Introduction
Globally, lung cancer has emerged as the predominant
malignant tumor with the highest rates of incidence and mortality,
experiencing a notable increase in the past 50 years (1). The classification of this cancer
predominantly includes non-small cell lung cancer (NSCLC) and small
cell lung cancer (SCLC). Among NSCLC types, lung adenocarcinoma
(LUAD) emerges as the most commonly identified pathological
subtype, accounting for ~40% of all lung cancer diagnoses, with an
incidence that continues to escalate (2). LUAD, which often lacks distinct early
clinical manifestations, tends to exhibit high metastatic capacity
from the onset, leading to a grim prognosis with 5-year survival
rates <20% (3,4). Despite the significant advances in
molecularly-targeted therapies, the long-term outcomes for LUAD
remain suboptimal (5). Identifying
unique tumor markers and novel therapeutic strategies, enhancing
early detection through systematic screening, and fostering the
development of precise and early intervention treatments are
imperative to enhance survival outcomes for patients with LUAD.
The armadillo protein family includes several
plakophilins (PKPs), specifically PKP1, PKP2 and PKP3, which are
integral to its structure. These proteins are involved in the
formation of bridging granules, playing a pivotal role in promoting
intercellular adhesion and communication. Alterations in PKP3
expression are implicated in various human pathologies,
particularly cancers (6). Emerging
studies highlight that the function of PKP3 can oscillate between a
tumor suppressor role and oncogenic activity, depending on the
stage and type of the cancer. Notably, heightened levels of PKP3
are associated with an enhancement in cell proliferation, migration
and invasion in specific types of cancer such as pancreatic,
ovarian and NSCLC (7–9). Conversely, a reduction in PKP3
expression in colon and bladder cancers correlates with poorer
prognoses (10,11). These findings indicate the dual and
variable impact of PKP3 within different cancer contexts. Previous
studies have shown that elevated PKP3 levels correlate with adverse
outcomes in NSCLC, enhancing tumor cell migration, invasion and
immune escape (9,12–14).
However, the specific mechanisms through which PKP3 influences
LUAD, a prevalent NSCLC subtype, remain to be elucidated.
Programmed cell death (PCD) is governed by genetic
regulation. Traditionally, apoptosis, which relies on cysteine
asparaginase, was regarded as the sole PCD pathway. As research
progresses, more types of PCD are gradually being identified,
including necroptosis (15).
Fundamentally differing from apoptosis, necroptosis is independent
of cysteine asparaginase and demonstrates characteristics similar
to those of necrosis, including increased cellular volume, swelling
of organelles, rupture of the plasma membrane, and the subsequent
expulsion of cellular constituents (16). The regulation of necroptosis is
stringent, requiring the activation of receptor-interacting protein
(RIP) kinases such as RIPK1 and RIPK3, along with the involvement
of the mixed lineage kinase domain-like protein (MLKL), which acts
as a substrate for RIPK3 (17).
This mode of cell death releases damage-associated molecular
patterns, which enhance antitumor immune responses and may trigger
further immune functions. Evidence increasingly supports the idea
that modulation of the necroptosis signaling pathway could provide
a viable strategy for cancer therapy. Nonetheless, cancer cells
often enhance their survival by modulating genes associated with
necroptosis, such as by reducing RIPK3 expression levels. The exact
regulatory mechanisms governing necroptosis in LUAD remain
unclear.
MicroRNAs (miRNAs or miRs) are small RNA molecules
that lack coding capability and exhibit significant conservation
across species. They play a crucial role in modulating diverse
cellular activities, either by regulating protein synthesis or
facilitating the degradation of mRNA (18). These miRNAs participate in the
regulation of PCD, which includes apoptosis, autophagy and
necroptosis. However, the literature provides limited details on
the specific mechanisms through which miRNAs influence necroptotic
cell death (19). According to
studies conducted by Harari-Steinfeld et al (20), miR-675 enhances necroptosis in
hepatocellular carcinoma cells by inhibiting the Fas-associated
protein with death domain (FADD), a key mediator in apoptotic
signaling. By contrast, research by Li et al (21) indicated that miR-204-3p reduces
necroptosis pathways, specifically MAPK and RIP1/MLK1, leading to
decreased proliferation and increased apoptosis in gastric cancer
cells. Elevated levels of miR-10b-5p have increasingly been
associated with worse patient outcomes in various cancers,
including gastric and hepatocellular carcinomas, as well as glioma
(22–24). However, the impact of miR-10b-5p on
necroptosis pathways in tumor cells remains unexplored.
The current investigation provides initial evidence
through both in vivo and ex vivo studies that
miR-10b-5p acts as an oncogene. It inhibits the activation of the
RIPK3/MLKL signalling pathway by directly targeting PKP3, thereby
promoting tumour proliferation and inhibiting necroptosis
development. Notably, reducing miR-10b-5p levels curbed the
progression of LUAD by facilitating necroptosis. These findings
elucidate the molecular mechanisms underlying LUAD and highlight
miR-10b-5p as a promising target for innovative therapeutic
strategies.
Materials and methods
Tissue samples
Over the course of 1 year, from July 2022 to July
2023, the Department of Thoracic Surgery at The Second Affiliated
Hospital of Kunming Medical University (KMUH) collected tissue
samples from six patients diagnosed with LUAD and were receiving
treatment, three of whom were female and three of whom were >60
years old. These samples included both cancerous tissues and
adjacent non-tumorous tissues. Each specimen was confirmed as LUAD
through histopathological evaluation. After collection, all samples
were immediately immersed in liquid nitrogen to ensure rapid
freezing and were then stored at −80°C for further examination. The
research protocols adhered to the ethical guidelines of the
Declaration of Helsinki and were approved by the KMUH Ethics
Committee (approval no. PJ-2024-125; Kunming, China). Written
informed consent was obtained from all participants.
Cell lines and cell culture
Both A549 and 293FT cell line was procured from
Brick Bio (https://www.brickbio.com), and
cultured using F-12K and DMEM medium (Gibco; Thermo Fisher
Scientific, Inc). This medium was supplemented with fetal bovine
serum at a 10% concentration and a 1% solution of
penicillin-streptomycin, with both additives sourced from Gibco;
Thermo Fisher Scientific, Inc. The protocol required the exclusive
use of F-12K medium. After 293FT cells were used as tool cells for
lentiviral packaging, they were cultured in IMDM medium containing
10% fetal bovine serum and 1% PSG. Both types of cells were
cultured in an incubator at 5% CO2 concentration and
37°C. Selection for further experimental use was restricted to
cells that demonstrated vigorous growth during the logarithmic
phase.
MiRNA differential expression
analysis
Fresh LUAD tissues, along with their matched
non-cancerous counterparts, were procured and classified into two
categories: One comprising LUAD tissues and the other consisting of
the adjacent non-malignant tissues. Three samples from each group
were randomly selected for sequencing. The tissue samples were cut
into fragments <0.5 cm, stored in an insulated container filled
with dry ice, and dispatched to Shanghai Ouyi Biomedical Technology
Co., Ltd. for processing. RNA extraction, quantification and
quality assessment were performed by the same company. Following
successful library quality checks, sequencing was carried out on
the Small RNA platform (illumina Hiseq X ten, illumina Co., Ltd.).
Differentially expressed miRNAs (DEmiRNAs) between LUAD and
paracancerous tissues were identified using the ‘Lima’ software
package in R (https://CRAN.R-project.org/package=limma), based on
the sequencing data with |log2FC| ≥2 and FDR<0.05 as selection
criteria. The ‘ggplot2’ package was employed to create a volcano
plot of these DEmiRNAs. Multiple databases, including TargetScan
Human 7.2 (https://www.targetscan.org/vert_72/), Starbase
(https://rnasysu.com/encori/), miRWalk
(http://mirwalk.umm.uni-heidelberg.de/) and miRmap
(https://mirmap.ezlab.org/), were used to
identify miRNAs that potentially bind to the PKP3 target. miRNAs
confirmed in each database as having a target-binding relationship
were cross-referenced with those identified through sequencing.
Cell transfection
From the 3rd to 5th generations post-recovery, A549
cells exhibiting robust growth were selected for experiments. The
cells were dissociated into a single-cell suspension using trypsin
digestion, followed by one to two phosphate-buffered saline (PBS)
washes and subsequent cell quantification. Prior to plating, the
concentration of the cell suspension was adjusted to
2×105/ml, and the suspension was then dispensed into
24-well plates to ensure a confluence of 30–50% at the onset of
transfection. During the transfection process, a miRNA mimic
(UACCCUGUAGAACCGAAUUUGUG) was first diluted by mixing 1.25 µl of a
20 µM stock solution into 30 µl of 1X riboFECT™ CP Buffer with
gentle stirring. A total of 3 µl of riboFECT™ CP Reagent (Guangzhou
RiboBio Co., Ltd.) were introduced into the solution, followed by
gentle pipetting to mix. The mixture was allowed to stand at room
temperature for 15 min. Afterward, the mixture was combined with
serum-free medium, and the setup was incubated at 37°C within a
CO2 incubator for 48 h to facilitate subsequent
experimental procedures.
