Global cancer statistics from 2022 indicate that ~20
million new cancer cases were diagnosed, with an estimated 9.7
million cancer-related deaths (1).
The complexity of the tumor microenvironment (TME) represents a
significant obstacle to effective cancer treatment. The TME
consists of diverse cellular and non-cellular components that drive
tumor progression and therapeutic resistance through intricate
molecular interactions (2). Among
them, cancer-associated fibroblasts (CAFs) are key regulators of
tumor initiation, progression, metastasis and resistance to therapy
(3). In certain cancer types, CAFs
may comprise up to 60% of the tumor stroma, with an elevated
stromal fraction being highly associated with poor prognosis
(4,5). Furthermore, inflammation is crucial in
tumorigenesis by inducing epithelial mutations, supporting tumor
stem cell maintenance and facilitating immune surveillance
(6). Tumor cells recruit
inflammatory cells via chemokine receptors, leading to
enhanced cytokine expression and contributing to invasion,
metastatic dissemination and suppression of antitumor immune
responses (7).
Inflammation and CAFs are inherently interconnected
through key signaling cascades, including interleukin-6 (IL-6),
transforming growth factor-β (TGF-β) and nuclear factor κβ (NF-κB),
which collectively establish a self-sustaining feedback loop that
promotes tumor progression and immune evasion. Although these
pathways are universally present across various cancers, their
activation patterns show cancer-specific distinctions. For
instance, in pancreatic and breast carcinomas, IL-1β and IL-6
promote the activation of inflammatory CAFs (iCAFs) (8,9),
whereas in lung and colorectal cancers, TGF-β-driven CAFs
differentiation enhances immunosuppressive signaling and regulates
extracellular matrix (ECM) remodeling (10–12).
Considering this complexity, only targeting inflammation may be
insufficient to effectively disrupt the CAF-driven pro-tumorigenic
microenvironment. Therefore, integrating CAF-targeted therapies
with anti-inflammatory interventions is proposed as a more
comprehensive strategy for modulating the TME.
Traditional anti-inflammatory drugs, such as aspirin
and celecoxib, have been shown to reduce cancer incidence and
mortality (13,14). Furthermore, therapeutic strategies
aimed at inhibiting IL-6, TGF-β and NF-κB are increasingly being
explored for their potential to enhance patient survival in various
clinical trials (15–18). However, only targeting inflammatory
pathways fails to completely neutralize the tumor-promoting
functions of CAFs. For instance, cyclooxygenase-2 (COX-2)
inhibitors lower IL-6 and prostaglandin E2 levels (19), yet CAF activity persists through
alternative mechanisms, including TGF-β signaling and ECM
remodeling. Similarly, IL-6 inhibition suppresses inflammation but
does not effectively prevent CAF-mediated immune suppression and
fibrosis (20).
Currently, two main immunotherapeutic strategies
targeting CAFs are being explored: i) Direct elimination of CAFs by
targeting surface markers, e.g., fibroblast activation protein
(FAP), and ii) suppression of CAF activation and function
via the modulation of key signaling molecules, e.g., TGF-β.
Although CAF-depleting therapies have demonstrated some efficacy in
preclinical animal models, their success in clinical trials remains
limited (21), with their
development progressing slower than therapies targeting
CAF-associated signaling pathways. Some emerging therapies
targeting CAF-associated signaling pathways (e.g., TGF-β
inhibitors) or inflammatory cytokines (e.g., IL-6 blockade) have
demonstrated the ability to regulate both CAF activity and the
inflammatory response (9,10). Combining TGF-β inhibitors with
gemcitabine and anti-PD-L1 antibodies has proven to yield better
anti-tumor efficacy (22–24). Furthermore, tocilizumab, an
inhibitor of the IL-6/JAK/STAT3 signaling pathway, has demonstrated
the potential to enhance immune responses and improve tumor control
(25). These findings indicate that
disrupting the crosstalk between CAFs and inflammation may improve
therapeutic efficacy by impairing stromal remodeling and
alleviating immune suppression.
A deeper understanding of the molecular mechanisms
underlying this interaction is essential to optimize such
combination strategies. A comprehensive examination of the
functions of key signaling pathways-such as IL-6/STAT3, TGF-β and
NF-κB-in regulating the inflammatory and stromal components of the
TME is crucial for the identification of novel therapeutic targets
and the development of rational and effective combinatorial
treatment strategies. The following sections examined the intricate
bidirectional crosstalk between CAFs and inflammation, emphasizing
the signaling mechanisms contributing to tumor progression and
therapeutic resistance.
Tumorigenesis is intrinsically associated with
inflammation, and tumor progression closely parallels the
advancement of inflammatory processes. Metabolic changes within the
TME, cellular death and microbial existence and their secreted
products collectively contribute to the establishment of
inflammation (26). Furthermore,
conventional cancer therapies such as chemotherapy and radiation
have been shown to induce IL-6 expression within tumors, thus
promoting chronic inflammation (27). Therefore, CAFs remain a persistent
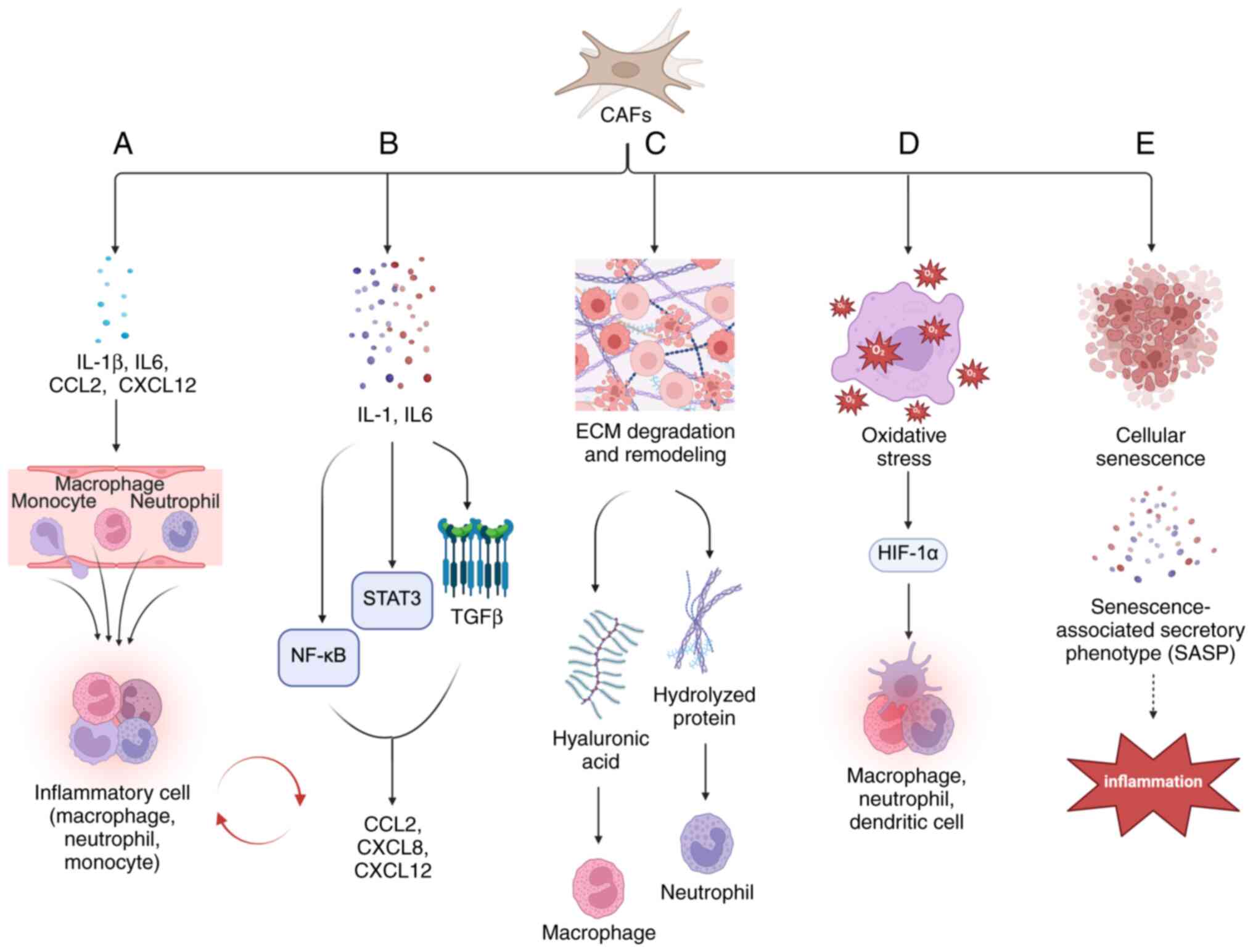
and active component in shaping the inflammatory TME (Fig. 1).
