks apoptosis by inhibiting p53.
To verify whether this protein had actually played a
central role in promoting tumor reversion processes, the
researchers inhibited its expression through antisense cDNA. In
fact, they could then observe the suppression of the malignant
phenotype. TCTP could therefore be a target for possible reversion
inducing drugs. However, the process is complex and, as Telerman
explains: ‘The gene expression profile suggests that it is not the
processes per se of cell cycle arrest, apoptosis and
terminal differentiation, that matter here, and that provide by
themselves the framework for reversion. It is rather a
‘reorganizing’ function of all these processes as a form of
rerouting and trigger of the whole machinery that enables the tumor
cells to quit the malignant pathway, even bypassing mutant or
wild-type p53’ (78).
Consistent with this hypothesis is the research by
Proietti et al (82) who, in
2019, managed to induce phenotypic reversion on two different
breast cancer cell lines (MCF-7 and MDA-MB-231) by using a
zebrafish embryo microenvironment. They obtained the phenotypic
reversion of cancer cells and highlighted some mechanisms through
which the reversion is activated. Specifically, these were a
remodeling of the cytoskeleton and a downregulation of TCTP.
Apoptosis and phenotypic differentiation immediately followed
(82). Subsequent studies confirmed
the same results with different cancer cell line cultures (liver
cancer, colon cancer and glioblastoma) and different embryo models,
specifically trout embryo (Bizzarri, forthcoming).
An interesting work by Weaver et al (83) highlighted the role of integrins and
cell adhesion processes in triggering phenotypic reversion. They
used human breast cancer cells cultured in 3D with inhibitory
β1-integrin antibodies. The reversion of tumor phenotype occurred
following the inhibition of cancer cells β1-integrins and the
subsequent re-normalization of adherens junction assembly. This
showed that, despite genetic mutations remaining in cells, it is
possible to re-establish normal phenotype and normal tissue
morphology. Moreover, from the aforementioned study emerged the
role of the basement membrane in controlling cell proliferation
thus highlighting the fundamental role of tissue architecture in
modulating the phenotypic expression of cells.
Another investigation focused on the molecular
mechanisms of the reversion/phenotypic differentiation of cancer
cells was carried out by Hendrix et al (84). This experimental model consisted of
exposing two different tumor cell lines (melanoma deriving from the
neural crest and breast carcinoma of epithelial origin) to factors
secreted by human embryonic stem cells. The result was a loss of
malignancy and a consequent reprogramming toward a benign phenotype
(84).
Previous research has identified that aggressive
melanoma and breast cancer cells exhibit high levels of expression
of the gene encoding the Nodal protein, an element that plays a
fundamental role in the process of embryonic morphogenesis, thus
evidencing possible correlation between embryogenesis and cancer.
Furthermore, melanoma and breast cancer also lack the Lefty
protein, a natural Nodal inhibitor. This is a molecular signal
belonging to the superfamily of transforming growth factor-β
(TGF-β). It is expressed in embryonic stem cells and antagonizes
Nodal, thus ensuring a balance between the different signals
underlying the morphogenetic processes (85).
The concomitant overexpression of Nodal and the
absence of its inhibitor Lefty, therefore, gave rise to an abnormal
and unregulated behavior in the tumor cells. As a result, the cells
underwent uncontrolled proliferation.
Consistent with this hypothesis, it was observed
that exposing these two tumor cell lines to the microenvironment of
Lefty-rich embryonic stem cells could induce an inhibition of Nodal
and a consequent reprogramming of the tumor cells. These thus
reacquired a benign phenotype. Cancer cell lines were also exposed
to microenvironment soluble factors from other types of stem cells,
such as those deriving from amniotic fluid, umbilical cords and
bone marrow. All these types of stem cells do not produce Lefty
and, consistent with the working hypothesis, their microenvironment
was not able to induce a reprogramming of tumor cells (86). This mechanism, similar to the one
identified by Telerman on TCTP, represents a possible target for
new therapeutic strategies on cancer.
In general, this research confirmed the presence of
profound similarities between the processes of embryogenesis and
those of tumor reversion and differentiation. In particular, the
authors explain how: ‘The phenotype of stem cells and cancer cells
is profoundly influenced by the microenvironment. During
embryogenesis, precursor cells are specified to particular fates
through the delivery of signaling molecules, and malignant cells
similarly release and receive cues that promote tumor growth and
metastasis’ (86). Among these
signals, the main ones are those belonging to the superfamilies of
Notch, Wnt, TGF-β and Nodal (87).
It must be stressed that all of these are epigenetic
processes. They control the gene expression through environmental
signals. These signals interact with DNA and its expression, for
example, via methylation and acetylation. Indeed, DNA methylation
and histone modification are among the most known epigenetic
events. When tumor promoters and tumor suppressors involve genes,
these events can, regardless of the presence of mutations, lead the
behavior of the cell toward a tumor path. This is why interest in
micro-RNAs (miRNAs) is growing. Micro-RNA is a type of non-coding
RNA that can play an important role in guiding the epigenetic
modulations of DNA (88). A single
miRNA can interact with hundreds of thousands of target proteins
and promote systemic action (89).
To investigate this hypothesis, an experimental
model was developed in which C8161 line melanoma cells were exposed
to the microenvironment of human embryonic stem cells. The
expression profiles of miRNAs within the tumor cells were studied
before and after exposure to the embryonic microenvironment. MiRNAs
were thus identified, some of which were upregulated in melanoma
cells, specifically miR-302a. Others, on the contrary, such as
miR-27b, were downregulated (90).
These miRNAs were also associated with embryogenesis processes,
thus confirming the thesis according to tumor reversion processes
have strong similarities with embryo genetic processes (91).
The aforementioned study also highlighted a
connection between miRNAs and Nodal regulation. In fact, it
identified a second Nodal inhibition mechanism that exploits the
Nodal-Notch4 axis. This axis is key in embryo genetic processes: It
drives the formation of the right-left axes (92). Lefty promotes an increase in miR302a
levels which, in turn, silences the Nodal and Notch4 signaling
circuit. The result is a loss of the malignant phenotype by
melanoma cells (90).
Cell cycle control is key in maintaining a correct
balance between cell proliferation and differentiation. Precisely
inside the cells, there are proteins called cyclin-dependent
kinases that have the task of blocking uncontrolled cell
proliferation. For this reason, they are also called ‘gatekeepers‘
since, in correspondence with the different checkpoints of the cell
cycle, they block abnormal cells (93). Clearly, cancer cells escape these
controls and continue to proliferate, completing the entire cycle
undisturbed. A possible therapeutic strategy for cancer, therefore,
could aim at restoring the correct functioning of the various cell
cycle checkpoints or forcibly blocking the tumor cells in the
initial G1 phase of the cell cycle. In 2009, Giuffrida et al
(94) developed an experimental
model exposing different cell lines of human epithelial tumors,
such as ovarian, prostate, and breast cancer, to substances
secreted by embryonic stem cells. The aforementioned study
confirmed the ‘antitumor’ activity of the embryonic
microenvironment and investigated the cell cycle modification
during phenotypic reversion processes.
The aforementioned study confirmed a selective
antitumor action of the embryonic stem cell extracts. It also
confirmed the selectivity of cancer cells by exposing human
fibroblasts to the same factors. In this case, no inhibition was
observed. Furthermore, following exposure to the embryonic
microenvironment, an important number of cancer cells remained
blocked in the G1 phase. At the same time, smaller quantities of
cancer cells were detected in the most advanced stages of the
cycle, S and G2/M, indicating that the soluble factors produced by
embryonic stem cells might have exerted an inhibitory action on the
cell cycle, selective toward cancer cells.