PKP3 lentiviral transfection was performed using
293T cells, interfering lentivirus and PGMLV-ZsGreen1-Puro vector
control. Interfering lentivirus and PGMLV-ZsGreen1-Puro vector
control was purchased from Honorgene (https://www.honorgene.com). The lentivirus was
constructed by Shanghai Jiman Biotechnology Co. Ltd. using a
second-generation viral packaging system, and the lentiviruses,
packaging plasmids, and envelope plasmids used were vsvg, gag, and
rev, respectively. In a 6-well plate, A549 cells were seeded at a
density of 3×105 cells per well. Upon achieving ~70%
confluence, the original culture medium was replaced with 1 ml of
fresh medium containing 8 µg/ml of polyethylene glycol, sourced
from Beijing Solarbio Science & Technology Co., Ltd.
Concurrently, 10 µg of lentiviral plasmid was added to each well to
achieve multiplicity of infection (MOI)=5. After 72 h of infection
at 37°C, cells expressing green fluorescent protein were visible
under a fluorescence microscope. The medium was then replaced with
selection medium containing 4 µg/ml puromycin from Beijing Solarbio
Science & Technology Co., Ltd., which allowed for 1–2
generations of selection to establish stable cell lines. The
cultures were propagated at a constant temperature of 37°C within a
cell culture incubator maintained at 5% CO2 and optimal
humidity levels.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated employing TRIzol reagent
(Thermo Fisher Scientific, Inc.). Spectrophotometry was utilized to
assess the purity and concentration of the extracted nucleic acids.
For converting RNA into cDNA, the following kits were used
according to the manufacturers' protocols: the FastKing RT Kit
(With gDNase) from Tiangen Biotech Co., Ltd., or the Bulge-Loop
miRNA RT-qPCR Starter Kit (Guangzhou RiboBio Co., Ltd.,) were
selected. For RT-qPCR analysis targeting both mRNA and miRNA, the
Taq Pro Universal SYBR qPCR Master Mix and the SYBR Green Mix
(Invitrogen; Thermo Fisher Scientific, Inc.) were applied.
According to the manufacturer's instructions, thermocycling
conditions were as follows: mRNA, pre-denaturation at 95°C for 30
sec, 40 cycles at 95°C for 10 sec, 60°C for 30 sec; miRNA:
pre-denaturation at 95°C for 10 min, 40 cycles at 95°C for 2 sec,
60°C for 20 sec, 70°C for 10 sec. The primer sequences are detailed
in Table SI. The relative gene
expression was quantified using the 2−ΔΔCq method
(25), with GAPDH serving as the
internal reference gene.
Immunoblotting
Tissue samples or cells from tumors were lysed using
RIPA buffer (Shanghai Beyotime Co., Ltd.), followed by
centrifugation to extract total proteins. Protein concentrations
were determined using an IMPLEN Ultra-Micro Spectrophotometer
(measuring absorbance at 280 nm). After quantification, proteins
were separated by SDS-PAGE through a 4% concentrate gel (adjust the
amount of protein sampled to 40 µg per lane) and transferred
electrophoretically to PVDF membranes (MilliporeSigma). The
membranes were blocked with 5% non-fat milk for 2 h at ambient
temperature, followed by overnight incubation at 4°C with primary
antibodies: PKP3 (1:2;000; cat. no. 109441), PCNA (1:2,000; cat.
no. 92552), Ki67 (1:1,000, cat. no. 16667), Bax (1:2,000; cat. no.
32503), Bcl-2 (1:2,000; cat. no. 182858), Caspase-3 (1:2,000; cat.
no. 184787; all from Abcam), RIPK3 (1:1:1,000, cat. no. 10188),
phosphorylated (p)-RIPK3 (1:1,000, 91702), MLKL (1:1,000; cat. No
14993), p-MLKL (1:1,000, cat. no. 18640), Caspase-8 (1:1,000, cat.
no. 9746; all from Cell Signaling Technology, Inc.) and GAPDH
(1:2,000, cat. no. OTI2D9; OriGene Technologies, Inc.). Membranes
were then incubated with HRP-conjugated secondary antibodies (goat
anti-rabbit; 1:4,000, cat. no. GB13063-50; Wuhan Servicebio
Technology Co., Ltd.; goat anti-mouse; 1:4,000, M21001S; Abmart
Pharmaceutical Technology Co., Ltd.) for 2 h at room temperature.
Protein bands were visualized using an Enhanced Chemiluminescence
(ECL) System Kit (Thermo Fisher Scientific, Inc.). Final analysis
was performed using ImageJ software (version 1.50b; National
Institutes of Health).
Dual-luciferase reporter assay
To investigate the specific interaction between
miR-10b-5p and the PKP3 3′-untranslated region (UTR), the region
predicted to contain the miR-10b-5p binding site was amplified and
subsequently cloned into the pGL3 control vector (Promega
Corporation). This construct was introduced into A549 cells via
co-transfection with either a miR-10b-5p mimic or a NC mimic using
Lipofectamine™ 3000 (Thermo Fisher Scientific, Inc.). Wild-type
(WT) and mutant (MUT) versions of the PKP3 reporter plasmid, which
included modifications at the miR-128 binding site, were generated
using the QuikChange II Targeted Mutation Kit (Agilent
Technologies, Inc.), following the manufacturer's protocol.
Relative luciferase activity was measured 48 h post-transfection
utilizing the dual luciferase reporter assay system from Promega
Corporation, following the protocol recommended by the
manufacturer. The experimental precision was upheld by comparing
the luminescence readings of firefly luciferase (fLuc) and
Renilla luciferase (rLuc), expressed as relative light
units. By utilizing Renilla luciferase as a control, the activation
of the target gene was analyzed by calculating the ratio of
luminescence between fLuc and rLuc across different samples.
Cell Counting Kit-8 (CCK-8)
To assess the viability of T cells, the CCK-8 assay
was utilized (Shanghai Beyotime Co., Ltd.). A549 cells were
subjected to 0.25% trypsin for digestion, collected, and the
supernatant was removed. The cells were resuspended in 1 ml of
complete medium. Following enumeration, the cells were plated at a
density of 5,000 cells per well in 96-well plates, with triplicate
samples for each experimental group. When cell confluence reached
~80%, transfections were carried out for 24 h. After this period,
10 µl of CCK-8 solution was added to each well. The plates were
then incubated in the dark at 37°C for 2 h. OD was measured at 450
nm using a microplate reader. Cell viability percentage was
calculated with the formula: (OD of experimental wells-OD of blank
wells)/(OD of control wells-OD of blank wells) ×100%.
Colony formation assay
Cells were inoculated into 6-well plates (500
cells/well) and cultured at 37°C, 5% CO2 and 90%
relative humidity for 2 weeks, and then fixed with 4%
paraformaldehyde (PFA) solution at ambient temperature (Shanghai
Aladdin Biochemical Technology Co., Ltd.) and stained with 0.1%
crystal violet staining solution at ambient temperature (Beijing
Solarbio Science & Technology Co., Ltd.) for 10 min. The
staining solution was then gently rinsed with tap water, and the
images were captured to record the cellular clone formation. When
each clone is >50 cells, it can be stained for observation and
subsequently images can be captured for manual counting. Rate of
clone formation (%) was determined using the following formula:
number of cell clones/number of inoculated cells ×100%.
Apoptosis detection
Apoptosis assays were performed using flow cytometry
techniques. A549 cells were enzymatically digested, and aliquots of
30,000 cells were dispensed into each well of 12-well plates.
Triplicate wells were prepared for each experimental condition.
After reaching the logarithmic phase growth, apoptosis was assessed
24 h post-transfection. Additionally, three control groups were
established: A blank control, a PI single positive control, and a
FITC single positive control. To facilitate resuspension, 400 µl of
Binding Buffer was added to each tube. Subsequently, 400 µl of
Binding Buffer was added to resuspend the cells. To the
experimental wells, 5 µl of FITC dye was added and incubated for 15
min, followed by the addition of 10 µl of PI dye. After an
additional 5-min incubation, apoptosis was quantified by flow
cytometry (Novocyte 2060Rl ACEA Bioscience, Inc.) using the
NovoExpress software (version 1.6.2; Agilent Technologies,
Inc.).
Tumor xenograft experiment
A total of 21 male BALB/c nude mice, 4–6 weeks old,
averaging 19.5 g, were obtained from Kunming Medical University
(Yunnan, China). The mice were housed in an SPF-grade sterile
laminar flow chamber, with a controlled humidity level of 55±5% and
temperatures ranging from 22–25≥C. They had free access to water
and were fed a standard laboratory diet. Cells were subjected to
trypsin treatment until they became rounded but remained attached
to the culture dish. After trypsin removal, a serum-free medium was
added to prepare a cell suspension. This suspension was then
centrifuged at at 1,200 × g for 5 min under ambient conditions,
rinsed once, and resuspended in PBS to a final concentration of
1×105 cells/ml. Each mouse received a subcutaneous
injection at a vascular-rich site on the upper right waist. After
the first injection, the mice were handled routinely following
three inoculations, with 1-day intervals between each. The weight
of the mice and the volume of subcutaneous tumors were documented;
the length and maximum diameter of the tumors were measured to plot
growth curves. A total of 5 weeks after inoculation, mice were
euthanized by cervical dislocation, subcutaneous tumours were
removed, weighed, and images were captured. Tumor dimensions were
carefully recorded before the samples were immediately preserved by
freezing in liquid nitrogen, then stored at −80°C for subsequent
molecular analyses. A subset of these tumor samples was randomly
selected for further analysis; half were fixed for 1 h at ambient
temperature in 2.5% glutaraldehyde for electron microscopy, while
the remainder were fixed for 30 min at ambient temperature in 4%
paraformaldehyde for morphological studies. The experimental
protocol was approved by the KMUH Animal Care Committee (approval
no. kyfeyxm2024127; Kunming, China).