These CAFs play a pivotal role in the recruitment
and polarization of inflammatory cells (Fig. 1A). Chemokines secreted by both tumor
cells and CAFs facilitate the infiltration of tumor-associated
macrophages (TAMs), tumor-associated neutrophils (TANs) and
lymphocytes into the TME, thus intensifying the inflammatory
response (28,29). CAFs contribute to macrophage
recruitment by secreting pro-inflammatory cytokines such as IL-1β
and IL-6, along with C-X-C motif chemokine ligand (CXCL)1 and −2,
exerting these effects even in the absence of tumor cells (30). Furthermore, CAFs produce extra
domain A fibronectin variants that bind to macrophage Toll-like
receptor 4, consequently inducing M2 macrophage polarization
(31). In hepatocellular carcinoma
(HCC), cardiotrophin-like cytokine factor 1 secreted by CAFs
enhances the production of chemokine ligands CXCL6 and TGF-β in
tumor cells, thus promoting tumor cell stemness through an
autocrine mechanism while facilitating TAN infiltration and
polarization via paracrine signaling (32). In lung cancer, CAFs secrete CCL2 and
CXCL12, which mediate the recruitment of monocytes and promote
their differentiation into myeloid-derived suppressor cells
(MDSCs), thus suppressing CD8+ T-cell proliferation and
interferon-γ (IFNγ) production (33). In addition, hyaluronic acid
(HA)-producing CAFs interact with MDSCs and epithelial tumor cells,
leading to HA degradation and the accumulation of pro-inflammatory
HA fragments, further exacerbating cancer-associated inflammation.
The HA-rich stromal environment promotes the differentiation of
tumor-infiltrating hyaluronan 2+ MDSCs into programmed death ligand
1 (PD-L1)+ TAMs, thus establishing an immunosuppressive and
tumor-favorable TME (34).
CAFs are the main cellular components of the stroma
and are the primary source of connective tissue and proteolytic
enzymes within the ECM (Fig. 1C).
The production of ECM by CAFs modifies and engages multiple
signaling pathways from the cell surface to the nucleus, resulting
in alterations in gene expression and cellular behavior. CAFs
promote ECM degradation and remodeling by secreting cytokines,
chemokines and other effector molecules (TGF-β, CXCL2), various
matrix proteins (fibronectin and type I collagen) and MMPs
(47,48). ECM deposition is intricately
associated with TGF-β, a relationship mediated by CAFs through the
production of activin A, which promotes epithelial cell migration
and induces EMT (49). These
fibroblasts synthesize significant amounts of laminin, which binds
to α6β4 integrin receptors on malignant cells, thus enhancing their
migration potential (50). They are
also the primary source of HA, a crucial stromal-derived component
that facilitates the recruitment of TAMs, which are predominantly
concentrated within the HA-rich tumor stroma (51). A single-cell RNA-sequencing study
revealed that CAFs interact with a tumor-specific keratinocyte
subpopulation that shows significant EMT features, with the
pleiotropic growth factor Midkine being upregulated in primary CAFs
(52).
The ECM is a crucial component of the TME, providing
structural support and regulating the microenvironment and cellular
interactions. Changes in its composition, density and rigidity are
closely associated with tumor progression. Increased ECM rigidity
influences cellular behavior by altering mechanotransduction
pathways, thus affecting the capacity of cells to perceive and
respond to external mechanical stimuli. The ECM also activates T
cells and promotes their differentiation through integrin-mediated
complexes to regulate immune cells (53). TGF-β plays a significant role in
regulating ECM stiffness, while MMPs promote ECM degradation and
remodeling, both of which are essential for tumor cell invasion.
TGF-β is predominantly secreted and deposited within the ECM as
latent complexes (54).
Pathological upregulation of TGF-β induces EMT, promotes ECM
deposition and drives the activation of CAFs, ultimately
contributing to fibrotic diseases and cancer progression (16). MMP is the most relevant protease for
primary tumors, regulating various physiological processes and
signal transduction events (55).
The most well-known function of MMP is to cleave ECM proteins to
regulate ECM remodeling. Certain hydrolyzed protein fragments of
the ECM are chemotactic, recruiting neutrophils, increasing their
chemotactic activity and exacerbating tumor inflammatory responses
(56). The interaction between the
tumor and ECM activates the Notch1 pathway through pro-inflammatory
signaling, leading to the induction of CXCL8, which promotes tumor
metastasis (57). Several proteins
associated with inflammation, stromal remodeling, TGF-β receptor
signaling and angiogenesis have been identified within the stromal
microenvironment (58).
Inflammatory fibroblasts (iCAFs) exhibit
hypoxia-associated gene expression and biochemical profiles. These
cells are predominantly localized in hypoxic regions of pancreatic
cancer, whereas myofibroblasts (myCAFs) are largely absent. Hypoxia
further enhances cytokine-induced iCAF phenotypes, contributing to
tumor progression (59). The
transcriptional target of hypoxia-inducible factor (HIF)-1α, the
G-protein estrogen receptor, establishes a feed-forward loop in
which IL-1β secretion by fibroblasts enhances IL1R1 expression in
breast cancer cells. Furthermore, IL-1β present in the conditioned
medium of triple-negative breast cancer cells under hypoxic
conditions reinforces the invasion of fibroblasts (Fig. 1D) (60). MyCAFs deficient in caveolin-1
activate HIF and NF-κB transcription factors, generating oxidative
stress that promotes aerobic glycolysis and inflammation, thus
creating a pseudo-hypoxic state. This phenomenon drives the
‘reverse Warburg effect’ within the TME, where aerobic glycolysis
occurs predominantly in tumor-associated fibroblasts rather than
malignant cells, facilitating metastasis (61,62).
The lactate-NAD+ axis further activates CAFs by downregulating p62,
which enhances tumorigenesis through inflammation and metabolic
reprogramming in both in vitro and in vivo models
(63). Furthermore, the dense ECM
exerts mechanical pressure on blood vessels, inducing hypoxia, with
collagen deposition contributing to the expression of
hypoxia-related aberrant factors (64).
The association between hypoxia and inflammation is
well established, with inflammatory diseases frequently showing
severe hypoxic conditions. Malignant tumor cell clones consume
substantial amounts of oxygen, inducing persistent hypoxia that
sustains chronic inflammation. This process is driven by the
release of reactive oxygen species (ROS) and nitric oxide alongside
NF-κB activation, which plays a pivotal role in the induction of
HIF. Elevated levels of HIF further promote the production of
multiple pro-inflammatory mediators, reinforcing the inflammatory
state within the TME (65,66). HIF-1α can interact with p53 to
inhibit its activity, thus reducing p53-induced apoptosis and
promoting tumor cell survival and metastasis (67). The inactivation of the tumor
suppressor p53 results in the upregulation of NF-κB, a key positive
regulator of inflammatory signaling, thus fostering a
pro-inflammatory microenvironment conducive to tumor metastasis
(68). HIF-1α modulates various
immune functions, including the polarization of M1 macrophages, the
maturation and migration of dendritic cells, and the formation and
survival of neutrophil extracellular traps. In comparison, another
transcription factor, HIF-2α, promotes M2 macrophage polarization
by inducing the expression of M2-associated markers, e.g., arginase
1 (69). HIF-1α has been shown to
influence the differentiation and function of different T-cell
subsets under both hypoxic and normoxic conditions (70,71).