In their research, Giuffrida et al (94) characterized these factors as
low-weight and thermostable molecules that allow embryonic stem
cells to exert paracrine and autocrine actions. These substances
could therefore represent a valid therapeutic strategy: ‘Instead of
using stem cells themselves for therapy, it will likely be possible
to identify and synthesize the specific tumor-suppressing factors
secreted by embryonic stem cells, thereby bypassing the practical
and ethical issues currently associated with embryonic stem cell
therapy’ (94).
Previous literature has already shown that
epigenetic alterations are pivotal in the early stages of breast
cancer development (95,96). For this reason, it was conceivable
that one of the mechanisms through which embryonic extracts could
exert their ‘tumor reversion’ action might be the modification of
chromatin. In fact, hypermethylation is one of the main silencing
processes of tumor suppressor genes. Therefore, the hypothesis to
be tested was whether the embryonic extracts were able to
demethylate these genes. For this purpose, a series of genes that
could be the targets of these actions, such as RARB, CST6, CCND2,
GAS2 and CDKN2A, were identified.
Further elements on the possible mechanisms
underlying the processes of tumor reversion and differentiation
were provided by Allegrucci et al (97). Their experimental model exposed a
breast cancer cell line, MCF7, to three different types of
‘embryonic extracts’ from three different sources, namely a Mexican
salamander embryonic (Axolotl), frog embryo (Xenopus
laevis), and mouse embryonic stem cell. The goal was to both
verify the differentiation potential of these extracts on cancer
cells and investigate possible epigenetic mechanisms underlying
these processes.
The results confirmed the initial hypotheses that
both salamander and frog embryo extracts favor the re-expression of
these genes, although not always restoring it to normal level.
Furthermore, only the extracts received during the initial stages
of embryogenesis exerted this action. Extracts received at a later
stage did not exhibit this activity. The extracts of mouse
embryonic cells, on the other hand, did not give satisfactory
results, inducing only the re-expression of the GAS2 gene. However,
since reprogramming actions were observed at the cellular level,
perhaps the mechanisms worked through other pathways in this case.
In general, the most efficient extracts were those from
salamanders.
To verify the stability of these epigenetic
modifications, the reprogrammed tumor cells were implanted in
immunosuppressed mice to be compared with ‘non-reprogrammed’ tumor
cells. Tumors from the ‘reprogrammed/reverted’ cells were markedly
smaller than the control group eight weeks after transplantation.
Even at the histological level, tumors from the reprogrammed cells
were more circumscribed, although not encapsulated, and had a lower
rate of mitotic division, which indicates a lower degree of
malignancy.
These results confirmed that certain embryonic
extracts were able to exert a stable reprogramming action on cancer
cells. This approach has opened up promising new avenues for
research: ‘It will now be important to identify the oocyte-specific
molecules involved in this process, and the molecular pathways
responsible for the arrest of tumor growth. In our view, the
identification of these molecules will provide a rich source of
information for the design of synthetic molecules that can be used
for pharmaceutical interventions’ (97). Further experiments performed by Saad
et al (98) on MCF-7 cells
treated with axolotl oocyte extract highlighted more mechanisms
involved in reprogramming/reversion processes such as cell cycle
arrest associated with upregulated expression of P27 and reduction
of RB phosphorylation. An interesting result was the induction of
tumor cell dormancy (98).
Proietti et al (82) also obtained the phenotypic reversion
of malignant cancer cells. He exposed an aggressive breast cancer
line, MDA-MB-231, to zebrafish and trout embryo extracts. His
research confirmed the reduction of cell proliferation and
highlighted some mechanisms through which the reversion was
activated: A remodeling of the cytoskeleton and a downregulation of
the TCTP protein that had already been studied by Telerman. This
downregulation promoted apoptotic processes and cell cycle
stabilization (82). A recent study
identified some of the most relevant genes involved in tumor
reversion processes, including STAT3, ROCK, BRCA1, SETDB1, MMP9,
YBX1, FASN, RARα, RB1, PRKD1, PP14, FAK, and P53 GM2/GM3. In some
cases, post-translational mechanisms have been highlighted. These
include phosphorylation, methylation, deglycosylation and silyation
(99).
All of this experimental evidence provided for a new
therapeutic rationale based on epigenetic regulation processes.
These processes are increasingly explored in the context of
cellular reprogramming. According to this model, the embryo
microenvironment can induce the differentiation of tumor cells,
consequently promoting a reversion of the malignant phenotype
through epigenetic regulation pathways. These are, in numerous
cases, similar to those involved in embryogenesis processes.
Therefore, the control of tumor development appears to take place
at the epigenetic and non-genetic level.
The contribution of cellular reprogramming
research in the context of tumor reversion
A big boost to tumor reversion research was provided
by the studies on cell reprogramming by Shinya Yamanaka and John
Gurdon, granting them the Nobel Prize for medicine in 2012.
Takahashi et al (100)
demonstrated the possibility of reprogramming totally
differentiated somatic cells by transforming them into pluripotent
stem cells (iPCS) due to the forced expression of four
transcription factors: Oct3/4, Sox2, Klf4 and c-Myc. Following the
aforementioned results, the concept of phenotypic reversibility and
cellular plasticity became substantiated (101).
Subsequent studies have shown that cellular
reprogramming is not strictly necessary to activate the expression
of Oct3/4, Sox2, Klf4 and c-Myc. For example, the overexpression of
E-cadherin is sufficient to activate the reprogramming with no need
for the activation of Oct4 (102).
Among these, the main molecular programs involved in cellular
reprogramming are the modulation of the Wnt/β-catenin and PI3K/Akt
pathway, the downregulation of Prenselina-1 (PSEN-1), Notch and
SNAI1, and the increase in the synthesis of E-cadherin (103,104).
Even the biophysical elements of the cell culture
environment, such as plate material (glass or graphene) can trigger
the reprogramming of fibroblasts in induced pluripotent cells
(105). This highlights the
importance of the microenvironment and the tissue in determining
the processes of cellular differentiation or de-differentiation. In
order to grasp the key factors involved in cellular reprogramming,
it is necessary to go beyond the study of molecular pathways and
look for the tissue level (106).
Despite progress in understanding these mechanisms,
clinical application remains far away. It is important to note that
when talking about cellular reprogramming, unlike the concept of
tumor reversion, the transformation of epithelial cells into
mesenchymal stem cells is generally meant. This process is known as
epithelial-mesenchymal transition (EMT) and it characterizes
neoplastic transformation. These cellular reprogramming processes
imply significant changes in cell morphology, such as the
acquisition of a rounder shape and the loss of cell-cell contacts
typical of somatic cells. All of these morphological
transformations favor the acquisition of properties such as
motility and invasiveness that are typical of tumor processes
(107). This is one of the main
reasons why cell reprogramming techniques have not found clinical
applications. In fact, the activation of regulatory factors that
initiate cell reprogramming often induces tumors (108,109).
Indeed, these data reinforce the hypothesis that
cancer may be an error in the process of cell development and
differentiation (12) and that, as
demonstrated in the past, molecular signals involved in cell
differentiation and embryonic development may also exert a role on
tumor cells (110,111). Based on these common biological
pathways between cellular reprogramming and oncogenesis, various
studies aimed at finding effective strategies to apply cellular
reprogramming techniques to cancer cells.