TUNEL staining
Sections of paraffin-embedded subcutaneous graft
tumor samples, measuring 4 µm in thickness, underwent
deparaffinization with xylene followed by rehydration via graduated
ethanol solutions. Proteinase K, free of DNase and at a
concentration of 20 µg/ml, was incrementally added and then
incubated at 42°C for a period of 25 min. To quench the activity of
endogenous peroxidase, tissues were treated with a 3% solution of
hydrogen peroxide for 20 min at ambient temperature. Following the
manufacturer's guidelines, TdTase was combined with Biorin-dUTP at
a ratio of 1:9 to ensure thorough integration before application.
The mixture of Biorin-dUTP was then applied in a stepwise manner
and incubated in obscurity at 37°C for 60 min. The process was
terminated by adding a stopping solution and permitting an
additional incubation of 10 min at ambient temperature. An
incremental addition of streptavidin-HRP solution was performed,
followed by a 30-min incubation at ambient temperature. Thereafter,
the sections underwent stain development using DAB chromogenic
substrate. To finalize the process, the samples were stained with
hematoxylin for a duration of 5 min and then differentiated with 1%
hydrochloric acid in ethanol. The slides underwent subsequent
dehydration, clearing, and mounting as aforementioned, and finally
observed using a light microscope with 5 fields of view per section
and counted. The scale of the displayed images is both 1 and 2
microns.
Transmission electron microscopy
(TEM)
Subcutaneous graft tumor samples were segmented into
blocks ranging from 0.5–1.0 mm3 and subsequently
immersed in a 2.5% glutaraldehyde solution for 1 h at ambient
temperature for fixation. After trypsin digestion, the cells were
centrifuged at 1,200 × g for 5 min under ambient conditions and
rapidly immersed in an electron microscope fixative at 4°C for 2–4
h. The samples underwent three washes, each utilizing 0.1 M PBS at
pH 7.4, with each wash lasting 15 min. Following exposure to 1%
osmium tetroxide until darkening occurred, the samples were washed
with 1 M PBS for 10 min. Dehydration was carried out using
progressively increasing concentrations of alcohol and acetone.
After dehydration, the specimens were embedded in an appropriate
medium. Ultrathin sections were then prepared for further analysis.
The specimens were subjected to a dual staining process, first with
uranyl acetate, followed by lead citrate, and were then allowed to
air dry overnight at room temperature. Observations were conducted
using transmission electron microscopy, and images were captured
for further analysis.
Statistical analysis
Data were analysed using GraphPad Prism software
version 10.02 (GraphPad Software; Dotmatics). Data from our study
are presented as the mean ± SEM. Data obtained from at least three
independent experiments were used for all analyses. Statistical
analyses were performed by one-way ANOVA. Then, Bonferroni's
multiple comparison test (for comparisons between more than 2
groups) or Student's t-test (for comparison of the two groups) was
performed. P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-10b-5p screening in LUAD
tissues
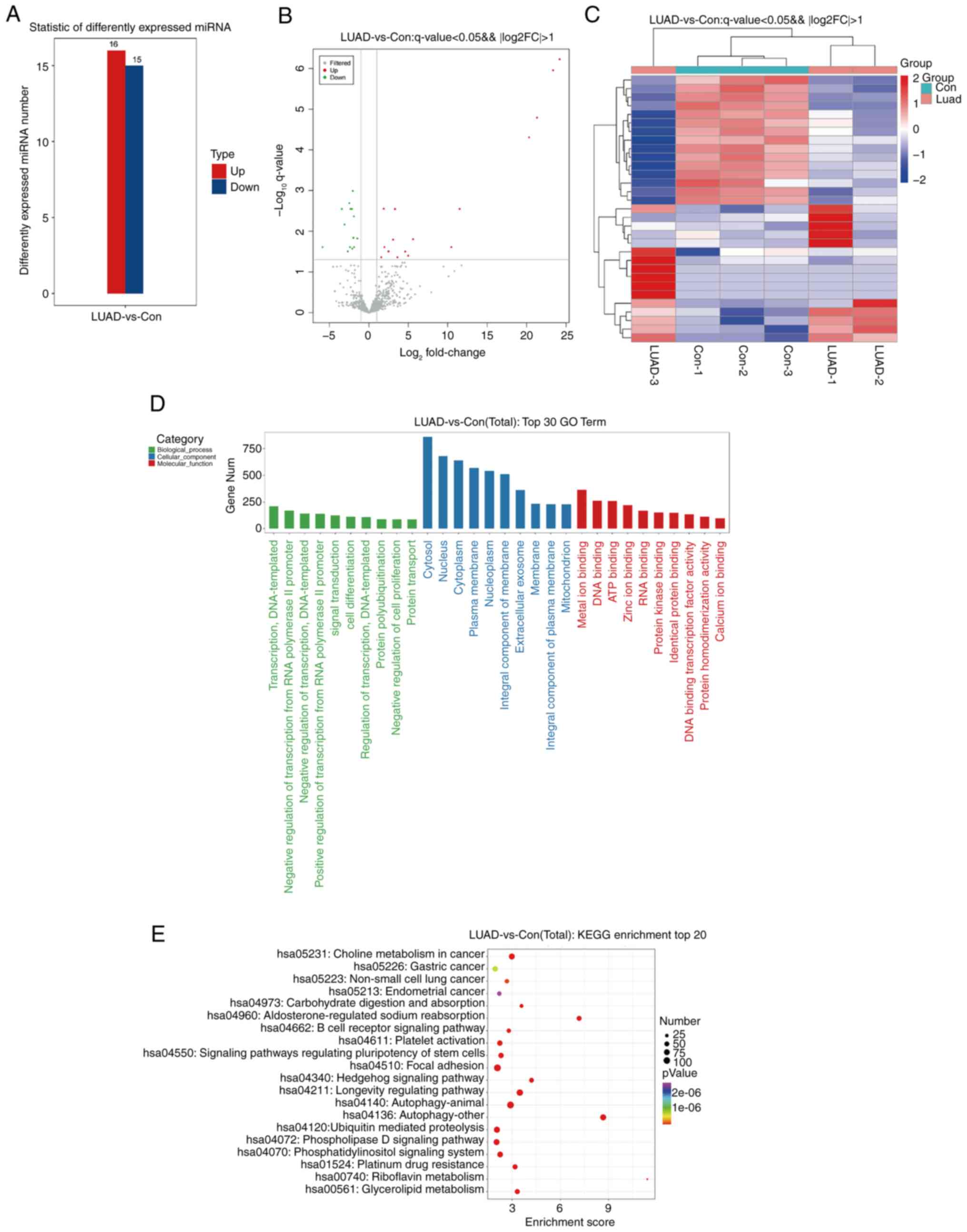
Sequencing analyses were performed on lung cancer
samples and nearby non-malignant tissues to pinpoint miRNAs that
display significant differences in expression in LUAD, utilizing a
threshold of P<0.05 and an absolute log2FC >2. The study
identified 16 miRNAs exhibiting increased expression and 15
displaying decreased expression in lung cancer specimens, as
enumerated in Table I.
 | Table I.Differential miRNAs. |
Table I.
Differential miRNAs.
| miRNA ID | Fold
regulation | P-value |
|---|
|
|---|
| Upregulated |
|---|
| hsa-miR-10b-5p | 1.565 | 0.001387 |
| hsa-miR-10b-3p | 1.8817 | 0.000020 |
|
hsa-miR-200b-5p | 1.97421 | 0.0005742 |
|
hsa-miR-365a-5p | 2.4891 | 0.0008496 |
|
hsa-miR-135b-5p | 3.069 | 0.000318 |
|
hsa-miR-135b-3p | 3.307 | 0.0000356 |
| hsa-miR-379-3p | 3.618 | 0.001409 |
| hsa-miR-105-5p | 20.30 | 0.0000002 |
|
hsa-miR-1912-5p | 24.168 | 0.000558 |
|
hsa-miR-1298-3p | 23.34 | 0.039984 |
| hsa-miR-767-5p | 21.305 | 0.017374 |
|
hsa-miR-1298-5p | 11.50 | 0.0000239 |
| hsa-miR-205-5p | 5.602 | 0.008949 |
|
hsa-miR-323a-3p | 4.9766 | 0.001205 |
|
hsa-miR-1911-5p | 10.46 | 0.004968 |
| hsa-miR-431-5p | 4.604 | 0.00092565 |
|
|
Downregulated |
|
|
hsa-miR-518a-3p | −5.868 | 0.001134 |
|
hsa-miR-30c-2-3p | −1.467 | 0.000267 |
| hsa-miR-126-5p | −1.890 | 0.00005858 |
| hsa-miR-486-5p | −1.976 | 0.000243 |
| hsa-miR-126-3p | −2.040 | 0.0005586 |
| hsa-miR-451a | −1.939 | 0.0002373 |
|
hsa-miR-135a-5p | −2.13 | 0.00003133 |
| hsa-miR-144-5p | −2.102 | 0.0006989 |
| hsa-miR-934 | −2.407 | 0.0006117 |
| hsa-miR-144-3p | −2.32 | 0.0000325 |
| hsa-miR-486-3p | −2.47 | 0.00001277 |
| hsa-miR-184 | −2.675 | 0.0008835 |
| hsa-miR-30a-5p | −1.870 | 0.000598 |
| hsa-miR-873-5p | −3.07 | 0.000099 |
|
hsa-miR-516b-5p | −3.43 | 0.0000299 |
Following the analysis, histograms of differential
miRNA and volcano plots were generated (Fig. 1A and B). Furthermore, heatmap
analysis was performed to delineate the expression patterns of
differential miRNAs in both the lung cancer and para-cancer groups
(Fig. 1C). To elucidate the
functions of the identified differential miRNAs, Gene Ontology (GO)
and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses
were conducted. GO analysis highlighted that the primary biological
functions of these miRNAs include inhibiting transcription via RNA
polymerase II promoters; their cellular localization was mainly in
the cytosol and the nucleus; additionally, their molecular roles
were closely linked to binding of metal ions and DNA (Fig. 1D). Analysis through KEGG pathways
showed that the miRNAs with varied expression significantly
participated in pathways such as aldosterone-regulated sodium
reabsorption, multiple autophagy mechanisms, Hedgehog signaling and
27 other pathways (Fig. 1E).