HIF-2α is also expressed in TAMs and its depletion in TAMs impairs
the expression of chemokine receptors and the migration and
infiltration of TAMs (69).
Cellular senescence is characterized by progressive
mitochondrial dysfunction, resulting in elevated production of ROS,
such as H2O2, which increases the risk of
carcinogenesis (72). Studies
suggest that alterations in senescence-associated secretory
phenotype (SASP) gene expression in senescent CAFs facilitate
malignant tumor proliferation (73). In tumor tissues,
H2O2-activated CAFs interact with tumor cells
to produce H2O2, mimicking the behavior of
immune cells like macrophages and neutrophils, driving local and
systemic inflammation through the innate immune response, mainly
via NF-κB activation (Fig.
1E) (74). Pro-inflammatory
cytokines mediate the epigenetic modification of H3K27me3 in CAFs,
thus sustaining the SASP and promoting peritoneal tumor formation
in gastric cancer by activating the JAK/STAT3 signaling pathway
(75).
Cellular senescence is distinguished by a reduced
proliferative potential, the activation of anti-apoptotic pathways
and the secretion of pro-inflammatory cytokines, chemokines and
interleukins (IL-6, IL-1α and IL-1β). It also involves releasing
growth factors, proteases and their inhibitors, angiogenic factors
and insoluble components (fibronectin, collagen and laminin). Other
inflammatory mediators, including growth differentiation factor-15,
TGFβ1 and IFNγ, also contribute to this secretory profile,
collectively called the SASP (76).
Senescent cells demonstrate high oxidative metabolism and ROS
production (76). The complex
interaction among the SASP, oxidative stress and inflammation
highlights the intrinsic association between cellular senescence
and the initiation and progressive accumulation of inflammatory
responses (77).
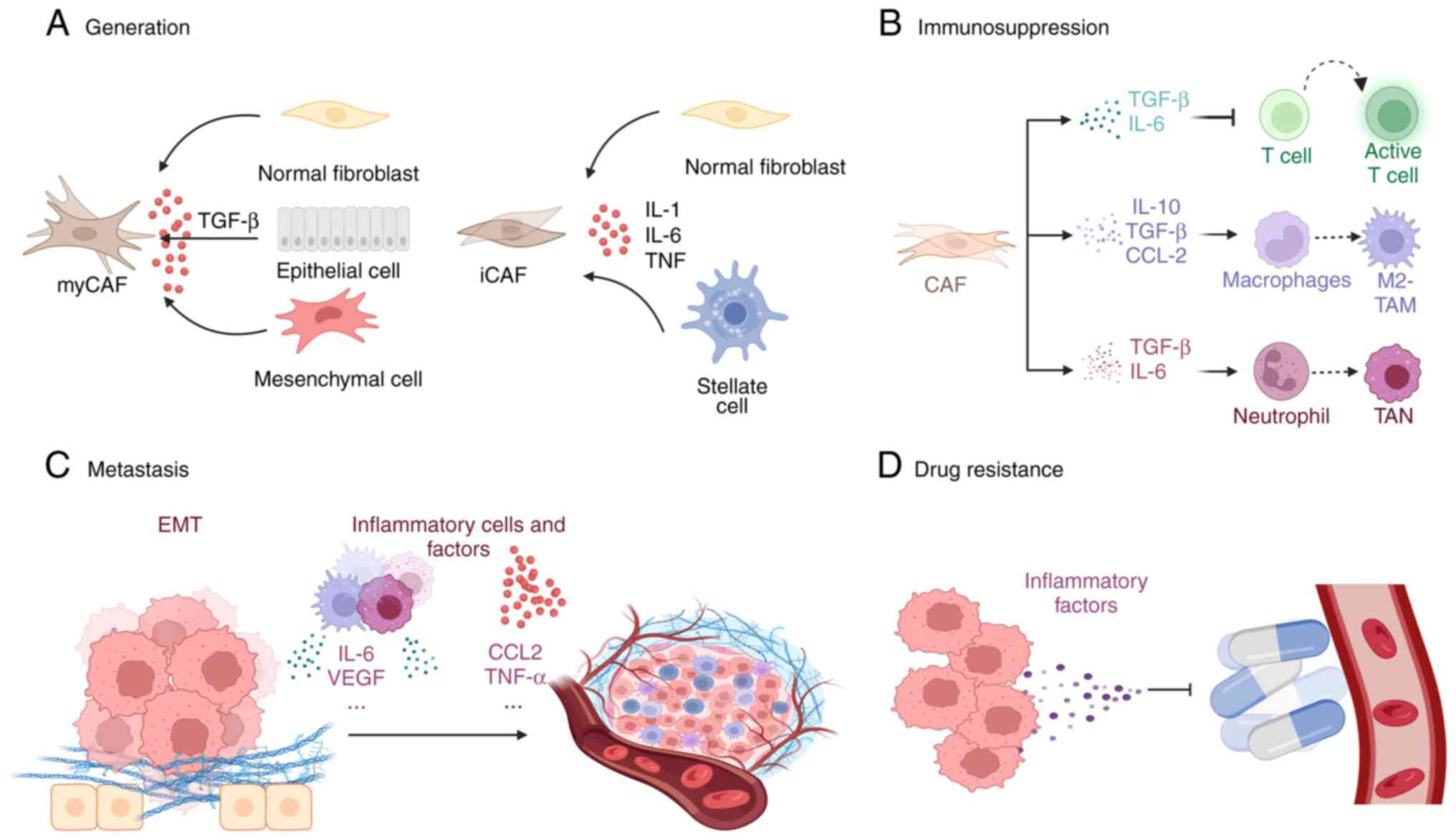
These CAFs represent a heterogeneous population with
distinct origins, phenotypic characteristics and functional
properties, contributing significantly to the complexity of the
TME-their heterogeneity results in particular functions for
different types of CAF. For instance, myCAFs, which show elevated
expression of α-smooth muscle actin (αSMA), promote fibrosis and
contribute to ECM remodeling to support tumor growth (21). However, iCAFs, characterized by low
expression of αSMA and the secretion of IL-6 and other
pro-inflammatory mediators, contribute to immune evasion and
resistance to chemotherapy (21).
Furthermore, antigen-presenting CAFs, distinguished by the
expression of major histocompatibility complex class II genes,
function as decoy receptors to promote immune suppression (78). Inflammation plays a crucial role in
shaping both the phenotype and functional properties of CAFs
(Fig. 2).
MyCAFs originate from various cell types, including
fibroblasts, smooth muscle cells and epithelial cells. Their
activation is primarily driven by inflammatory signals and tissue
injury (Fig. 2A). These cells
functionally resemble contractile fibroblasts involved in wound
healing. TGF-β is a key regulator of myCAF differentiation,
inducing their activation from multiple cell types. Upon
activation, TGF-β binds to its receptor, leading to the
phosphorylation of Smad2/3, which then form a complex with Smad4
and translocate into the nucleus to regulate gene transcription
(12); TGF-β promotes CAF formation
primarily through the Smad-dependent signaling pathway, where
Smad2/3 phosphorylation leads to CAF differentiation with high
expression of αSMA and SDF1/CXCL12 (79–81).