Unlike the tumor reversion approach, one of the
first strategies developed to induce tumor reprogramming involved
the de-differentiation of tumor cells. The goal was their
transformation into pluripotent cells that could subsequently
re-differentiate into non-malignant somatic cells. Promising
results have been obtained on skin tumor cell lines treated with a
specific microRNA (miRNA3025) (112) and on murine melanoma cells
(113). However, this research is
difficult to translate into clinic procedures because the
non-selective cell de-differentiation step could favor the onset of
tumors in other cells of the body.
In 2013, Rapino et al (114) reprogrammed human lymphoma and
leukemia B cells. They transformed them into macrophage-like cells
by introducing a transcription factor C/EBPα. Huang et al
(115) provided further
confirmations in 2014. It was demonstrated how a combination of
transcription factors can play an important role in reprogramming
human fibroblasts into pseudo-hepatocytes (115). McClellan et al (116) applied the same concept and managed
to reprogram lymphoblastic leukemia cell lines using small
molecules.
Exosomes also appear to be involved in tumor
reprogramming. In 2017, Zhou et al (117) inhibited tumor proliferation in
vitro and slowed down oncogenesis in vivo by using
exosomes extracted from embryonic stem cells. A further
contribution to this innovative cancer treatment approach was
obtained in 2018 by Ishay-Rosen et al (118), who managed to transform breast
cancer cells into adipocytes by means of a mix of molecules
including insulin, dexamethasone and BMP2. Cancer cells were first
treated with TGF-β to bring them back to a pluripotent stage. They
were then differentiated by exposure to a mix of molecules that
directed these cells toward the acquisition of normal phenotype
(118).
In 2019, Cheng et al (119) managed to induce direct
reprogramming, that is, without going through the reactivation of
the pluripotent stage. In addition, in this case, hepatocellular
carcinoma cells were exposed to small molecules (a mix of
transcription factors). This treatment induced their
differentiation into normal liver cells (119).
A recent study on ‘oncogene addicted’ cancer cells
provided for further evidence of the possibility of the phenotypic
reversion of cancer cells. Li et al (120) obtained tumor reversion by
inhibiting specific oncogenes of tumor hepatocytes in xmrk
transgenic zebrafish model. By genetic recombination, tumor cells
were marked and it was confirmed that tumor hepatocytes
morphologically and molecularly converted into normal hepatocytes
(120).
Theoretical implications of ‘tumor
reversion’
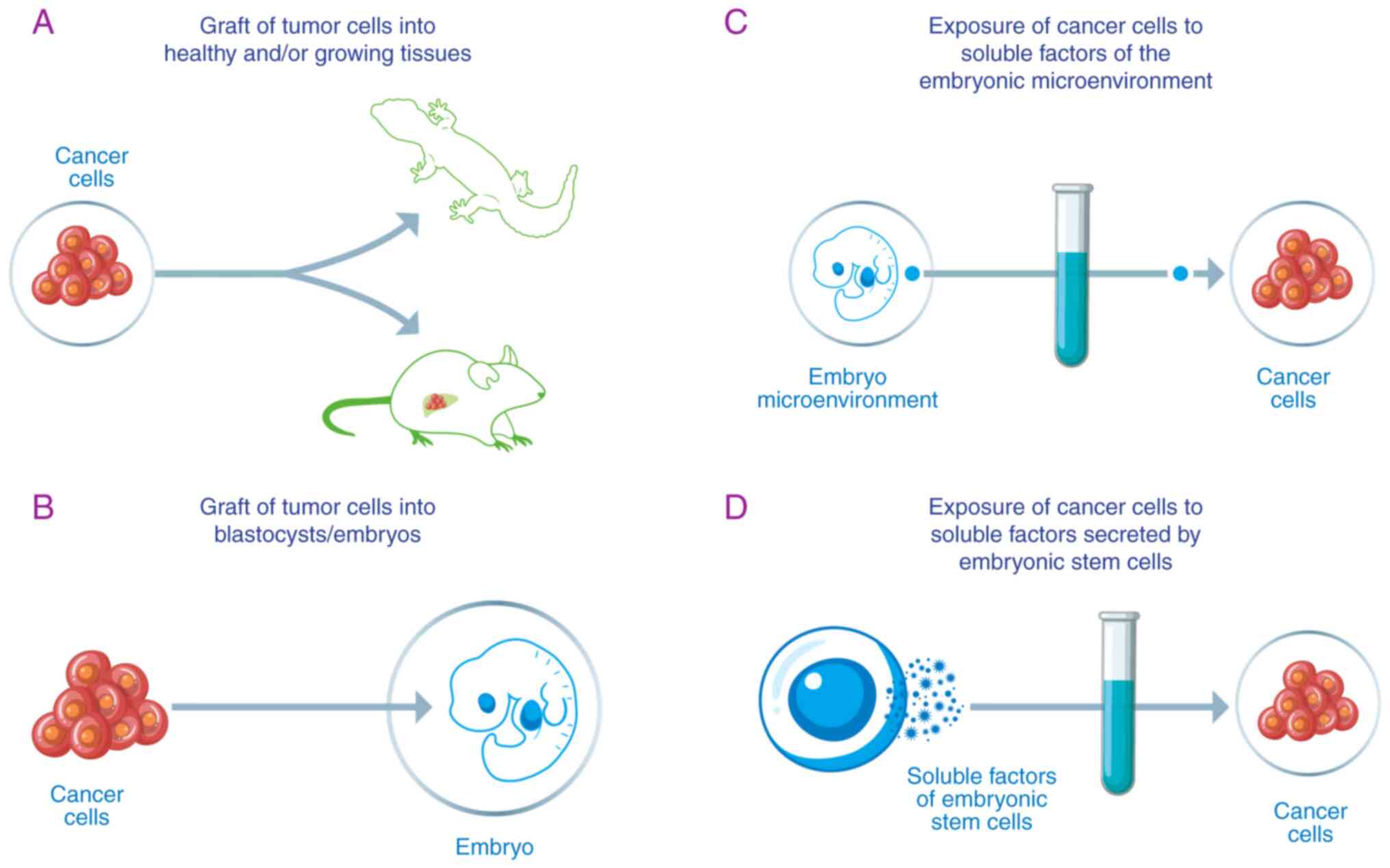
Fig. 1
A non-trivial issue deals with the lexicon and the
conceptual tools used to describe experimental and clinical
observations (121). Four main
terms occur most frequently in the literature, referring to ‘tumor
reversion’ processes, namely, regression, differentiation,
reversion and reprogramming.
The term ‘regression’ appeared in the first half of
the 20th century and was mostly used until the 1970s. It mainly
refers to the clinical observation of a ‘regressing’ tumor that
decreases in mass. In this sense, this term says nothing about the
malignant or benign nature of cancer cells. The term
‘differentiation’ was introduced by Pierce and has been used-albeit
less frequently-until today. It mainly refers to the
characteristics of the cancer cells that differentiate and return
to integrate into the tissues, losing their malignancy. Pierce's
use of this term took shape from observations on teratocarcinomas
in which undifferentiated embryonic tumor cells progressively gave
rise to differentiated cells following a kind of embryogenesis
processes. This term recalls the conceptual framework of embryonic
development and indicates the processes affecting individual
cells.