To identify target miRNAs effectively, a
comprehensive search was conducted using the Starbase, Targetscan,
miRWalk and miRmap databases to discover miRNAs that potentially
interact with PKP3. Prior experimental data indicated a low
expression of PKP3 in LUAD tissues, associating with necroptosis.
Consequently, the search prioritized highly expressed miRNAs. Among
these, only miR-10b-5p fulfilled all specified criteria, leading to
its selection for subsequent clinical validation.
PKP3 is a direct regulatory target
gene of miR-10b-5p
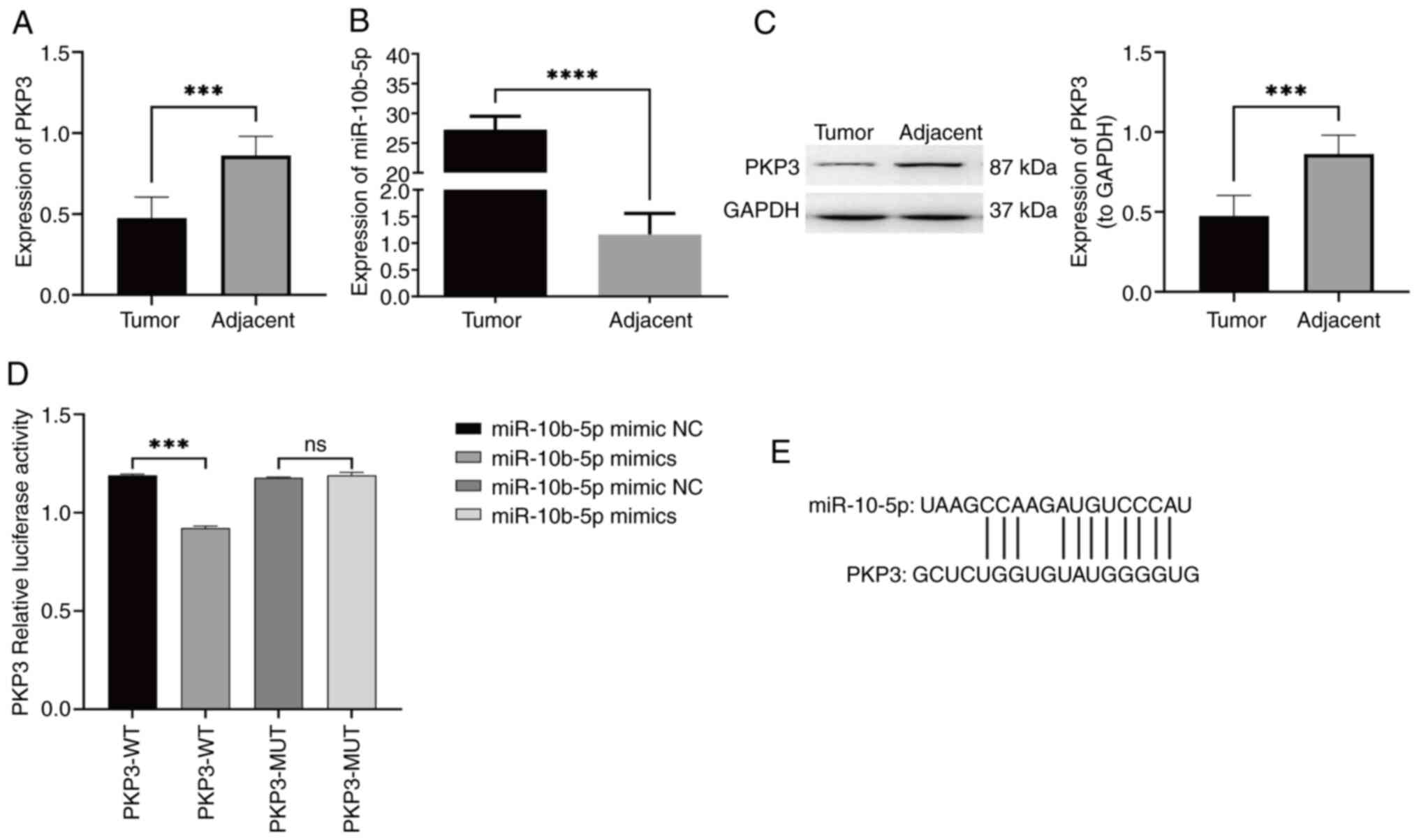
To corroborate the preliminary results and the
screening of miRNAs, the expression levels of PKP3 mRNA and
miR-10b-5p in LUAD tissues vs. non-tumorous counterparts were
quantified using RT-qPCR (Fig. 2A and
B). An analysis conducted on six paired samples indicated a
significant reduction in PKP3 mRNA in LUAD tissues relative to the
non-cancerous tissue, whereas miR-10b-5p showed a significant
elevation. Additionally, the protein expression of PKP3 was
assessed using western blot analysis in both cancerous and adjacent
non-cancerous tissues (Fig. 2C). A
significant reduction in PKP3 protein was observed in the LUAD
samples. These results corroborate the initial data mining and
sequencing findings. Further experiments involved co-transfecting
A549 cells with luciferase reporter vectors embedded with either
the WT PKP3 3′-UTR (PKP3-WT) or its mutant version (PKP3-MUT),
along with either a miR-10b-5p mimic or mimic NC. The luminescence
assay revealed diminished luciferase activity for the PKP3-WT
reporter in the presence of miR-10b-5p compared with the control, a
suppression that was reversed with mutations in the binding sites
of PKP3 3′-UTR, indicating direct interaction and downregulation of
PKP3 by miR-10b-5p (Fig. 2D). A
plot of PKP3 binding site to miR-10b-5p is illustrated in Fig. 2E.
Effect of upregulation of miR-10b-5p
expression on proliferation and necroptosis in LUAD cells
The investigation into the role of miR-10b-5p in
LUAD involved A549 cells transfected with mimics, inhibitors, or a
control vector. A total of 48 h after transfection, RT-qPCR
analysis showed a significant increase in miR-10b-5p levels in
cells treated with mimics compared with controls. Cells treated
with inhibitors exhibited a significant reduction in miR-10b-5p,
validating the efficacy of the transfection approach (Fig. 3A). A CCK-8 and cell cloning assay
were conducted to evaluate the effect of miR-10b-5p level
modulation on cell viability. The results revealed enhanced
proliferation in cells treated with mimics and a substantial
decrease in proliferation in those treated with inhibitors
(Fig. 3B and C).