The upregulation of microRNA-21 (miR-21) in CAFs, mediated by the
TGF-β signaling pathway, enhances their CAF-like morphology and
migratory capacity (82). Smad7, a
negative feedback regulator that antagonizes receptor/Smad
signaling, plays a complementary role in modulating this pathway.
Either depletion of Smad7 or increased expression of miR-21
contributes to the sustained activation of CAFs, ultimately
promoting tumor progression (82).
The EMT process with increased expression of FAP, α-SMA and
vimentin, primarily driven by TGF-β, further facilitates the
differentiation of myCAFs, contributing to fibrosis and metastasis
(83,84). In addition to Smad signaling, TGF-β
activates non-Smad pathways, including PI3K/AKT and MAPK/ERK, which
collectively control fibroblast proliferation, contractility and
ECM deposition (85). Furthermore,
macrophage recruitment can activate hematopoietic stem cells,
maintained by TNF and IL-1, which promote myCAFs activation
via ROS and NF-κB-dependent pathways (39,86).
MyCAFs are contractile and ECM-remodeling cells that highly express
αSMA, transgelin, periostin and collagen-related genes (21). They play a crucial role in
modulating the mechanical and structural properties of the tumor
stroma. By promoting desmoplasia, myCAFs increase tissue stiffness
and contribute to the formation of a dense ECM that acts as a
physical barrier, thereby limiting immune cell infiltration and
reducing the efficacy of drug delivery.
Growing evidence suggests that the IL-6/STAT3 and
TGF-β signaling pathways interact synergistically, forming a
positive feedback loop that sustains inflammation-driven activation
of CAF. IL-6, a pro-inflammatory cytokine, activates the JAK/STAT3
pathway, which in turn enhances TGF-β signaling by promoting the
phosphorylation of Smad3, a key mediator of TGF-β-induced
transcriptional responses (90).
Treatment with IL-6 increased the expression of TGF-β type I
receptor in A549, NCI-H358 and NHLF cells, thus enhancing TGF-β
signaling and fibroblast activation (91). However, certain findings indicate
that the simultaneous presence of cytokines may lead to
TGF-β-mediated suppression of IL-6-induced proliferative effects,
highlighting more intricate regulatory dynamics (92,93).
The immunosuppressive properties of CAFs are
modulated by inflammatory signaling cascades, cytokines and
chemokines (Fig. 2B). IL-6 and
TGF-β are crucial mediators through which CAFs suppress cytotoxic T
lymphocyte infiltration, and blocking IL-6 has been shown to
enhance T-cell function (94,95).
The CXCL12-CXCR4 signaling axis promotes the interaction between
CAFs and monocytes, inducing the reprogramming of monocytes into an
immunosuppressive phenotype (96).
Furthermore, CAFs also promote the recruitment of monocytes and
their differentiation into M2-TAMs by secreting IL-8, IL-10, TGF-β
and CCL2, thus impairing effector T-cell responses and inducing
immunosuppression within the TME (97). CAFs also mediate neutrophil
chemotaxis and activation of TANs through the IL-6/STAT3/ERK1/2
axis (98). IL-6 stimulation
activates the STAT3 signaling pathway in TANs, suppressing T-cell
activity and inducing immune tolerance via PD-L1 expression
(99). In melanoma and colorectal
cancer, CXCL5 facilitates the upregulation of PD-L1 expression on
tumor cells via a PI3K/AKT-dependent mechanism, thus
enhancing immune tolerance and promoting tumor immune evasion
(100).
The ECM, comprising structural components such as
collagen and HA, plays a pivotal role in tumor chemoresistance by
establishing a dense matrix that serves as a physical barrier, thus
hindering the effective penetration of therapeutic agents (107). CAFs contribute to this resistance
by secreting cytokines, chemokines, growth factors and exosomes,
which engage their respective signaling pathways, ultimately
protecting cancer cells from apoptosis induced by therapeutic
interventions (Fig. 2D).
Inflammation plays a crucial role in tumor
formation. During tumor treatment, a reduction in the
neutrophil-to-lymphocyte ratio induces the reprogramming of iCAFs,
leading to a marked decrease in IL-6/STAT-3 expression and
enhancing chemotherapy sensitivity in preclinical models of
pancreatic cancer (9). Hypoxia
within the TME, driven by COX-2 secretion from CAFs, M2 macrophages
and cancer cells, and its positive interaction with Yes-associated
protein 1 and anti-apoptotic mediators, fosters cancer cell
resistance to chemotherapy (14).
Furthermore, exosomal miRNA-20a secreted by CAFs inhibits the
phosphatase and tensin homolog/PI3K-AKT pathway, promoting
non-small cell lung cancer progression and inducing resistance to
cisplatin (108).
Multiple therapeutic strategies targeting CAFs and
their role in tumor-associated inflammation have been explored in
preclinical and clinical research (Table I). For instance, inhibition of TGF-β
receptor 1 (300 mg/day) has been associated with prolonged patient
survival in phase II clinical trials for pancreatic cancer and HCC
(22,23). However, considering the functional
heterogeneity of CAFs, broad inhibition of TGF-β may inadvertently
induce immunosuppressive effects. To overcome this bottleneck,
combination approaches, such as co-administering TGF-β inhibitors
with immune checkpoint blockade therapies (e.g., anti-PD-L1
antibodies), have demonstrated superior anti-tumor immune responses
compared to monotherapy (24).
Similarly, in a phase I clinical trial, IL-6 inhibitors (1, 2, 4 or
8 mg/kg intravenously, every 4 weeks) have been shown to stimulate
CD8+ T-cell activation and increase levels of anti-tumor effectors,
such as IFN-γ and TNF-α (25). In a
dual recombinase-driven model of pancreatic ductal adenocarcinoma
(PDAC), the knockdown of IL-6 on αSMA+ CAFs markedly enhanced the
efficacy of gemcitabine, whereas its deletion from FAP+ CAFs did
not yield a similar effect (109).
Furthermore, although IL-6 blockade failed to demonstrate synergy
with anti-PD-1 immunotherapy, it significantly accelerated
gemcitabine-induced tumor suppression, ultimately prolonging
survival in PDAC mouse models (109). These findings highlight the
intricate and context-dependent roles of CAF-derived IL-6 in tumor
progression and therapeutic resistance.
Recent therapeutic strategies increasingly emphasize
the role of stromal proteins and HIF-1α in mediating the crosstalk
between inflammation and CAFs. Structural proteins within the tumor
stroma, such as collagen and fibronectin, contribute to forming a
fibrotic ECM that facilitates tumor progression and immune evasion.
Targeting these stromal components offers a promising approach to
modulating CAF activity and suppressing inflammation. The findings
from a phase III clinical trial demonstrated that integrating
stromal protein-targeting agents with gemcitabine and paclitaxel
prolonged patient survival and reduced adverse effects associated
with treatment (112). Similarly,
HIF-1α, a key mediator of hypoxia and inflammation, has emerged as
a dual-action therapeutic mediator. Inhibiting HIF-1α disrupts the
tumor-promoting functions of CAFs and enhances immune responses,
underscoring its clinical potential (113,114).
Nanoparticle-based drug delivery systems have become
effective platforms for improving drug penetration within tumors.
However, CAFs pose substantial challenges to the efficacy of
nanomedicine by establishing physical and biochemical barriers
within the TME. To overcome these obstacles, researchers have
designed CAF-targeted nanoparticle delivery systems capable of
either modulating CAF activity to enhance drug diffusion or
directly transporting therapeutic agents to the tumor stroma, thus
improving treatment efficacy (115). A sequential nanomedicine approach
using dasatinib to remodel the ECM and enhance epirubicin
penetration has demonstrated promising results in breast cancer
models. This strategy effectively reduces ECM deposition,
facilitates improved drug delivery, enhances anti-tumor immune
responses and works synergistically with anti-programmed death-1
therapy. Thus, it significantly inhibits tumor growth and prevents
lung metastasis while minimizing systemic toxicity (116).