The term ‘reversion’ evokes a markedly more radical
concept. It was used for the first time in relation to cancer by
Macpherson in 1965, then later structured by Telerman. Macpherson,
like Braun, observed the renormalization of cells in their
morphology and function and used the term reversion to indicate
this recovery of normal characteristics. At the etymological level,
the term ‘reversion,’ from the Latin reversio, means to go
back and implicitly alludes to a restoration of the original
conditions. Such a process in its entirety has not been confirmed
experimentally in the sense that the tumor cells have not been
induced to ‘go back’ to their original characteristics: in the
transformation of the cell from state A (normal) to B (tumor) to C
(revertant), it cannot be said that A is equal to C. As
aforementioned, Telerman explains this point and the term ‘tumor
reversion,’ defined as: ‘The process by which some cancer cells
lose their malignant phenotype (…) tumor reversion can be defined
at the molecular level, not just as the reversal of malignant
transformation, but as a biological process in its own right
involving a cellular reprogramming mechanism, overriding genetic
changes in cancer, by triggering an alternative pathway leading to
suppression of tumorigenicity’ (78). This is very important because it
clarifies that the term reversion refers exclusively to the
malignant phenotypic characteristics of cancer cells (such as
invasiveness and migration) and that the pathways through which
cancer cells undergo this transformation can be different. It is
therefore more precise to use the expression ‘phenotypic
reversion’.
The last term used, which Telerman himself uses in
clarifying the definition of tumor reversion, is that of
reprogramming; a term that took shape from Yamanaka's work on
cellular reprogramming and that is borrowed from the world of
information technology. This word refers to the control,
programming and activity of the cell's genes and implicitly alludes
to an external action, typically human, of the ‘manipulation’ of
biological processes. In this sense, it is more difficult to
associate the word ‘spontaneous’ with the one of ‘reprogramming.’
It should also be noted that the translation of the concept of
programming borrowed from the world of information technology for
biology has some application limits (122). Therefore, it should be specified
that by cellular reprogramming, it is meant the induction of a
controlled transformation of the cell phenotype typically by the
path a) de-differentiation, b) re-differentiation, namely in cancer
a) EMT-b) MET.
This relative variety in terms reflects the variety
of experimental models within which the
regression/differentiation/reversion/reprogramming processes have
been observed. The various experimental models and results are
outlined in Table I.
 | Table I.Relevant experimental models and
results (literature is presented in chronological order). |
Table I.
Relevant experimental models and
results (literature is presented in chronological order).
| Author's/year | Experimental
model | Description and
results | (Refs.) |
|---|
| Braun, 1959 | Graft of tumor
cells into healthy and/or growing tissues | Succession ofgrafts
of plant teratoma clonal cells on healthy tobacco plant.
Disappearance of the teratoma and plant generation with seeds
capable of giving life to a new plant | (26) |
| Pierce, 1961 |
| Transplantation of
Murine Embryonic Tumor Cells into mice healthy tissues results in
cancer cells differentiation re- | (16) |
| Macpherson,
1965 |
| Hamster sarcoma
cells. Succession of cell cultures and platings. Transformation of
19% of cells, which return to orienting themselves in an orderly
manner, as in healthy tissues | (21) |
| Rose &
Wallingford, 1948 |
| Lucke renal tumor
cells. Planting on regenerative salamander limbs. Block of tumor
growth and subsequent differentiation of cells. Failed to determine
whether the differentiated cells came from cancer cells or healthy
tissue | (27,28) |
| Coleman, 1993 |
| Liver cancer cells.
Injected into liver tissue. Reduction of malignancy and, in some
cases, differentiation of cancer cells. | (36) |
| Brinster, 1974 | Graft of tumor
cells into blastocysts/embryos | Murine testicular
teratocarcinoma cells. Injection into murine blastocyst implanted
in albino femalemice. Development of healthy mice | (17) |
| Mintz &
Illmensee, 1974 |
| Embryonic carcinoma
cells from black mice. Blastocyst injection implanted in brown
female mice. Normal fetal development; normal newborn mice feature
hybrid traits between black and brown mice | (18) |
| Podesta, 1984 |
| Neuroblastoma
cells. Injected into 8 ½ day old murine blastocyst. Differentiation
of tumor cells. | (41,42) |
| Gootwine, 1982 |
| Leukemia cells.
Injected into 10-day old murine blastocyst. Correct hematopoietic
maturation. | (43) |
| Bissell, 1984 |
| Rous sarcoma virus.
Injected into chicken embryos. No tumor development. | (24) |
| Gerschenson,
1986 |
| Mouse melanoma
cells. Implanted into embryos in the murine uterus. Cell
differentiation and normal embryonic development. Differentiation
occurs when cells are implanted into a 14-day embryo. | (44) |
| Hendrix, 2005 |
| Human melanoma
cells. Implanted in zebrafish embryos in the early stages of
development. Suppression of malignant tumor phenotype and birth of
healthy fish. | (50) |
| Gersch, 1951 | Induction of tumors
in healthy animal tissues and monitoring of their evolution | Spontaneous tumors
in animals. Observations on the rate of onset. Reduced occurrence
of tumors in animals with high regenerative capacities | (29) |
| Seilern-Aspang
& Kratochwil, 1962 |
| Triton-induced
epithelial tumors. Monitoring the spontaneous evolution of tumors.
Tendency to tumor regression in anatomical areas with high
regenerative potential. Results confirmed by histological
analysis | (32) |
| DeCosse, 1973 | Exposure of cancer
cells to solublefactors of the embryonic microenvironment | Murine breast
adenocarcinoma cells. Exposure to diffusible substances of murine
embryonic mesenchyme. Differentiation of tumor cells. | (46) |
| Biava, 1988 |
| Primary murine lung
cancer. Administration (in vivo) of homogenates of pregnant
murine uteri. Suppression of tumor development | (47) |
| Biava, 2001;
2002 |
| Glioblastoma,
melanoma, renal adenocarcinoma, breast cancer, and lymphoblastic
leukemia cells. Exposure to embryonic extracts of zebrafish taken
before gastrulation. Reduction of cell proliferation rates. | (48,49) |
| Bizzarri, 2006 |
| Human colon cancer
cells. Exposure to factors extracted from zebrafish embryos prior
to gastrulation. Reduced rate of cell proliferation. In addition,
the activation of p53, of the cell cycle blocking system pRb/E2F1,
and of a synergistic effect with 5FU was observed. | (51) |
| Allegrucci,
2011 |
| Breast cancer
cells. Exposure to axolotl, frog, and mouse embryonic cell
extracts. Stable reversal of malignant phenotype (confirmed with
subsequent implantation of reprogrammed cells in immunosuppressed
mice). | (97) |
| Saad, 2018 |
| Breast cancer
cells. Exposure to axolotl oocyte extracts. Stable reversal of
cancer phenotype, cell cycle arrest mediated by upregulation of p27
and reduction of RB phosphorylation, induction of tumor
dormancy. | (98) |
| Bizzarri, 2019 |
| Breast cancer
cells. Exposure to embryonic extracts of zebrafish taken at
different times of embryogenesis. Reduction of invasiveness,
migration, and proliferation parameters; action on cytoskeleton and
TCTP downregulation. An activation method of reversion was
identified, implying the down-regulation of TCTP by exposing the
cells to a specific embryonic microenvironment composition that
corresponds to a specific phase of embryogenesis. | (82) |
| Henrix, 2007
Postovit, 2008 | Exposure of cancer
cells to soluble factors secreted by embryonic stem cells | Melanoma cells and
breast cancer cells. Exposed to embryonic stem cell factors.
Reversal of the malignant phenotype and activation of apoptotic
processes (nodal signal inhibition was also observed). If cells are
exposed to factors extracted from umbilical cord and bone marrow
stem cells, then no phenotypic reversion is observed. | (84, 86) |
| Giuffrida,
2009 |
| Ovarian, prostate,
and breast cancer cells. Microenvironmental exposure of human
embryonic stem cells. Reversion of malignant phenotype block of
cancer cells in phase G1 | (94) |
| Costa, 2009 |
| Melanoma cells.
Microenvironmental exposure of human embryonic stem cells.