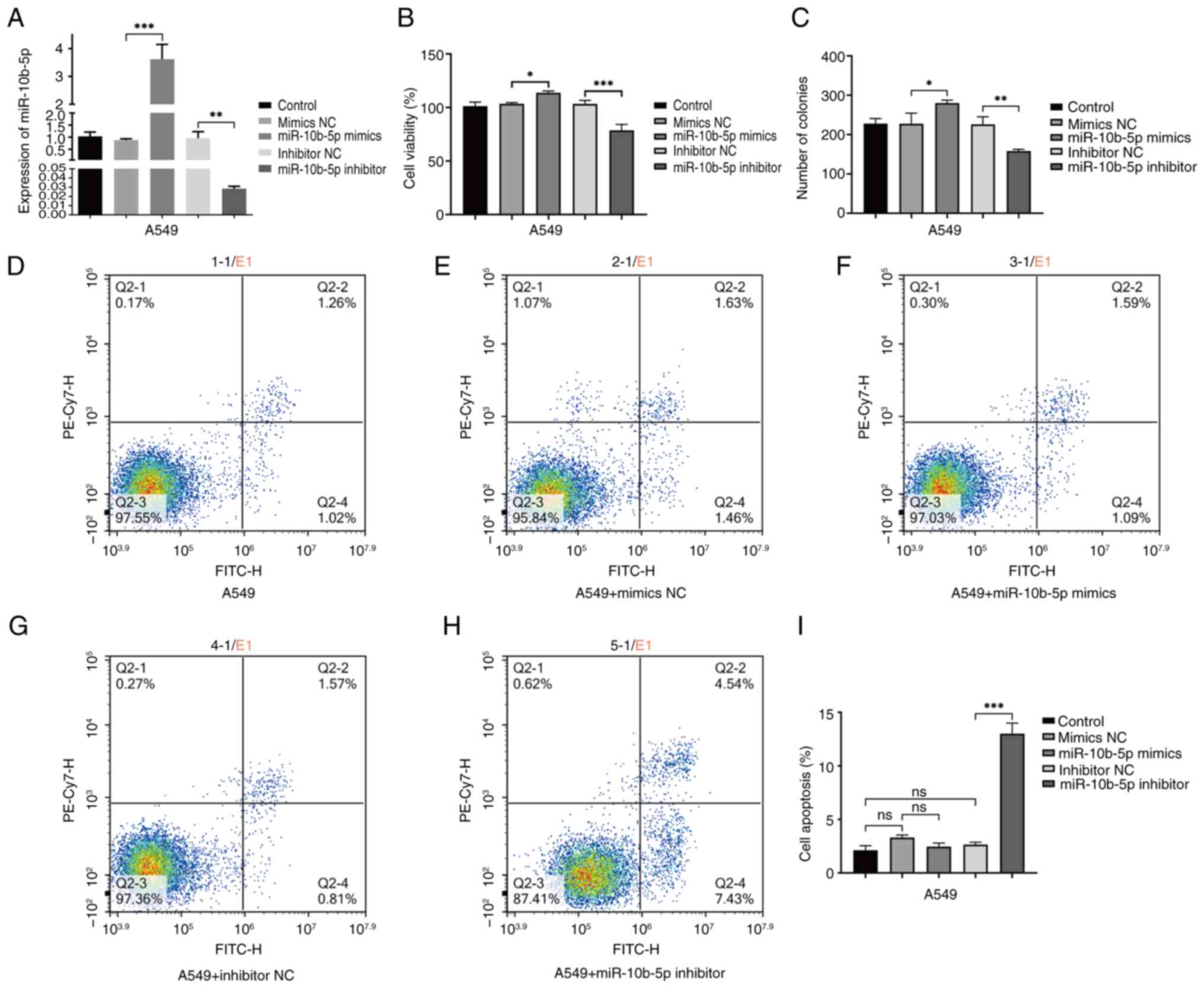 | Figure 3.Examination of miR-10b-5p expression
and its effects on A549 cell viability and apoptotic processes. (A)
Verification of miR-10b-5p expression levels was conducted across
different treatment groups in A549 cells using reverse
transcription-quantitative PCR. (B) Cell viability was assessed in
different A549 cell treatment groups using the Cell Counting Kit-8
assay. (C) Cell clones were assayed for cell proliferation in
different A549 cell treatment groups. These groups include the
control, mimics NC, miR-10b-5p mimics, inhibitor NC and miR-10b-5p
inhibitor. (D-H) Flow cytometric analysis for (D) A549 cells, (E)
mimics NC group, (F) miR-10b-5p mimics group, (G) inhibitor NC
group and (H) miR-10b-5p inhibitor group, with Q2-2 quadrant
representing late apoptotic cells and Q2-4 quadrant indicating
early apoptotic cells. (I) A bar graph presents the flow cytometry
findings, which are depicted as the mean ± SD from no less than
three independent experiments for each group. *P<0.05,
**P<0.01 and ***P<0.001. miR, microRNA; NC, negative control;
ns, not significant, (P>0.05). |
To investigate the role of miR-10b-5p in the process
of necroptosis, A549 cells were stained with Annexin V-FITC and PI,
followed by analysis via flow cytometry. This approach
differentiated and quantified the cells into four categories:
Viable (Annexin V-FITC-negative/PI-negative), early apoptotic
(Annexin V-FITC-positive/PI-negative), late apoptotic or necrotic
(Annexin V-FITC-positive/PI-positive), and non-viable (Annexin
V-FITC-negative/PI-positive). The results demonstrated that the
introduction of miR-10b-5p mimics did not significantly impact
necroptosis compared with the control. However, interestingly,
treatment of A549 cells with miR-10b-5p inhibitors significantly
promoted the occurrence of necroptosis. Inhibition of miR-10b-5p
led to a notable increase in necroptotic activity within A549
cells, suggesting that miR-10b-5p inhibition promotes necroptotic
processes (Fig. 3D-H).
miR-10b-5p inhibits PKP3 expression
and RIPK3/MLKL signalling pathway activation and promotes Caspase-8
expression
Research indicates that upregulated miR-10b-5p
facilitates proliferation while reducing necroptosis in LUAD cells.
To explore the underlying molecular mechanisms of miR-10b-5p
actions, the effects of both miR-10b-5p mimics and inhibitors on
the expression of PKP3 and apoptosis-related genes, as well as
their effects on the RIPK3/MLKL signaling pathway, were
investigated using RT-qPCR and western blotting. RT-qPCR analysis
indicated that miR-10b-5p mimics suppressed mRNA levels of PKP3,
RIPK3 and MLKL while elevating those of Caspase-8, relative to the
control. By contrast, miR-10b-5p inhibitors resulted in elevated
mRNA levels of PKP3, RIPK3 and MLKL, and reduced levels of
Caspase-8 (Fig. 4A-D). Western blot
analysis (Fig. 4E) demonstrated a
reduction in the protein levels of PKP3, p-RIPK3 and p-MLKL in the
group treated with miR-10b-5p mimics relative to the control group,
accompanied by an elevation in Caspase-8 protein levels.
Conversely, the use of miR-10b-5p inhibitors was associated with an
increase in the protein levels of PKP3, p-RIPK3 and p-MLKL, along
with a reduction in Caspase-8 protein levels.
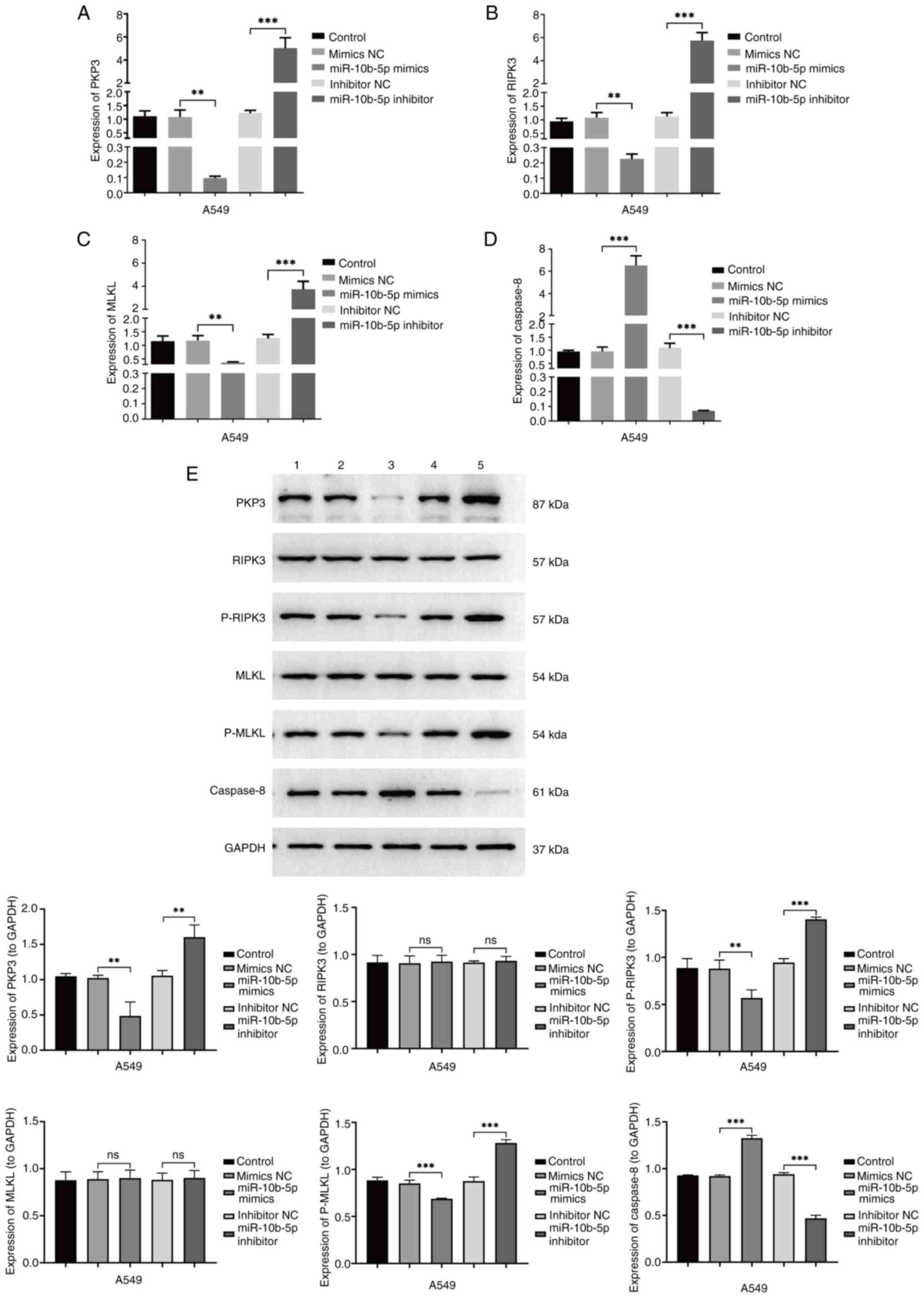 | Figure 4.Effect of miR-10b-5p on A549 cellular
pathways. (A) Reverse transcription-quantitative PCR confirmed PKP3
mRNA expression across various A549 cell treatment groups. (B)
RIPK3 mRNA levels quantified in diverse treatment cohorts. (C)
Assessment of MLKL mRNA levels in different experimental groups.