Based on the existing methods, the combined
therapies are becoming increasingly prevalent. These strategies
target CAFs while addressing inflammation or immune suppression,
aiming to interfere with multiple tumor-supportive mechanisms and
strengthen anti-cancer immunity. For instance, the combined
administration of TGF-β, WNT, COX, PD-1/PD-L1 inhibitors and
cytotoxic chemotherapy have demonstrated enhanced therapeutic
efficacy (24,117–119). Such multifaceted approaches
provide a comprehensive framework for overcoming tumor resistance
and optimizing therapeutic efficacy.
This review provides a comprehensive analysis of the
involvement of CAFs in tumor progression and therapy resistance,
with a particular focus on recent therapeutic advancements.
However, the findings presented are predominantly derived from
preclinical models and early-phase clinical trials, with limited
long-term clinical validation of multiple CAF-targeted therapies
(120–123). Furthermore, the heterogeneity of
CAFs across different tumor types suggests that certain findings
may not be universally applicable. Future research should
prioritize adding data from large-scale clinical studies and
patient-derived models to refine the understanding and optimization
of CAF-targeted therapeutic approaches.
Understanding the intricate crosstalk between CAFs
and inflammation is crucial for identifying precise therapeutic
targets and enhancing treatment efficacy. Inflammation is a basic
regulator of CAF-mediated tumor progression; however, current
therapeutic strategies often address these factors independently,
potentially limiting their effectiveness. Dual-targeted approaches
that simultaneously modulate CAF-associated signaling pathways and
inflammatory cytokines may lead to improved clinical outcomes.
Specifically, pathways such as IL-6/STAT3 and TGF-β, which regulate
inflammatory responses and CAF activation, represent promising
targets for more effective anti-tumor interventions. In addition,
further investigation into the roles of hypoxia, oxidative stress
and cellular senescence in modulating CAF behavior may uncover
novel therapeutic opportunities.
Targeting the stromal microenvironment also holds
significant therapeutic promise. Stromal proteins and HIF-1α, as
key mediators of the crosstalk between inflammation and CAFs,
represent promising therapeutic targets. Expanding clinical trials
to evaluate inhibitors of these pathways, either as monotherapies
or in combination with chemotherapy and immunotherapy, will be
necessary for translating preclinical insights into clinical
applications. Furthermore, identifying reliable biomarkers
associated with the CAF-inflammation axis could enable more precise
patient stratification, facilitating the development of
personalized treatment strategies.
Further advancements in therapeutic strategies
should integrate innovations in nanoparticle-based drug delivery,
metabolic modulation and the design of combination therapies.
Incorporating these approaches into clinical trials will optimize
therapeutic efficacy while minimizing adverse effects. Therefore,
developing strategies targeting the CAF-inflammation axis will
improve cancer treatment outcomes and facilitate the emergence of
more personalized and effective therapeutic modalities.
This review synthesizes the bidirectional interplay
between CAFs and inflammatory processes, demonstrating how CAFs
drive pro-inflammatory signaling while inflammatory mediators
reinforce CAF activation. The study highlighted novel therapeutic
approaches targeting stromal elements and employing
nanotechnologies to disrupt CAF-tumor crosstalk. Notably,
pharmacological interventions simultaneously addressing CAF
signaling and inflammatory cytokines exhibit enhanced antitumor
effects compared to mono-targeted therapies. The present analysis
underscores the critical advantage of dual-pathway strategies that
concurrently modulate both CAF functionality and inflammatory
microenvironments, proposing this combined targeting approach as a
promising paradigm for optimizing cancer therapeutics.
Not applicable.
This study was supported by the National Natural Science
Foundation of China (grant no. 82274640).
Not applicable.
Conceptualization, XL and JW. Writing-original draft
preparation, XL. Review and editing, CW, HM and JW. Funding
acquisition, JW. All authors have read and agreed to the published
version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Duan Q, Zhang H, Zheng J and Zhang L:
Turning cold INTO hot: Firing up the tumor microenvironment. Trends
Cancer. 6:605–618. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalluri R: The biology and function of
fibroblasts in cancer. Nat Rev Cancer. 16:582–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shin N, Son GM, Shin DH, Kwon MS, Park BS,
Kim HS, Ryu D and Kang CD: Cancer-associated fibroblasts and
desmoplastic reactions related to cancer invasiveness in patients
with colorectal cancer. Ann Coloproctol. 35:36–46. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee D, Ham IH, Son SY, Han SU, Kim YB and
Hur H: Intratumor stromal proportion predicts aggressive phenotype
of gastric signet ring cell carcinomas. Gastric Cancer. 20:591–601.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Greten FR and Grivennikov SI: Inflammation
and cancer: Triggers, mechanisms, and consequences. Immunity.
51:27–41. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y
and Li Y: Inflammation and tumor progression: Signaling pathways
and targeted intervention. Signal Transduct Target Ther. 6:2632021.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rubinstein-Achiasaf L, Morein D,
Ben-Yaakov H, Liubomirski Y, Meshel T, Elbaz E, Dorot O, Pichinuk
E, Gershovits M, Weil M and Ben-Baruch A: Persistent inflammatory
stimulation drives the conversion of MSCs to inflammatory CAFs that
promote pro-metastatic characteristics in breast cancer cells.
Cancers (Basel). 13:14722021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Castro Silva I, Bianchi A, Deshpande
NU, Sharma P, Mehra S, Garrido VT, Saigh SJ, England J, Hosein PJ,
Kwon D, et al: Neutrophil-mediated fibroblast-tumor cell
il-6/stat-3 signaling underlies the association between
neutrophil-to-lymphocyte ratio dynamics and chemotherapy response
in localized pancreatic cancer: A hybrid clinical-preclinical
study. Elife. 11:e789212022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saito A, Horie M and Nagase T: TGF-beta
signaling in lung health and disease. Int J Mol Sci. 19:24602018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caja L, Dituri F, Mancarella S,
Caballero-Diaz D, Moustakas A, Giannelli G and Fabregat I: TGF-β
and the tissue microenvironment: Relevance in fibrosis and cancer.
Int J Mol Sci. 19:12942018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hawinkels LJ, Paauwe M, Verspaget HW,
Wiercinska E, van der Zon JM, van der Ploeg K, Koelink PJ, Lindeman
JH, Mesker W, ten Dijke P and Sier CF: Interaction with colon
cancer cells hyperactivates TGF-β signaling in cancer-associated
fibroblasts. Oncogene. 33:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rothwell PM, Wilson M, Price JF, Belch JF,
Meade TW and Mehta Z: Effect of daily aspirin on risk of cancer
metastasis: A study of incident cancers during randomised
controlled trials. Lancet. 379:1591–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goradel NH, Najafi M, Salehi E, Farhood B
and Mortezaee K: Cyclooxygenase-2 in cancer: A review. J Cell
Physiol. 234:5683–5699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herbertz S, Sawyer JS, Stauber AJ,
Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba
SC, Benhadji KA, et al: Clinical development of galunisertib
(LY2157299 monohydrate), a small molecule inhibitor of transforming
growth factor-beta signaling pathway. Drug Des Devel Ther.
9:4479–4499. 2015.PubMed/NCBI
|
|
16
|
Peng D, Fu M, Wang M, Wei Y and Wei X:
Targeting TGF-β signal transduction for fibrosis and cancer
therapy. Mol Cancer. 21:1042022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yao X, Huang J, Zhong H, Shen N, Faggioni
R, Fung M and Yao Y: Targeting interleukin-6 in inflammatory
autoimmune diseases and cancers. Pharmacol Ther. 141:125–139. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5:2092020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fosslien E: Molecular pathology of
cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 30:3–21.