Reversion of malignant phenotype. The study identified some mRNAs
involved in these cellular reprogramming processes. | (90) |
It is best to find the correct lexical expression
for the observed reversion processes so as not to induce, through
imprecise terms, erroneous interpretations of experimental data.
Nowadays, the literature is still not homogeneous in naming these
processes. For the present review, the term ‘phenotypic reversion’
was proposed because it is more precise. It is underscored that
some authors use the expression ‘tumor dormancy’ (83,120).
This highlights the need for improved comprehension on the nature
of these processes as well as their disambiguation. For example, is
‘phenotypic reversion’ a unique process, or do different processes
drive cells to different types of benignant states?
From a deeper analysis of this research, it is
possible to identify and classify the following experimental
models: i) Transplantation of tumor cells into healthy and/or
growing tissues; ii) transplantation of tumor cells into
blastocysts/embryos; iii) induction of tumors in healthy animal
tissues and monitoring of their evolution; iv) exposure of cancer
cells to substances of the embryonic microenvironment; and v)
exposure of cancer cells to substances secreted by embryonic stem
cells (Fig. 2). Within these
models, the various variables taken into consideration are: i) Type
of tumor; ii) specific phase of embryonic development in which the
tumor is implanted or from which the contained substances are
extracted; and iii) anatomical site of tumor implantation.
Conclusions
The present review aimed to present the complex
‘state of the art‘ of this rather unknown field in cancer research.
Despite the experimental evidence on phenotypic tumor reversion is
based on different experimental models and laboratories, a
systematic study program in this field is still missing.
Consequently, research programs on cancer research still miss the
opportunity to investigate the effectiveness of tumor reversion on
cancer therapies. The experimental and interpretative framework is
complex and needs a systemic approach to be modeled. The present
review only presented a general picture of the ‘state of the art’
and does not try to propose a theoretical model of tumor reversion.
However, some conclusions can be advanced on the recurrent
elements. First, the fate of cancer cells is not irreversible. This
statement implies the ‘proof of principle’ of new therapeutic
strategies for cancer. However, it should be noted that
‘non-irreversibility’ does not automatically correspond to
reversibility understood as a return to the original state of the
cells. It would therefore be more correct to state that the fate of
cancer cells is not unchangeable and that cancer cells display
relevant plasticity. Second, it is possible to inhibit the
phenotypic expression of the malignant characteristics of cancer
cells mostly through epigenetic processes, although other
mechanisms are likely to participate. This statement implicitly
contains the concept of ‘chronicization’ of the tumor. When talking
about ‘phenotypic expression,‘ it is necessary to clarify what is
meant by phenotype. Specifically, a modification in the behavior of
cells that pass from presenting aggressive functions such as the
ability to migrate and invade new tissues to have more controlled,
and therefore benign, characteristics, is indicated. Third,
specific embryonic and tissue morphogenetic fields are capable of
exerting a direct action on the phenotypic expression of cancer
cells. Within the embryo (at certain stages of development) and in
specific healthy tissues, there are substances/mechanisms capable
of directing malignant tumor cells toward the re-expression of
benign characteristics. This statement implies a reconsideration of
the hierarchical relationships between genes and the context of the
cellular microenvironment, between genetics and epigenetics. The
concept of morphogenetic field is also used to indicate the complex
network of biological and biophysical signals that can exert
regulatory (and binding) actions on cells. Clearly, in order to
describe these processes, new conceptual tools are needed to grasp
the complexity and dynamism of a biological network. Fourth, the
pivotal role of genes in explanatory models of cancer should be
reconsidered and integrated with new conceptual tools such as
morphogenetic field, biological network, cell reprogramming and
phenotypic reversion. These terms are ‘foreign’ to the conceptual
arsenal developed in the reductionist current paradigm within which
cancer has been interpreted.
Fifth, new techniques such as single-cell
transcriptomics or cell state transition assessment and regulation
(cSTAR) may offer important cues on understanding the
malignant-benign dynamic processes of cell state transition
(123). Sixth, numerous questions
remain open, such as those related to the nature of these
‘transformed’ cancer cells, their stability, the biological
pathways that lead to these transformations, and the specificity of
the interactions between cancer cells and the microenvironment. In
fact, not all types of cancer cells respond to embryonic signals
and, in light of the heterogeneity of the tumor masses, it is
logical to ask how a renormalization of the entire tumor complexity
can occur. Furthermore, it is worth mentioning that the most
relevant results in tumor reversion experiments derive from the
exposure of cancer cells to a raw extract of the embryo
microenvironment. In this form, it is difficult to standardize and
industrialize the extracts. Issues such as pharmacodynamics,
bioavailability and stability are needed for a clinical translation
of these approaches.
Seventh, differentiation therapies represent a
promising research field for cancer treatments but the way remains
long. Several issues should be addressed, not only of a scientific
nature, but also technological, industrial and probably regulatory.
Do these extracts work in their entirety or there are few active
molecules? How should an oocyte extract be juridically classified?
What kind of patent is allowed regarding a natural extract? How to
measure pharmacodynamics, bioavailability and stability? Eighth,
occasionally scientific research opens new ways that are still not
integrated under the socio-economic framework and this can
represent a further obstacle to the research development.
Scientists (and pioneers) should therefore acquire a systemic view
not only for their specific research model but also for a wider and
more effective integration of their work into society.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AP carried out the research and classification of
the material, wrote the initial manuscript and draw the figures. M
Be and MBi contributed to the organization of the work and revising
the manuscript. All authors contributed to the manuscript through
revisions of earlier versions, and read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Recamier JCA: Recherches sur le Traitement
du Cancer, etc. Chez Gabo; Paris: 1829
|
|
2
|
Oberling CH: The Riddle of Cancer. Yale
University Press; New Haven: pp. p1961944
|
|
3
|
Oberling C: The riddle of cancer. Yale
University Press; New Haven: pp. 26–27. 1946
|
|
4
|
Müller J: Über den feinern Bau und die
Formen der krankhaften Geschwülste. G. Reimer, Berlin. Nat Cancer
Inst Mnogr Spontaneous Regression Cancer. 1976:441838.
|
|
5
|
Virchow R: Editoral Archiv fuer
pathologische Anatomie und Physiologie und fuer klinische. Medizin.
8:231855.
|
|
6
|
Virchow R: Cellular Pathology. Hirschwald
A: August Hirschwald; Berlin: 1858, PubMed/NCBI
|
|
7
|
Durante F: Nesso fisiopatologico tra la
struttura dei nei materni e la genesi di alcuni tumori maligni.
Arch Memorie ed Osservazioni di Chirurgia Pratica. 1874:217–226.
1874.
|
|
8
|
Cohnheim J: Congenitales, quergestreiftes
muskelsarkon der nireren. Virchows Arch. 65:64–69. 1875. View Article : Google Scholar
|
|
9
|
Wilms M: Die Mischgeschwuelste. Leipzing;
Arthur Georgi: 1899
|
|
10
|
Ribbert H: Ueber Rückbildung an Zellen und
Geweben und über die Entstehung der Geschwülste. Erwin Nägele;
Stuttgart: 1897
|
|
11
|
Ribbert, op. cit., Rückbildung (note 51).
pp42–43, idem, op. cit., Beiträge (note 51). pp8–13, See also
Johach, op. cit. (note 11). pp246–267
|
|
12
|
Soto AM, Maffini MV and Sonnenschein C:
Neoplasia as development gone awry: The role of endocrine
disruptors. Int J Androl. 31:288–293. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Askanazy M: Die Teratome nach ihrem Bau,
ihrem Verlauf, ihrer Genese und im Vergleich zum experimentellen
Teratoid. Verhandl Deutsch Pathol. 11:39–82. 1907.
|
|
14
|
Stevens LC and Little CC: Spontaneous
testicular teratomas in an inbred strain of mice. Proc Natl Acad
Sci USA. 40:1080–1087. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pierce GB and Dixon FJ: Testicular
teratomas: I. The demonstration of teratogenesis by metamorphosis
of multipotent cells. Cancer. 12:573–583. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pierce GB and Verney EL: An in vitro and
in vivo study of differentiation in teratocarcinomas. Cancer.