(D) Analysis of caspase-8 mRNA expression among the groups. (E)
Protein levels of PKP3, RIPK3, p-RIPK3, MLKL and p-MLKL, along with
Caspase-8 were evaluated via western blotting in varying A549 cell
treatment contexts. These groups include the A549 control, mimics
NC, miR-10b-5p mimics, inhibitor NC and miR-10b-5p inhibitor. Data
are presented as the mean ± SD from a minimum of three independent
experiments for each group. **P<0.01 and ***P<0.001. miR,
microRNA; PKP, plakophilin; RIPK, receptor-interacting protein
kinase; MLKL, mixed lineage kinase domain-like protein; p-,
phosphorylated; NC, negative control; ns, not significant,
(P>0.05). |
miR-10b-5p affects LUAD cell
proliferation and necroptosis by regulating the PKP3/RIPK3/MLKL
signalling pathway
Experimental findings suggest that an increase in
miR-10b-5p expression reduces PKP3 levels and decreases activation
within the RIPK3/MLKL signaling pathway, while simultaneously
increasing Caspase-8 expression. To confirm that miR-10b-5p
inhibits proliferation and necroptosis in LUAD cells by targeting
PKP3, a stable PKP3 knockdown was established in the A549 cell
line. RT-qPCR and western blot analysis showed significantly
reduced PKP3 mRNA and protein levels in sh-PKP3 cells compared with
controls (Fig. 5A and B).
Functional assays, including CCK-8 and flow cytometry, indicated
that miR-10b-5p inhibitor treatment significantly lowered A549 cell
viability and increased apoptotic activity (Fig. 5C-I). The reduction of PKP3 mitigated
the effects of the miR-10b-5p inhibitor on cell viability and
apoptosis induction (Fig. 5C-I).
Further qPCR analysis of mRNA levels for RIPK3, MLKL and Caspase-8
among treatment groups revealed an increase in RIPK3 and MLKL mRNA
and a decrease in Caspase-8 mRNA following miR-10b-5p inhibitor
treatment, an effect that was reversed with PKP3 knockdown
(Fig. 5J-L). Subsequently, qPCR
detected the expression of proliferation and apoptosis related
genes (Fig. 5M-Q). Additionally,
western blot results verified that miR-10b-5p inhibitor treatment
markedly upregulated p-RIPK3, p-MLKL, Bax and Caspase-3 protein
levels, while decreasing Caspase-8, proliferating cell nuclear
antigen, Ki-67 and Bcl-2 protein levels, effects that were negated
by PKP3 knockdown (Fig. 5R). These
findings suggest that miR-10b-5p disrupts RIPK3/MLKL pathway
activation by directly targeting PKP3, thereby fostering
proliferation and curtailing necroptosis in A549 cells.
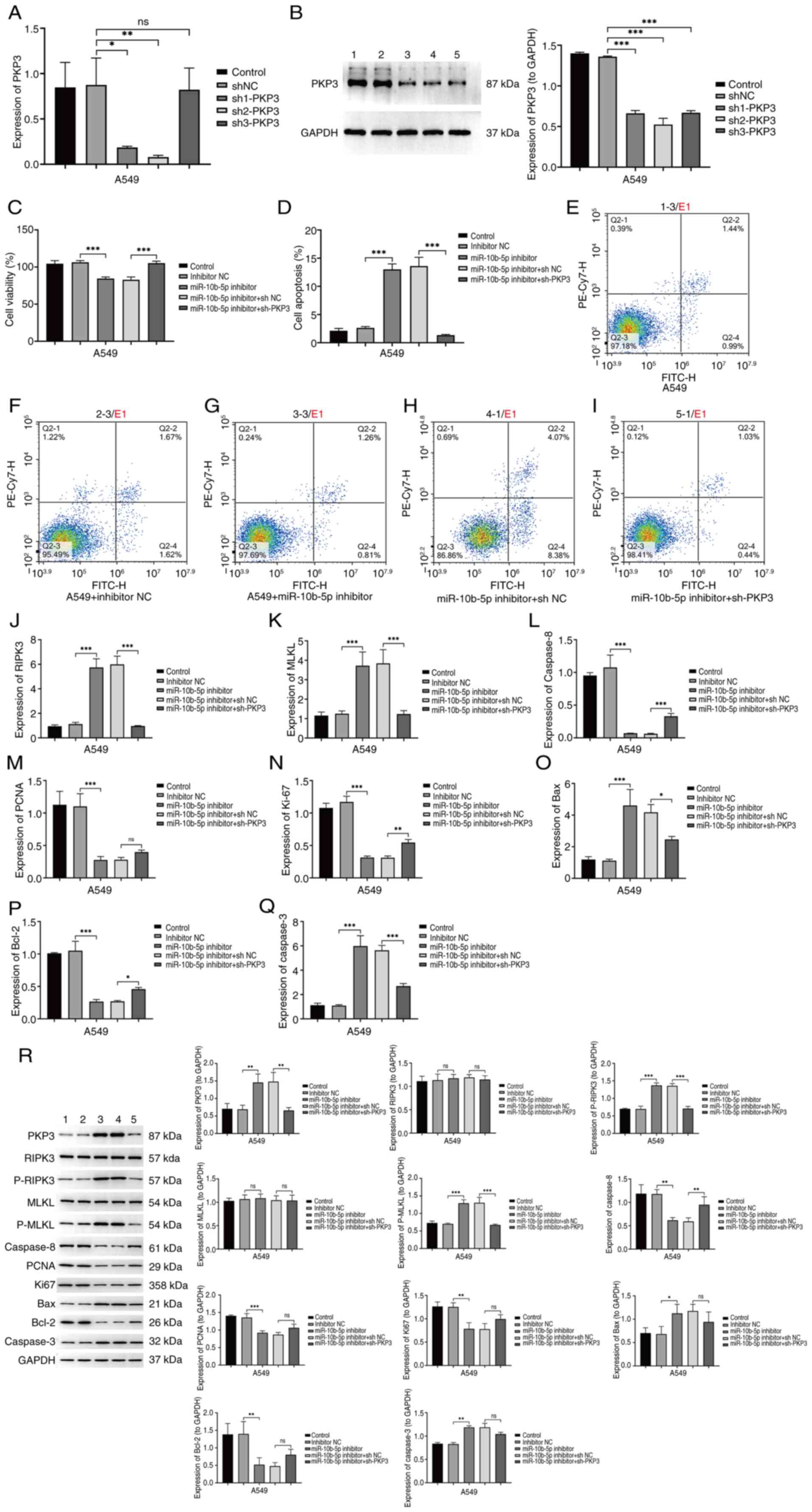 | Figure 5.Modulation of cell proliferation and
apoptosis via the PKP3/RIPK3/MLKL pathway (A) Validation of PKP3
mRNA levels in PKP3-silenced cells was performed by RT-qPCR. (B)
PKP3 protein levels in PKP3-silenced cells were confirmed via
western blotting. These groups include the control, shNC, sh1-PKP3,
sh2-PKP3 and sh2-PKP3. (C) Cell Counting Kit-8 assay assessed the
viability of A549 cells across various treatment groups. (D-I)
Apoptotic rates in A549 cells were analyzed through flow cytometry
across different treatment modalities. (D) Statistical histogram,
(E) A549 group, (F) inhibitor NC group, (G) miR-10b-5p inhibitor
group, (H) miR-10b-5p inhibitor + shNC group and (I) miR-10b-5p
inhibitor + sh-PKP3 group. Late-stage apoptotic cells were marked
in the Q2-2 quadrant, and early apoptotic cells in the Q2-4
quadrant. (J-Q) The expression levels of RIPK3, MLKL, Caspase-8,
PCNA, Ki67, Bax, Bcl-2 and Caspase-3 mRNA were detected by RT-qPCR
in A549 cells of different treatment groups. (R) Western blot
analysis quantified PKP3 mRNA levels across varied treatment groups
in A549 cells. Protein expression levels of PKP3, RIPK3, p-RIPK3,
MLKL, p-MLKL, Caspase-8, PCNA, Ki67, Bax, Bcl-2 and Caspase-3 in
A549 cells across the treatment groups were documented. Group
identifiers included the A549 group, inhibitor NC group, miR-10b-5p
inhibitor group, miR-10b-5p inhibitor + shNC group and miR-10b-5p
inhibitor + sh-PKP3 group. Representative images or data are
presented as the mean ± SD from a minimum of three independent
experiments per group. *P<0.05, **P<0.01 and ***P<0.001.