2000.PubMed/NCBI
|
|
20
|
Scheller J, Garbers C and Rose-John S:
Interleukin-6: From basic biology to selective blockade of
pro-inflammatory activities. Semin Immunol. 26:2–12. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mao X, Xu J, Wang W, Liang C, Hua J, Liu
J, Zhang B, Meng Q, Yu X and Shi S: Crosstalk between
cancer-associated fibroblasts and immune cells in the tumor
microenvironment: New findings and future perspectives. Mol Cancer.
20:1312021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Faivre S, Santoro A, Kelley RK, Gane E,
Costentin CE, Gueorguieva I, Smith C, Cleverly A, Lahn MM, Raymond
E, et al: Novel transforming growth factor beta receptor I kinase
inhibitor galunisertib (LY2157299) in advanced hepatocellular
carcinoma. Liver Int. 39:1468–1477. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Melisi D, Garcia-Carbonero R, Macarulla T,
Pezet D, Deplanque G, Fuchs M, Trojan J, Oettle H, Kozloff M,
Cleverly A, et al: Galunisertib plus gemcitabine vs. gemcitabine
for first-line treatment of patients with unresectable pancreatic
cancer. Br J Cancer. 119:1208–1214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holmgaard RB, Schaer DA, Li Y, Castaneda
SP, Murphy MY, Xu X, Inigo I, Dobkin J, Manro JR, Iversen PW, et
al: Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI
small molecule inhibitor, promotes anti-tumor immunity leading to
durable, complete responses, as monotherapy and in combination with
checkpoint blockade. J Immunother Cancer. 6:472018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dijkgraaf EM, Santegoets SJ, Reyners AK,
Goedemans R, Wouters MC, Kenter GG, van Erkel AR, van Poelgeest MI,
Nijman HW, van der Hoeven JJ, et al: A phase I trial combining
carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal
antibody, and interferon-α2b in patients with recurrent epithelial
ovarian cancer. Ann Oncol. 26:2141–2149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mantovani A, Ponzetta A, Inforzato A and
Jaillon S: Innate immunity, inflammation and tumour progression:
double-edged swords. J Intern Med. 285:524–532. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu CT, Chen MF, Chen WC and Hsieh CC: The
role of IL-6 in the radiation response of prostate cancer. Radiat
Oncol. 8:1592013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lazennec G and Richmond A: Chemokines and
chemokine receptors: New insights into cancer-related inflammation.
Trends Mol Med. 16:133–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Balachander GM, Talukdar PM, Debnath M,
Rangarajan A and Chatterjee K: Inflammatory role of
cancer-associated fibroblasts in invasive breast tumors revealed
using a fibrous polymer scaffold. ACS Appl Mater Interfaces.
10:33814–33826. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Erez N, Truitt M, Olson P, Arron ST and
Hanahan D: Cancer-associated fibroblasts are activated in incipient
neoplasia to orchestrate tumor-promoting inflammation in an
NF-kappaB-dependent manner. Cancer Cell. 17:135–147. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jain S, Rick JW, Joshi RS, Beniwal A,
Spatz J, Gill S, Chang AC, Choudhary N, Nguyen AT, Sudhir S, et al:
Single-cell RNA sequencing and spatial transcriptomics reveal
cancer-associated fibroblasts in glioblastoma with protumoral
effects. J Clin Invest. 133:e1470872023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song M, He J, Pan QZ, Yang J, Zhao J,
Zhang YJ, Huang Y, Tang Y, Wang Q, He J, et al: Cancer-associated
fibroblast-mediated cellular crosstalk supports hepatocellular
carcinoma progression. Hepatology. 73:1717–1735. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiang H, Ramil CP, Hai J, Zhang C, Wang H,
Watkins AA, Afshar R, Georgiev P, Sze MA, Song XS, et al:
Cancer-associated fibroblasts promote immunosuppression by inducing
ROS-generating monocytic MDSCs in lung squamous cell carcinoma.
Cancer Immunol Res. 8:436–450. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Donelan W, Dominguez-Gutierrez PR and
Kusmartsev S: Deregulated hyaluronan metabolism in the tumor
microenvironment drives cancer inflammation and tumor-associated
immune suppression. Front Immunol. 13:9712782022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ershaid N, Sharon Y, Doron H, Raz Y, Shani
O, Cohen N, Monteran L, Leider-Trejo L, Ben-Shmuel A, Yassin M, et
al: NLRP3 inflammasome in fibroblasts links tissue damage with
inflammation in breast cancer progression and metastasis. Nat
Commun. 10:43752019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fang Z, Meng Q, Xu J, Wang W, Zhang B, Liu
J, Liang C, Hua J, Zhao Y, Yu X and Shi S: Signaling pathways in
cancer-associated fibroblasts: Recent advances and future
perspectives. Cancer Commun (Lond). 43:3–41. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W,
Dang Y, Chu Y, Fan J and He R: FAP promotes immunosuppression by
cancer-associated fibroblasts in the tumor microenvironment via
STAT3-CCL2 signaling. Cancer Res. 76:4124–4135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Biffi G, Oni TE, Spielman B, Hao Y, Elyada
E, Park Y, Preall J and Tuveson DA: IL1-induced JAK/STAT signaling
is antagonized by TGFβ to shape CAF heterogeneity in pancreatic
ductal adenocarcinoma. Cancer Discov. 9:282–301. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fukui H, Zhang X, Sun C, Hara K, Kikuchi
S, Yamasaki T, Kondo T, Tomita T, Oshima T, Watari J, et al: IL-22
produced by cancer-associated fibroblasts promotes gastric cancer
cell invasion via STAT3 and ERK signaling. Br J Cancer.
111:763–771. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ebbing EA, van der Zalm AP, Steins A,
Creemers A, Hermsen S, Rentenaar R, Klein M, Waasdorp C, Hooijer
GKJ, Meijer SL, et al: Stromal-derived interleukin 6 drives
epithelial-to-mesenchymal transition and therapy resistance in
esophageal adenocarcinoma. Proc Natl Acad Sci USA. 116:2237–2242.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brunetto E, De Monte L, Balzano G, Camisa
B, Laino V, Riba M, Heltai S, Bianchi M, Bordignon C, Falconi M, et
al: The IL-1/IL-1 receptor axis and tumor cell released
inflammasome adaptor ASC are key regulators of TSLP secretion by
cancer associated fibroblasts in pancreatic cancer. J Immunother
Cancer. 7:452019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Servais C and Erez N: From sentinel cells
to inflammatory culprits: Cancer-associated fibroblasts in
tumour-related inflammation. J Pathol. 229:198–207. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mueller L, Goumas FA, Affeldt M, Sandtner
S, Gehling UM, Brilloff S, Walter J, Karnatz N, Lamszus K, Rogiers
X and Broering DC: Stromal fibroblasts in colorectal liver
metastases originate from resident fibroblasts and generate an
inflammatory microenvironment. Am J Pathol. 171:1608–1618. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mueller L, von Seggern L, Schumacher J,
Goumas F, Wilms C, Braun F and Broering DC: TNF-alpha similarly
induces IL-6 and MCP-1 in fibroblasts from colorectal liver
metastases and normal liver fibroblasts. Biochem Biophys Res
Commun. 397:586–591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kojima Y, Acar A, Eaton EN, Mellody KT,
Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg
RA and Orimo A: Autocrine TGF-beta and stromal cell-derived
factor-1 (SDF-1) signaling drives the evolution of tumor-promoting
mammary stromal myofibroblasts. Proc Natl Acad Sci USA.