14:1017–1029. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brinster RL: The effect of cells
transferred into the mouse blastocyst on subsequent development. J
Exp Med. 140:1049–1056. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mintz B and Illmensee K: Normal
genetically mosaic mice produced from malignant teratocarcinoma
cells. Proc Natl Acad Sci USA. 72:3585–3589. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grobstein C: The differentiation of such
tissues may depend on inductive interactions between embryonic
components. 13th Symposium of the Society for Development and
Growth. Rudnick D: Princeton University Press; Princeton, NJ: pp.
233–256. 1954
|
|
20
|
Rous P: A Sarcoma of the Fowl
Transmissible by an Agent Separable from the Tumor Cells. J Exp
Med. 13:397–411. 1911. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Macpherson I: Reversion in hamster cells
transformed by Rous sarcoma virus. Science. 148:1731–1733. 1965.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pollack RE, Green H and Todaro GJ: Growth
control in cultured cells: Selection of sublines with increased
sensitivity to contact inhibition and decreased tumor-producing
ability. Proc Natl Acad Sci USA. 60:126–133. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duran-Reynals F and Milford JJF: Growth of
a chicken sarcoma virus in the chick embryo in the absence of
neoplasia. Cancer Res. 3:578–584. 1943.
|
|
24
|
Dolberg DS and Bissell MJ: Inability of
Rous sarcoma virus to cause sarcomas in the avian embryo. Nature.
309:552–556. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Braun AC: Bacterial and host factors
concerned in determining tumor morphology in crown gall. Bot Gaz.
114:363–371. 1953. View
Article : Google Scholar
|
|
26
|
Braun AC: A Demonstration of the recovery
of the crown-gall tumor cell with the use of complex tumors of
single-cell origin. Proc Natl Acad Sci USA. 45:932–938. 1959.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rose SM: Epidermal dedifferentiation
during blastema formation in regeneration limbs of Triturus
viridescens. J Exp Zool. 108:337–362. 1948. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wallingford HM: Transformations of renal
tumors to normal tissue in regenerating limbs of salamanders.
Science. 107:4571948.PubMed/NCBI
|
|
29
|
Gersch M: Zellentartung und Zellwucherung
bei wirbellosen Tieren. Arch. Geschwulst-Forschung. 3:1–18.
1951.
|
|
30
|
Waddington CH: Cancer and the theory of
organizers. Nature. 135:606–608. 1935. View Article : Google Scholar
|
|
31
|
Needham J: New advances in the chemistry
and biology of organized growth. Proc R Soc London B Biol Sci.
29:1577–1626. 1936.PubMed/NCBI
|
|
32
|
Seilern-Aspang F and Kratochwil K:
Induction and differentiation of an epithelial tumour in the newt
(Triturus cristatus). J Embryol Exp Morphol. 10:337–356.
1962.PubMed/NCBI
|
|
33
|
McMichael H: Inhibition of growth of Shope
rabbit papilloma by hypervitaminosis A. Cancer Res. 25:947–955.
1965.PubMed/NCBI
|
|
34
|
Saffiotti J, Montesano R, Sellakumar AR
and Borg SA: Experimental cancer of the lung, inhibition by vitamin
a of the induction of tracheobronchial squamous metaplasia and
squamous cell tumors. Cancer. 20:857–864. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Davies RE: Effect of vitamin A on 7,
12-di-methylbenz(alpha) anthracene-induced papillomas in rhino
mouse skin. Cancer Res. 27:237–241. 1967.PubMed/NCBI
|
|
36
|
Coleman WB, Wennerberg AE, Smith GJ and
Grisham JW: Regulation of the differentiation of diploid and some
aneuploid rat liver epithelial (stemlike) cells by the hepatic
microenvironment. Am J Pathol. 142:1373–1382. 1993.PubMed/NCBI
|
|
37
|
Pierce GB: The cancer cell and its control
by the embryo. Rous-Whipple Award lecture. Am J Pathol.
113:115–124. 1983.
|
|
38
|
Pierce GB, Lewis SH, Miller GJ, Moritz E
and Miller P: Tumorigenicity of embryonal carcinoma as an assay to
study control of malignancy by the murine blastocyst. Proc Natl
Acad Sci USA. 76:6649–6651. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pierce GB, Pantazis CG, Caldwell JE and
Wells RS: Specificity of tumor formation by the blastocyst. Cancer
Res. 42:1082–1087. 1982.PubMed/NCBI
|
|
40
|
Wells RS: An in vitro assay for regulation
of embryonal carcinoma by the blastocyst. Cancer Res. 42:2736–2741.
1982.PubMed/NCBI
|
|
41
|
Podesta A, Beddington RSP, Wells RS and
Pierce GB: The neurula stage mouse embryo in control of
neuroblastoma. Proc Natl Acad Sci USA. 81:7608–7611. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Podesta AN, Mullins J, Pierce GB and Sells
RS: The neurula state mouse embryos in control of neuroblastomas.
Proc Natl Acad Sci USA. 81:7608–7611. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gootwine E, Webb CG and Sachs L:
Participation of myeloid leukaemia cells injected into embryos in
haematopoietic differentiation in adult mice. Nature. 299:63–65.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gerschenson M, Graves K, Carson SD, Wells
RS and Pierce GB: Regulation of melanoma by the embryonic skin.
Proc Natl Acad Sci USA. 83:7307–7310. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pierce GB, Aguilar D, Hood G and Wells RS:
Trophectoderm in control of murine embryonal carcinoma. Cancer Res.
44:3987–3996. 1984.PubMed/NCBI
|
|
46
|
DeCosse JJ, Gossens CL, Kuzma JF and
Unsworth BR: Breast cancer: Induction of differentiation by
embryonic tissue. Science. 181:1057–1058. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Biava PM, Fiorito A, Negro C and Mariani
M: Effects of treatment with embryonic and uterine tissue
homogenates on Lewis lung carcinoma development. Cancer Lett.
41:265–270. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Biava PM, Bonsignorio D and Hosha M: Cell
proliferation curves of different human tumor lines after in vitro
treatment with Zebrafish embryonic extracts. J Tumor Marker Oncol.
16:195–201. 2001.
|
|
49
|
Biava PM and Bonsignorio D: Cancer and
cell differentiation: A model to explain malignancy. J Tumor Marker
Oncol. 17:47–53. 2002.
|
|
50
|
Lee LM, Seftor EA, Bonde G, Cornell RA and
Hendrix MJ: The fate of human malignant melanoma cells transplanted
into zebrafish embryos: Assessment of migration and cell division
in the absence of tumour formation. Dev Dyn. 233:1560–1570. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cucina A, Biava PM, D'Anselmi F, Coluccia
P, Conti F, di Clemente R, Miccheli A, Frati L, Gulino A and
Bizzarri M: Zebrafish embryo proteins induce apoptosis in human
colon cancer cells (Caco2). Apoptosis. 11:1617–1628.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pierce GB and Johnson LD: Differentiation
and cancer. In Vitro. 7:140–145. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pierce GB and Wallace C: Differentiation
of malignant to benign cells. Cancer Res. 31:127–134.