PKP, plakophilin; RIPK, receptor-interacting protein kinase; MLKL,
mixed lineage kinase domain-like protein; RT-qPCR, reverse
transcription-quantitative PCR; sh-, short hairpin; NC, negative
control; PCNA, proliferating cell nuclear antigen; p-,
phosphorylated; ns, not significant, (P>0.05). |
miR-10b-5p promotes the tumorigenic
potential of LUAD cells in vivo
To investigate the role of miR-10b-5p in tumor
growth in vivo, A549 cells were implanted, including those
transfected with an inhibitor NC and a miR-10b-5p inhibitor, into
the right lumbar region of nude mice at a vascular-rich site. By
day 41 post-inoculation, tumors from the miR-10b-5p inhibitor
cohort were significantly smaller compared with those in the
control group (Fig. 6A-C). Analysis
post-euthanasia revealed that suppression of miR-10b-5p led to
diminished tumor growth rates relative to the controls (Fig. 6D). Subsequent studies explored the
regulatory mechanisms of miR-10b-5p in vivo. Gene expression
analysis related to apoptosis in tumor specimens was conducted via
RT-qPCR and western blotting. The RT-qPCR results demonstrated a
significant upregulation in the mRNA levels of PKP3, RIPK3 and MLKL
in tumors treated with the miR-10b-5p inhibitor, while the mRNA
levels of Caspase-8 were significantly reduced (Fig. 6E-H). Western blot analysis
corroborated the increases in PKP3, p-RIPK3 and p-MLKL proteins,
alongside a decrease in Caspase-8 protein levels in miR-10b-5p
inhibitor-treated tumors relative to controls (Fig. 6I). TUNEL assays indicated enhanced
apoptotic rates in the miR-10b-5p inhibitor-treated tumors
(Fig. 6J). Enhanced necroptosis in
cells from the miR-10b-5p inhibitor group compared with the control
group was observed under TEM (Fig.
6K). These findings suggest that inhibiting miR-10b-5p
activates the RIPK3/MLKL signaling pathway via PKP3 upregulation,
leading to diminished tumor growth and increased cellular
necroptosis, thereby impeding the progression of LUAD.
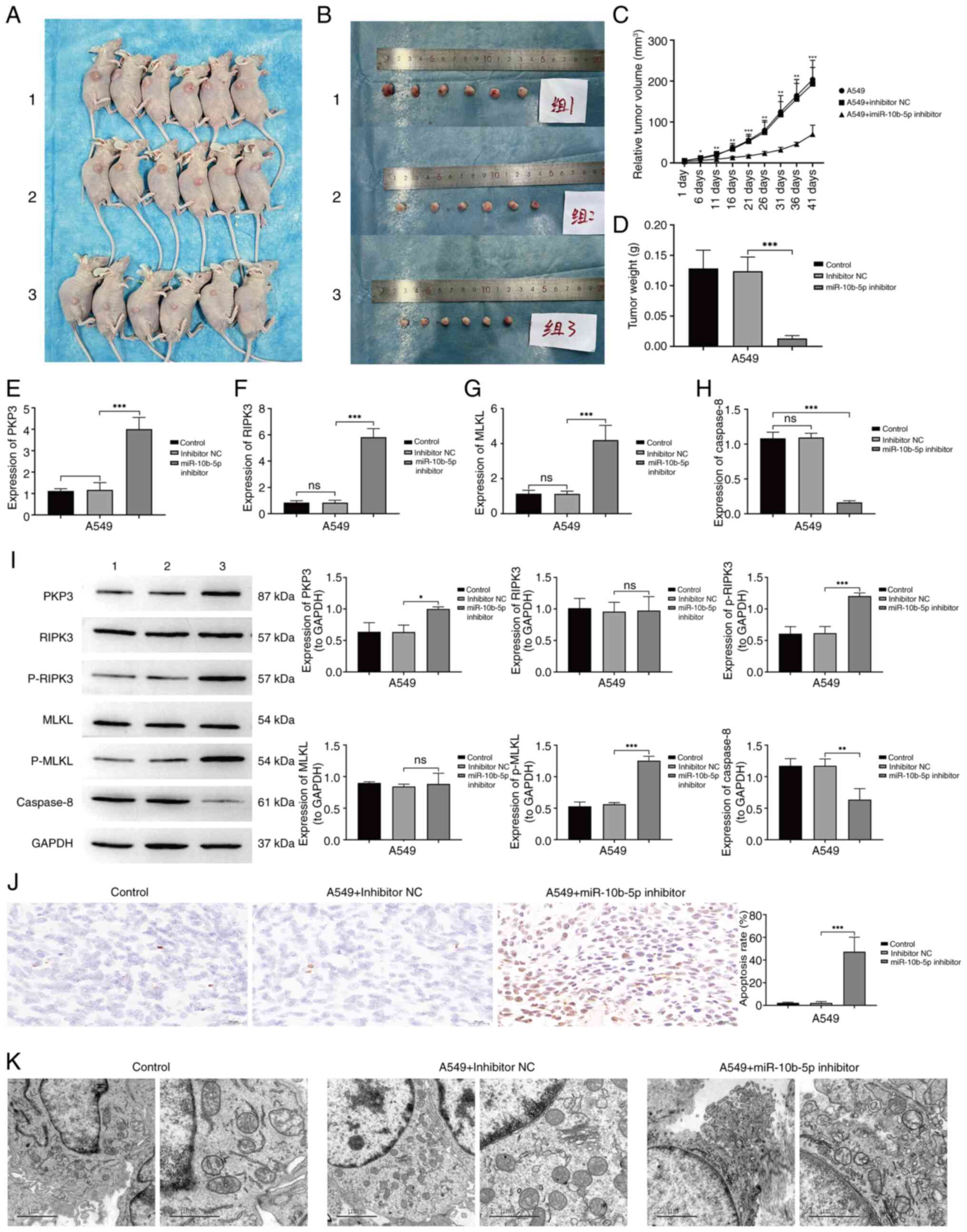 | Figure 6.Effect of miR-10b-5p inhibition on
the oncogenic capabilities of A549 cells in vivo (A-C)
BALB/c nude mice were randomly assigned to receive either A549
cells, a non-specific control inhibitor, or miR-10b-5p
inhibitor-treated A549 cells. By day 41 post-inoculation, tumors in
the miR-10b-5p inhibitor group exhibited a significant reduction in
size compared with the other groups. (D) Graph depicting tumor
growth trajectories following cell inoculation. (E-H) Expression
levels of (E) PKP3, (F) RIPK3, (G) MLKL and (H) Caspase-8 were
quantified by reverse transcription-quantitative PCR across the
various treatment cohorts. (I) Western blot analysis was conducted
to quantify the protein levels of PKP3, RIPK3, p-RIPK3, MLKL,
p-MLKL and Caspase-8 across different treatment groups. (J)
Apoptosis in cells from different treatment groups was assessed
using TUNEL staining. (K) Transmission electron microscopy provided
visual evidence of cellular morphology across the treatment groups.
Group designations were as follows: A549 group, inhibitor NC group,
and miR-10b-5p inhibitor group. Data are presented as the mean ±
SD, derived from a minimum of three independent experiments for
each group. *P<0.05, **P<0.01 and ***P<0.001. miR,
microRNA; PKP, plakophilin; RIPK, receptor-interacting protein
kinase; MLKL, mixed lineage kinase domain-like protein; p-,
phosphorylated; NC, negative control; ns, not significant,
(P>0.05). |
Discussion
Lung cancer continues to be a leading cause of
cancer-related deaths, with a 5-year survival rate of only 15%, and
adenocarcinoma represents the most prevalent pathological variant
(26). Despite progress in
immunotherapy and precision medicine, LUAD still presents
significant public health issues worldwide. These challenges stem
from its complex genetic diversity, common delays in diagnosis, and
widespread resistance to existing medications, contributing to poor
prognoses (27). In addition, gaps
in knowledge concerning its pathogenesis and ongoing changes in
tumor genomic expressions hinder the rapid development of effective
treatments for LUAD (28).
Therefore, genomic medicine is increasingly recognized as a vital
area to augment and enhance research into LUAD.
Initially identified in 1993 during studies on
Cryptobacterium hidradii nematodes (29), miRNAs are diminutive non-coding
RNAs, crucial for a wide range of biological functions and diseases
in various organisms. These include cell differentiation,
development, aging, neurotransmission, immune reactions, apoptosis,
and notably, cancer progression and tumorigenesis (30). miR-10b-5p has emerged as being
significantly influential in various cancers. Research indicates
that in glioblastoma, miR-10b-5p facilitates immune evasion by
downregulating Ten-eleven translocation 2, which leads to decreased
PD-L1 transcriptional repression (31). Another investigation revealed that
in hepatocellular carcinoma, miR-10b-5p targets SLC38A2 to modulate
cellular metabolism, thereby enhancing tumor growth (23). While this research offers
significant insights, the exact functions and processes by which
miR-10b-5p influences LUAD remain to be fully elucidated.
In the current investigation, miRNA sequencing
technology was employed to identify miRNAs that are expressed
differentially between LUAD and surrounding non-malignant tissues.
By integrating sequencing results, initial experimental findings,
and external database information, miR-10b-5p was identified.
Validation confirmed that LUAD tissues exhibited reduced PKP3
expression alongside significantly elevated miR-10b-5p levels.
Subsequent investigations demonstrated that the elevation in
miR-10b-5p levels was a consequence of its specific interaction
with the 3′-UTR of PKP3, resulting in a suppression of PKP3
expression.
Additional studies employed both enhancement and
inhibition strategies to elucidate the role and mechanism of
miR-10b-5p in LUAD. These experiments revealed that upregulation of
miR-10b-5p promotes proliferative activity and inhibits necroptosis
in A549 cells. On the other hand, downregulation of miR-10b-5p
inhibited the proliferative activity and promoted necrosis in A549
cells. The dysregulated activation of the RIPK3/MLKL signaling
pathway plays a crucial role in tumor progression and can trigger
necroptosis (32). However,
Caspase-8 has been shown to mitigate necroptosis by cleaving RIPK3
(33). The results demonstrated
that miR-10b-5p overexpression led to a decrease in PKP3 levels and
suppression of the RIPK3/MLKL pathway, while concurrently elevating
Caspase-8 levels. By contrast, reducing miR-10b-5p expression was
associated with increased PKP3 levels and activation of the
RIPK3/MLKL pathway, coupled with a decrease in Caspase-8
levels.