107:20009–20014. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ziani L, Chouaib S and Thiery J:
Alteration of the antitumor immune response by cancer-associated
fibroblasts. Front Immunol. 9:4142018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim R, Emi M and Tanabe K: Cancer
immunosuppression and autoimmune disease: Beyond immunosuppressive
networks for tumour immunity. Immunology. 119:254–264. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bauer J, Emon MAB, Staudacher JJ, Thomas
AL, Zessner-Spitzenberg J, Mancinelli G, Krett N, Saif MT and Jung
B: Author Correction: Increased stiffness of the tumor
microenvironment in colon cancer stimulates cancer associated
fibroblast-mediated prometastatic activin A signaling. Sci Rep.
10:76062020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fullar A, Dudas J, Olah L, Hollósi P, Papp
Z, Sobel G, Karászi K, Paku S, Baghy K and Kovalszky I: Remodeling
of extracellular matrix by normal and tumor-associated fibroblasts
promotes cervical cancer progression. BMC Cancer. 15:2562015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kobayashi N, Miyoshi S, Mikami T, Koyama
H, Kitazawa M, Takeoka M, Sano K, Amano J, Isogai Z, Niida S, et
al: Hyaluronan deficiency in tumor stroma impairs macrophage
trafficking and tumor neovascularization. Cancer Res. 70:7073–7083.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li X, Zhao S, Bian X, Zhang L, Lu L, Pei
S, Dong L, Shi W, Huang L, Zhang X, et al: Signatures of EMT,
immunosuppression, and inflammation in primary and recurrent human
cutaneous squamous cell carcinoma at single-cell resolution.
Theranostics. 12:7532–7549. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lu P, Weaver VM and Werb Z: The
extracellular matrix: A dynamic niche in cancer progression. J Cell
Biol. 196:395–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Minton K: Extracellular matrix:
Preconditioning the ECM for fibrosis. Nat Rev Mol Cell Biol.
15:766–767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: Regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Van den Steen PE, Proost P, Wuyts A, Van
Damme J and Opdenakker G: Neutrophil gelatinase B potentiates
interleukin-8 tenfold by aminoterminal processing, whereas it
degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2
intact. Blood. 96:2673–2681. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liubomirski Y, Lerrer S, Meshel T, Morein
D, Rubinstein-Achiasaf L, Sprinzak D, Wiemann S, Körner C, Ehrlich
M and Ben-Baruch A: Notch-mediated tumor-stroma-inflammation
networks promote invasive properties and CXCL8 expression in
triple-negative breast cancer. Front Immunol. 10:8042019.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Drev D, Bileck A, Erdem ZN, Mohr T,
Timelthaler G, Beer A, Gerner C and Marian B: Proteomic profiling
identifies markers for inflammation-related tumor-fibroblast
interaction. Clin Proteomics. 14:332017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Schworer S, Cimino FV, Ros M, Tsanov KM,
Ng C, Lowe SW, Carmona-Fontaine C and Thompson CB: Hypoxia
potentiates the inflammatory fibroblast phenotype promoted by
pancreatic cancer cell-derived cytokines. Cancer Res. 83:1596–1610.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lappano R, Talia M, Cirillo F,
Rigiracciolo DC, Scordamaglia D, Guzzi R, Miglietta AM, De
Francesco EM, Belfiore A, Sims AH and Maggiolini M: The IL1β-IL1R
signaling is involved in the stimulatory effects triggered by
hypoxia in breast cancer cells and cancer-associated fibroblasts
(CAFs). J Exp Clin Cancer Res. 39:1532020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pavlides S, Tsirigos A, Vera I, Flomenberg
N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn
UE, Howell A, et al: Transcriptional evidence for the ‘Reverse
Warburg Effect’ in human breast cancer tumor stroma and metastasis:
Similarities with oxidative stress, inflammation, Alzheimer's
disease, and ‘Neuron-Glia Metabolic Coupling’. Aging (Albany NY).
2:185–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pavlides S, Tsirigos A, Vera I, Flomenberg
N, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG,
et al: Loss of stromal caveolin-1 leads to oxidative stress, mimics
hypoxia and drives inflammation in the tumor microenvironment,
conferring the ‘reverse Warburg effect’: A transcriptional
informatics analysis with validation. Cell Cycle. 9:2201–2219.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Linares JF, Cid-Diaz T, Duran A, Osrodek
M, Martinez-Ordoñez A, Reina-Campos M, Kuo HH, Elemento O, Martin
ML, Cordes T, et al: The lactate-NAD(+) axis activates
cancer-associated fibroblasts by downregulating p62. Cell Rep.
39:1107922022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rossow L, Veitl S, Vorlova S, Wax JK, Kuhn
AE, Maltzahn V, Upcin B, Karl F, Hoffmann H, Gätzner S, et al:
LOX-catalyzed collagen stabilization is a proximal cause for
intrinsic resistance to chemotherapy. Oncogene. 37:4921–4940. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Korbecki J, Siminska D,
Gassowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D and
Baranowska-Bosiacka I: Chronic and cycling hypoxia: Drivers of
cancer chronic inflammation through HIF-1 and NF-κB activation: A
review of the molecular mechanisms. Int J Mol Sci. 22:107012021.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ravenna L, Principessa L, Verdina A,
Salvatori L, Russo MA and Petrangeli E: Distinct phenotypes of
human prostate cancer cells associate with different adaptation to
hypoxia and pro-inflammatory gene expression. PLoS One.
9:e962502014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Leszczynska KB, Foskolou IP, Abraham AG,
Anbalagan S, Tellier C, Haider S, Span PN, O'Neill EE, Buffa FM and
Hammond EM: Hypoxia-induced p53 modulates both apoptosis and
radiosensitivity via AKT. J Clin Invest. 125:2385–2398. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schwitalla S, Ziegler PK, Horst D, Becker
V, Kerle I, Begus-Nahrmann Y, Lechel A, Rudolph KL, Langer R,
Slotta-Huspenina J, et al: Loss of p53 in enterocytes generates an
inflammatory microenvironment enabling invasion and lymph node
metastasis of carcinogen-induced colorectal tumors. Cancer Cell.
23:93–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
McGettrick AF and O'Neill LAJ: The role of
HIF in immunity and inflammation. Cell Metab. 32:524–536. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Michalek RD, Gerriets VA, Jacobs SR,
Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG and
Rathmell JC: Cutting edge: Distinct glycolytic and lipid oxidative
metabolic programs are essential for effector and regulatory CD4+ T
cell subsets. J Immunol. 186:3299–3303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shi LZ, Wang R, Huang G, Vogel P, Neale G,
Green DR and Chi H: HIF1alpha-dependent glycolytic pathway
orchestrates a metabolic checkpoint for the differentiation of TH17
and Treg cells. J Exp Med. 208:1367–1376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Woo DK and Shadel GS: Mitochondrial stress
signals revise an old aging theory. Cell. 144:11–12. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Higashiguchi M, Murakami H, Akita H,
Kobayashi S, Takahama S, Iwagami Y, Yamada D, Tomimaru Y, Noda T,
Gotoh K, et al: The impact of cellular senescence and
senescence-associated secretory phenotype in cancer-associated
fibroblasts on the malignancy of pancreatic cancer. Oncol Rep.
49:982023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lisanti MP, Martinez-Outschoorn UE, Lin Z,
Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A and Sotgia F:
Hydrogen peroxide fuels aging, inflammation, cancer metabolism and
metastasis: The seed and soil also needs ‘fertilizer’. Cell Cycle.