1971.PubMed/NCBI
|
|
54
|
Kenny PA and Bissell MJ: Tumor reversion:
Correction of malignant behavior by microenvironmental cues.
International J Cancer. 107:688–695. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Camacho LH: Clinical application of
retinoids in cancer medicine. J Biol Regul Homeost Agents.
17:98–114. 2003.PubMed/NCBI
|
|
56
|
Pitha-Rowe I, Petty WJ, Kitareewan S and
Dmitrovsky E: Retinoid target genes in acute promyelocytic
leukemia. Leukemia. 17:1723–1730. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Segalla S, Rinaldi L, Kilstrup-Nielsen C,
Badaracco G, Minucci S, Pelicci PG and Landsberger N: Retinoic acid
receptor alpha fusion to PML affects in transcriptional and
chromatin-remodeling properties. Mol Cell Biol. 23:8795–808. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Alcalay M, Meani N, Gelmetti V, Fantozzi
A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E,
Casciari C, et al: Acute myeloid leukemia fusion proteins
deregulate genes involved in stem cell maintenance and DNA repair.
J Clin Invest. 112:1751–1761. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Strickland S and Madavi V: The induction
of differentiation in teratocarcinoma stem cells by retinoic acid.
Cell. 15:393–403. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Trump DL: Retinoids in bladder, testes and
prostate cancer: Epidemiologic, preclinical and clinical
observations. Leukemia. 8 (Suppl 3):S50–S54. 1994.PubMed/NCBI
|
|
61
|
Breitman TR, Selonick SE and Collins SJ:
Induction of differentiation of the human promyelocytic leukemia
cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA.
77:2936–2940. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Huang ME, Ye YC, Chen SR, Chai JR, Lu JX,
Zhoa L, Gu LJ and Wang ZY: Use of all-trans retinoic acid in the
treatment of acute promyelocytic leukemia. Blood. 72:567–572. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dragnev KH, Petty WJ and Dmitrovsky E:
Retinoid targets in cancer therapy and chemoprevention. Cancer Biol
Ther. 2 (Suppl 1):S150–S156. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Melnick A and Licht JD: Deconstruction a
disease: RARalpha, its fusion partners, and their roles in the
pathogenesis of acute promyelocytic leukemia. Blood. 99:3167–3215.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Warrell RP Jr, Frankel SR, Miller WH Jr,
Scheinberg DA, Itri LM, Hittelman WN, Vyas R, Andreeff M, Tafuri A
and Jakubowski A: Differention therapy of acute promyelocytic
leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med.
324:1385–1393. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 6127. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bertolaso M: Philosophy of Cancer-A
Dynamic and Relational View. Springer; New York, NY: 2016,
PubMed/NCBI
|
|
68
|
Rohdenburg GL: Fluctuations in the growth
of malignant tumors in man, with especial reference to spontaneous
regression. J Cancer Res. 3:192–221. 1918.
|
|
69
|
Cushing H and Wollbach S: The
transformation of malignant paravertebral Sympathicoblastoma into a
benign ganglioneuroma. Am J Pathol. 3:203–216.7. 1927.PubMed/NCBI
|
|
70
|
Bumpus HC: The apparent disappearance of
pulmonary metastasis in a case of hypernephroma following
nephrectomy. J Urol. 20:185–191. 1927. View Article : Google Scholar
|
|
71
|
Everson TC and Cole WH: Spontaneous
Regression of Cancer. W.B Saunders; Philadelphia, PA: 1966,
PubMed/NCBI
|
|
72
|
Cole WH: Spontaneous regression of cancer
and the importance of finding its cause. Nat Cancer Inst Mnogr.
44:5–9. 1976.PubMed/NCBI
|
|
73
|
Challis GB and Stam HJ: The spontaneous
regression of cancer. A review of cases from 1900 to 1987. Acta
Oncol. 29:545–549. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
O'Regan B and Hirschberg C: Spontaneous
Regression. An Annotated Bibliography Sausalito CA: Institute of
Noetic Science; 1993
|
|
75
|
Papac RJ: Spontaneous regression of
cancer: Possible mechanisms. In Vivo. 12:571–578. 1998.PubMed/NCBI
|
|
76
|
Livraghi T, Meloni F, Frosi A, Lazzaroni
S, Bizzarri TM, Frati L and Biava PM: Treatment with stem cell
differentiation stage factors of intermediate-advanced
hepatocellular carcinoma: An open randomized clinical trial. Oncol
Res. 15:399–408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Telerman A, Tuynder M, Dupressoir T,
Robaye B, Sigaux F, Shaulian E, Oren M, Rommelaere J and Amson R: A
model for tumor suppression using H-1 parvovirus. Proc Natl Acad
Sci USA. 90:8702–8706. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tuynder M, Susini L, Prieur S, Besse S,
Fiucci G, Amson R and Telerman A: Biological models and genes of
tumor reversion: Cellular reprogramming through tpt1/TCTP and
SIAH-1. Proc Natl Acad Sci USA. 99:14976–1481. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Telerman A and Amson R: The molecular
programme of tumour reversion: The steps beyond malignant
transformation. Nat Rev Cancer. 9:206–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tuynder M, Fiucci G, Prieur S, Lespagnol
A, Géant A, Beaucourt S, Duflaut D, Besse S, Susini L, Cavarelli J,
et al: Translationally controlled tumor protein is a target of
tumor reversion. Proc Natl Acad Sci USA. 101:15364–15369. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Thaw P, Baxter NJ, Hounslow AM, Price C,
Waltho JP and Craven CJ: Structure of TCTP reveals unexpected
relationship with guanine nucleotide-free chaperones. Nat Struct
Biol. 8:701–704. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
82
|
Proietti S, Cucina A, Pensotti A, Biava
PM, Minini M, Monti N, Catizone A, Ricci G, Leonetti E, Harrath AH,
et al: Active fraction from embryo fish extracts induces reversion
of the malignant invasive phenotype in breast cancer through
down-regulation of TCTP and modulation of E-cadherin/β-catenin
pathway. Int J Mol Sci. 20:21512019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Weaver VM, Petersen OW, Wang F, Larabell
CA, Briand P, Damsky C and Bissell MJ: Reversion of the malignant
phenotype of human breast cells in three-dimensional culture and in
vivo by integrin blocking antibodies. J Cell Biol. 137:231–245.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hendrix MJ, Seftor EA, Seftor RE,
Kasemeier-Kulesa J, Kulesa PM and Postovit LM: Reprogramming
metastatic tumour cells with embryonic microenvironments. Nat Rev
Cancer. 7:246–255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tabibzadeh S and Hemmati-Brivanlou A:
Lefty at the crossroads of ‘stemness’ and differentiative events.
Stem Cells. 24:1998–2006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Postovit LM, Margaryan NV, Seftor EA,
Kirschmann DA, Lipavsky A, Wheaton WW, Abbott DE, Seftor RE and
Hendrix MJ: Human embryonic stem cell microenvironment suppresses
the tumorigenic phenotype of aggressive cancer cells. Proc Natl
Acad Sci USA. 105:4329–4334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Topczewska JM, Postovit LM, Margaryan NV,
Sam A, Hess AR, Wheaton WW, Nickoloff BJ, Topczewski J and Hendrix
MJ: Embryonic and tumorigenic pathways converge via Nodal
signaling: Role in melanoma aggressiveness. Nat Med. 12:925–932.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
88
|
Costa FF: Non-coding RNAs: Lost in
translation? Gene. 386:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Garzon R, Fabbri M, Cimmino A, Calin GA
and Croce CM: MicroRNA expression and function in cancer. Trends
Mol Med. 12:580–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Costa FF, Seftor EA, Bischof JM,
Kirschmann DA, Strizzi L, Arndt K, Bonaldo Mde F, Soares MB and
Hendrix MJ: Epigenetically reprogramming metastatic tumor cells
with an embryonic microenvironment. Epigenomics. 1:387–398. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Card DA, Hebbar PB, Li L, Trotter KW,
Komatsu Y, Mishina Y and Archer TK: Oct4/Sox2-regulated miR-302
targets cyclin D1 in human embryonic stem cells. Mol Cell Biol.