To further examine whether miR-10b-5p exerts its
necroptotic inhibitory effects in A549 cells by targeting and
regulating PKP3, thereby constraining the activation of the
RIPK3/MLKL pathway, PKP3 was silenced. This silencing demonstrated
that the reduction of PKP3 countered the decrease in A549 cell
viability and the enhancement of necroptosis initially induced by
miR-10b-5p knockdown. In a similar fashion, reducing PKP3 levels
mitigated the enhanced activation of the RIPK3/MLKL pathway due to
miR-10b-5p suppression. These findings suggest that miR-10b-5p
mitigates apoptosis in LUAD cells by targeting PKP3, leading to
suppressed activation of the RIPK3/MLKL pathway. Additional in
vivo validations were conducted, revealing that miR-10b-5p
knockdown curbed LUAD progression and enhanced necroptosis via the
PKP3-mediated activation of the RIPK3/MLKL pathway.
In conclusion, through in vivo and ex
vivo experiments, it was found for the first time to the best
of our knowledge, that miR-10b-5p, as an oncogene, promotes the
proliferation of LUAD cells and inhibits necroptosis by suppressing
PKP3-mediated activation of the RIPK3/MLKL signalling pathway.
Notably, knockdown of miR-10b-5p inhibited LUAD development by
promoting the onset of necroptosis. In addition, the present study
has certain limitations. The sample size was small, only 6 cases,
which may affect the wide applicability of the results and the
reliability of statistical analyses, but a large number of
experiments were conducted at a later stage to prove and validate
them. In conclusion, the current findings provide new insights into
the mechanisms of LUAD progression and suggest that targeting
miR-10b-5p has the potential to be a new effective therapeutic
tool.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Yunnan health training
project of high-level talents (grant no. D-2018041).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YH, XL, ZY, RM, YW, GY and JH contributed to the
study conception and design, prepared materials, collected and
analyzed the data. YH and ZY wrote the first draft of the
manuscript. YW and GY confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The research protocols for human studies adhered to
the ethical guidelines of the Declaration of Helsinki and were
approved by The Second Affiliated Hospital of Kunming Medical
University (KMUH) Ethics Committee (approval no. PJ-2024-125;
Kunming, China). Written informed consent was obtained from all
participants. Animal experimental protocol was approved by the KMUH
Animal Care Committee (approval no. kyfeyxm2024127; Kunming,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhao B, Xu H, Ai X, Adalat Y, Tong Y,
Zhang J and Yang S: Expression profiles of long noncoding RNAs in
lung adenocarcinoma. Onco Targets Ther. 11:5383–5390. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei X, Li X, Hu S, Cheng J and Cai R:
Regulation of ferroptosis in lung adenocarcinoma. Int J Mol Sci.
24:146142023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Denisenko TV, Budkevich IN and Zhivotovsky
B: Cell death-based treatment of lung adenocarcinoma. Cell Death
Dis. 9:1172018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen H, Xia R, Jiang L, Zhou Y, Xu H, Peng
W, Yao C, Zhou G, Zhang Y, Xia H and Wang Y: Overexpression of RhoV
promotes the progression and EGFR-TKI resistance of lung
adenocarcinoma. Front Oncol. 11:6190132021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li S, Choi YL, Gong Z, Liu X, Lira M, Kan
Z, Oh E, Wang J, Ting JC, Ye X, et al: Comprehensive
characterization of oncogenic drivers in asian lung adenocarcinoma.
J Thorac Oncol. 11:2129–2140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Chen J, Tian J, Zhou Y and Liu Y:
Role and function of plakophilin 3 in cancer progression and skin
disease. Cancer Sci. 115:17–23. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du Y, Hou S, Chen Z, Li W, Li X and Zhou
W: Comprehensive analysis identifies PKP3 overexpression in
pancreatic cancer related to unfavorable prognosis. Biomedicines.
11:24722023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim V, Zhu H, Diao S, Hu L and Hu J: PKP3
interactions with MAPK-JNK-ERK1/2-mTOR pathway regulates autophagy
and invasion in ovarian cancer. Biochem Biophys Res Commun.
508:646–653. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu B, Feng Y, Xie N, Yang Y and Yang D:
FERMT1 promotes cell migration and invasion in non-small cell lung
cancer via regulating PKP3-mediated activation of p38 MAPK
signaling. BMC Cancer. 24:582024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee LYW, Woolley C, Starkey T, Biswas S,
Mirshahi T, Bardella C, Segditsas S, Irshad S and Tomlinson I:
Serum- and Glucocorticoid-induced kinase sgk1 directly promotes the
differentiation of colorectal cancer cells and restrains
metastasis. Clin Cancer Res. 25:629–640. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takahashi H, Nakatsuji H, Takahashi M,
Avirmed S, Fukawa T, Takemura M, Fukumori T and Kanayama H:
Up-regulation of plakophilin-2 and Down-regulation of plakophilin-3
are correlated with invasiveness in bladder cancer. Urology.
79:240.e1–e8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Furukawa C, Daigo Y, Ishikawa N, Kato T,
Ito T, Tsuchiya E, Sone S and Nakamura Y: Plakophilin 3 oncogene as
prognostic marker and therapeutic target for lung cancer. Cancer
Res. 65:7102–7110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Z, Wang T, She Y, Wu K, Gu S, Li L,
Dong C, Chen C and Zhou Y: N6-methyladenosine-modified circIGF2BP3
inhibits CD8+ T-cell responses to facilitate tumor immune evasion
by promoting the deubiquitination of PD-L1 in non-small cell lung
cancer. Mol Cancer. 20:1052021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim HJ, Roh MS, Son CH, Kim AJ, Jee HJ,
Song N, Kim M, Seo SY, Yoo YH and Yun J: Loss of Med1/TRAP220
promotes the invasion and metastasis of human non-small-cell lung
cancer cells by modulating the expression of metastasis-related
genes. Cancer Lett. 321:195–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qin X, Ma D, Tan YX, Wang HY and Cai Z:
The role of necroptosis in cancer: A double-edged sword? Biochim
Biophys Acta Rev Cancer. 1871:259–266. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu F, Zhang W, Yang T and He SD: Complex
roles of necroptosis in cancer. J Zhejiang Univ Sci B. 20:399–413.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Philipp S, Sosna J and Adam D: Cancer and
necroptosis: Friend or foe? Cell Mol Life Sci. 73:2183–2193. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayes J, Peruzzi PP and Lawler S:
MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol
Med. 20:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget.
6:8474–8490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harari-Steinfeld R, Gefen M, Simerzin A,
Zorde-Khvalevsky E, Rivkin M, Ella E, Friehmann T, Gerlic M,
Zucman-Rossi J, Caruso S, et al: The lncRNA H19-Derived
MicroRNA-675 promotes liver necroptosis by targeting FADD. Cancers
(Basel). 13:4112021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Tibenda JJ, Nan Y, Huang SC, Ning N,
Chen GQ, Du YH, Yang YT, Meng FD and Yuan L: MiR-204-3p
overexpression inhibits gastric carcinoma cell proliferation by
inhibiting the MAPK pathway and RIP1/MLK1 necroptosis pathway to
promote apoptosis. World J Gastroenterol. 29:4542–4556. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan T, Wang X, Wei G, Li H, Hao L, Liu Y,
Yu X, Zhu W, Liu P, Zhu Y and Zhou X: Exosomal miR-10b-5p mediates
cell communication of gastric cancer cells and fibroblasts and
facilitates cell proliferation. J Cancer. 12:2140–2150. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xia M, Chen J, Hu Y, Qu B, Bu Q and Shen
H: miR-10b-5p promotes tumor growth by regulating cell metabolism
in liver cancer via targeting SLC38A2. Cancer Biol Ther.
25:23156512024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li S, Mao L, Song L, Xia X, Wang Z, Cheng
Y, Lai J, Tang X and Chen X: Extracellular vesicles derived from
glioma stem cells affect glycometabolic reprogramming of glioma
cells through the miR-10b-5p/PTEN/PI3K/Akt pathway. Stem Cell Rev
Rep. 20:779–796. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song Y, Kelava L and Kiss I: MiRNAs in
lung adenocarcinoma: Role, diagnosis, prognosis, and therapy. Int J
Mol Sci. 24:133022023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu J, Zhang F, Wang J and Wang Y:
MicroRNA-mediated regulation in lung adenocarcinoma: Signaling
pathways and potential therapeutic implications (Review). Oncol
Rep. 50:2112023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Du W, Chen D, Wei K, Yu D, Gan Z, Xu G and
Yao G: MiR-10b-5p impairs TET2-Mediated inhibition of PD-L1
transcription thus promoting immune evasion and tumor progression
in glioblastoma. Tohoku J Exp Med. 260:205–214. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou Y, Xiang Y, Liu S, Li C, Dong J, Kong
X, Ji X, Cheng X and Zhang L: RIPK3 signaling and its role in
regulated cell death and diseases. Cell Death Discov. 10:2002024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yuan J and Ofengeim D: A guide to cell
death pathways. Nat Rev Mol Cell Biol. 25:379–395. 2024. View Article : Google Scholar : PubMed/NCBI
|