28:6426–6438. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Krebs LT, Iwai N, Nonaka S, Welsh IC, Lan
Y, Jiang R, Saijoh Y, O'Brien TP, Hamada H and Gridley T: Notch
signaling regulates left-right asymmetry determination by inducing
Nodal expression. Genes Dev. 17:1207–1212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Morgan DO: Cyclin-dependent kinases:
Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Giuffrida D, Rogers IM, Nagy A, Calogero
AE, Brown TJ and Casper RF: Human embryonic stem cells secrete
soluble factors that inhibit cancer cell growth. Cell Prolif.
42:788–798. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Novak P, Jensen TJ, Garbe JC, Stampfer MR
and Futscher BW: Stepwise DNA methylation changes are linked to
escape from defined proliferation barriers and mammary epithelial
cell immortalization. Cancer Res. 69:5251–5258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hinshelwood RA and Clark SJ: Breast cancer
epigenetics: Normal human mammary epithelial cells as a model
system. J Mol Med. 86:1315–1328. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Allegrucci C, Rushton MD, Dixon JE,
Sottile V, Shah M, Kumari R, Watson S, Alberio R and Johnson AD:
Epigenetic reprogramming of breast cancer cells with oocyte
extracts. Mol Cancer. 10:72011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Saad N, Alberio R, Johnson AD, Emes RD,
Giles TC, Clarke P, Grabowska AM and Allegrucci C: Cancer reversion
with oocyte extracts is mediated by cell cycle arrest and induction
of tumour dormancy. Oncotarget. 9:16008–16027. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tripathi A, Kashyap A, Tripathi G, Yadav
J, Bibban R, Aggarwal N, Thakur K, Chhokar A, Jadli M, Sah AK, et
al: Tumor reversion: A dream or a reality. Biomark Res. 9:312021.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Feinberg AP: Phenotypic plasticity and the
epigenetics of human disease. Nature. 447:433–440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Redmer T, Diecke S, Grigoryan T,
Quiroga-Negreira A, Birchmeier W and Besser D: E-cadherin is
crucial for embryonic stem cell pluripotency and can replace OCT4
during somatic cell reprogramming. EMBO Rep. 12:720–726. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Feng B, Ng JH, Heng JC and Ng HH:
Molecules that promote or enhance reprogramming of somatic cells to
induced pluripotent stem cells. Cell Stem Cell. 4:301–312. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Smith ZD, Sindhu C and Meissner A:
Molecular features of cellular reprogramming and development. Nat
Rev Mol Cell Biol. 17:139–154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yoo J and Kim J, Baek S, Park Y, Im H and
Kim J: Cell reprogramming into the pluripotent state using graphene
based substrates. Biomaterials. 35:8321–8329. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bizzarri M, Palombo A and Cucina A:
Theoretical aspects of systems biology. Prog Biophys Mol Biol.
112:33–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Abad M, Mosteiro L, Pantoja C, Cañamero M,
Rayon T, Ors I, Graña O, Megías D, Domínguez O, Martínez D, et al:
Reprogramming in vivo produces teratomas and iPS cells with
totipotency features. Nature. 502:340–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ohnishi K, Semi K, Yamamoto T, Shimizu M,
Tanaka A, Mitsunaga K, Okita K, Osafune K, Arioka Y, Maeda T, et
al: Premature termination of reprogramming in vivo leads to cancer
development through altered epigenetic regulation. Cell.
156:663–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Inman JL, Robertson C, Mott JD and Bissell
MJ: Mammary gland development: Cell fate specification, stem cells
and the microenvironment. Development. 142:1028–1042. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Bizzarri M and Giuliani A: Representing
cancer cell trajectories in a phase-space diagram: Switching
cellular states by biological phase transitions. Applied Statistics
for Network Biology: Methods in Systems Biology. Dehmer M,
Emmert-Streib F, Graber A and Salvador A: Wiley-VCH Verlag GmbH
& Co.; pp. 377–403. 2011, View Article : Google Scholar
|
|
112
|
Lin SL, Chang DC, Chang-Lin S, Lin CH, Wu
DT, Chen DT and Ying SY: Mir-302 reprograms human skin cancer cells
into a pluripotent ES-cell-like state. RNA. 14:2115–2124. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Utikal J, Maherali N, Kulalert W and
Hochedlinger K: Sox2 is dispensable for the reprogramming of
melanocytes and melanoma cells into induced pluripotent stem cells.
J Cell Sci. 122:3502–3510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Rapino F, Robles EF, Richter-Larrea JA,
Kallin EM, Martinez-Climent JA and Graf T: C/EBPα induces highly
efficient macrophage transdifferentiation of B lymphoma and
leukemia cell lines and impairs their tumorigenicity. Cell Rep.
3:1153–1163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Huang P, Zhang L, Gao Y, He Z, Yao D, Wu
Z, Cen J, Chen X, Liu C, Hu Y, et al: Direct reprogramming of human
fibroblasts to functional and expandable hepatocytes. Cell Stem
Cell. 14:370–384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
McClellan JS, Dove C, Gentles AJ, Ryan CE
and Majeti R: Reprogramming of primary human Philadelphia
chromosome-positive B cell acute lymphoblastic leukemia cells into
nonleukemic macrophages. Proc Natl Acad Sci USA. 112:4074–4079.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhou S, Abdouh M, Arena V, Arena M and
Arena GO: Reprogramming malignant cancer cells toward a benign
phenotype following expo-sure to human embryonic stem cell
microenvironment. PLoS One. 12:e01698992017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ishay-Ronen D, Diepenbruck M, Kalathur
RKR, Sugiyama N, Tiede S, Ivanek R, Bantug G, Morini MF, Wang J,
Hess C and Christofori G: Gain fat-lose metastasis: Converting
invasive breast cancer cells into adipocytes inhibits cancer
metastasis. Cancer Cell. 35:17–32.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Cheng Z, He Z, Cai Y, Zhang C, Fu G, Li H,
Sun W, Liu C, Cui X, Ning B, et al: Conversion of hepatoma cells to
hepatocyte-like cells by defined hepatocyte nuclear factors. Cell
Res. 29:124–135. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Li Y, Agrawal I and Gong Z: Reversion of
tumor hepatocytes to normal hepatocytes during liver tumor
regression in an oncogene-expressing transgenic zebrafish model.
Dis Model Mech. 12:dmm0395782019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Pensotti A, Bertolaso M and Bizzarri M: Is
cancer reversible? Rethinking carcinogenesis models-a new
epistemological tool. Biomolecules. 13:7332023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Longo G, Miquel PA, Sonnenschein C and
Soto AM: Is information a proper observable for biological
organization? Prog Biophys Mol Biol. 109:108–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kholodenko BN, Kolch W and Rukhlenko OS:
Reversing pathological cell states: The road less travelled can
extend the therapeutic horizon. Trends Cell Biol. 33:913–923. 2023.
View Article : Google Scholar : PubMed/NCBI
|