10:2440–2449. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yasuda T, Koiwa M, Yonemura A, Miyake K,
Kariya R, Kubota S, Yokomizo-Nakano T, Yasuda-Yoshihara N, Uchihara
T, Itoyama R, et al: Inflammation-driven senescence-associated
secretory phenotype in cancer-associated fibroblasts enhances
peritoneal dissemination. Cell Rep. 34:1087792021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Suryadevara V, Hudgins AD, Rajesh A,
Pappalardo A, Karpova A, Dey AK, Hertzel A, Agudelo A, Rocha A,
Soygur B, et al: SenNet recommendations for detecting senescent
cells in different tissues. Nat Rev Mol Cell Biol. 25:1001–1023.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li X, Li C, Zhang W, Wang Y, Qian P and
Huang H: Inflammation and aging: Signaling pathways and
intervention therapies. Signal Transduct Target Ther. 8:2392023.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Elyada E, Bolisetty M, Laise P, Flynn WF,
Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS,
et al: Cross-species single-cell analysis of pancreatic ductal
adenocarcinoma reveals antigen-presenting cancer-associated
fibroblasts. Cancer Discov. 9:1102–1123. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jena BC, Sarkar S, Rout L and Mandal M:
The transformation of cancer-associated fibroblasts: Current
perspectives on the role of TGF-β in CAF mediated tumor progression
and therapeutic resistance. Cancer Lett. 520:222–232. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gu J, Qian H, Shen L, Zhang X, Zhu W,
Huang L, Yan Y, Mao F, Zhao C, Shi Y and Xu W: Gastric cancer
exosomes trigger differentiation of umbilical cord derived
mesenchymal stem cells to carcinoma-associated fibroblasts through
TGF-β/Smad pathway. PLoS One. 7:e524652012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ and
Feng YM: Cancer-associated fibroblasts induce
epithelial-mesenchymal transition of breast cancer cells through
paracrine TGF-β signalling. Br J Cancer. 110:724–732. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li Q, Zhang D, Wang Y, Sun P, Hou X,
Larner J, Xiong W and Mi J: MiR-21/Smad 7 signaling determines
TGF-β1-induced CAF formation. Sci Rep. 3:20382013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Barcellos-de-Souza P, Comito G,
Pons-Segura C, Taddei ML, Gori V, Becherucci V, Bambi F, Margheri
F, Laurenzana A, Del Rosso M and Chiarugi P: Mesenchymal stem cells
are recruited and activated into carcinoma-associated fibroblasts
by prostate cancer microenvironment-derived TGF-β1. Stem Cells.
34:2536–2547. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wei M, Yang T, Chen X, Wu Y, Deng X, He W,
Yang J and Wang Z: Malignant ascites-derived exosomes promote
proliferation and induce carcinoma-associated fibroblasts
transition in peritoneal mesothelial cells. Oncotarget.
8:42262–42271. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Heneberg P: Paracrine tumor signaling
induces transdifferentiation of surrounding fibroblasts. Crit Rev
Oncol Hematol. 97:303–311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Pradere JP, Kluwe J, De Minicis S, Jiao
JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman
R, et al: Hepatic macrophages but not dendritic cells contribute to
liver fibrosis by promoting the survival of activated hepatic
stellate cells in mice. Hepatology. 58:1461–1473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Somerville TD, Biffi G, Daßler-Plenker J,
Hur SK, He XY, Vance KE, Miyabayashi K, Xu Y, Maia-Silva D,
Klingbeil O, et al: Squamous trans-differentiation of pancreatic
cancer cells promotes stromal inflammation. Elife. 9:e533812020.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bianchi A, De Castro Silva I, Deshpande
NU, Singh S, Mehra S, Garrido VT, Guo X, Nivelo LA, Kolonias DS,
Saigh SJ, et al: Cell-autonomous Cxcl1 sustains tolerogenic
circuitries and stromal inflammation via neutrophil-derived TNF in
pancreatic cancer. Cancer Discov. 13:1428–1453. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Schauer IG, Zhang J, Xing Z, Guo X,
Mercado-Uribe I, Sood AK, Huang P and Liu J: Interleukin-1beta
promotes ovarian tumorigenesis through a p53/NF-κB-mediated
inflammatory response in stromal fibroblasts. Neoplasia.
15:409–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Abulaiti A, Shintani Y, Funaki S, Nakagiri
T, Inoue M, Sawabata N, Minami M and Okumura M: Interaction between
non-small-cell lung cancer cells and fibroblasts via enhancement of
TGF-β signaling by IL-6. Lung Cancer. 82:204–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Srivastava A, Sharma H, Khanna S,
Balasundaram TS, Chowdhury S, Chowdhury R and Mukherjee S:
Interleukin-6 induced proliferation is attenuated by transforming
growth factor-β-induced signaling in human hepatocellular carcinoma
cells. Front Oncol. 11:8119412021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wiegertjes R, van Caam A, van Beuningen H,
Koenders M, van Lent P, van der Kraan P, van de Loo F and Davidson
EB: TGF-β dampens IL-6 signaling in articular chondrocytes by
decreasing IL-6 receptor expression. Osteoarthritis Cartilage.
27:1197–1207. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kato T, Noma K, Ohara T, Kashima H,
Katsura Y, Sato H, Komoto S, Katsube R, Ninomiya T, Tazawa H, et
al: Cancer-associated fibroblasts affect intratumoral CD8(+) and
FoxP3(+) T cells via IL6 in the tumor microenvironment. Clin Cancer
Res. 24:4820–4833. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Thomas DA and Massague J: TGF-beta
directly targets cytotoxic T cell functions during tumor evasion of
immune surveillance. Cancer Cell. 8:369–380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Timperi E, Gueguen P, Molgora M, Magagna
I, Kieffer Y, Lopez-Lastra S, Sirven P, Baudrin LG, Baulande S,
Nicolas A, et al: Lipid-associated macrophages are induced by
cancer-associated fibroblasts and mediate immune suppression in
breast cancer. Cancer Res. 82:3291–3306. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nagarsheth N, Wicha MS and Zou W:
Chemokines in the cancer microenvironment and their relevance in
cancer immunotherapy. Nat Rev Immunol. 17:559–572. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Cheng Y, Li H, Deng Y, Tai Y, Zeng K,
Zhang Y, Liu W, Zhang Q and Yang Y: Cancer-associated fibroblasts
induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster
immune suppression in hepatocellular carcinoma. Cell Death Dis.
9:4222018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan
X, Mao F, Wang M, Zhu W, Qian H and Xu W: The IL-6-STAT3 axis
mediates a reciprocal crosstalk between cancer-derived mesenchymal
stem cells and neutrophils to synergistically prompt gastric cancer
progression. Cell Death Dis. 5:e12952014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li Z, Zhou J, Zhang J, Li S, Wang H and Du
J: Cancer-associated fibroblasts promote PD-L1 expression in mice
cancer cells via secreting CXCL5. Int J Cancer. 145:1946–1957.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liubomirski Y, Lerrer S, Meshel T,
Rubinstein-Achiasaf L, Morein D, Wiemann S, Körner C and Ben-Baruch
A: Tumor-Stroma-inflammation networks promote pro-metastatic
chemokines and aggressiveness characteristics in triple-negative
breast cancer. Front Immunol. 10:7572019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Comito G, Giannoni E, Segura CP,
Barcellos-de-Souza P, Raspollini MR, Baroni G, Lanciotti M, Serni S
and Chiarugi P: Cancer-associated fibroblasts and M2-polarized
macrophages synergize during prostate carcinoma progression.
Oncogene. 33:2423–2431. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Hashimoto O, Yoshida M, Koma Y, Yanai T,
Hasegawa D, Kosaka Y, Nishimura N and Yokozaki H: Collaboration of
cancer-associated fibroblasts and tumour-associated macrophages for
neuroblastoma development. J Pathol. 240:211–223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ocana A, Nieto-Jimenez C, Pandiella A and
Templeton AJ: Neutrophils in cancer: prognostic role and
therapeutic strategies. Mol Cancer. 16:1372017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chen H, Han X, Zhang Y, Wang K, Liu D, Hu
Z and Wang J: Bruceine D suppresses CAF-promoted TNBC metastasis
under TNF-α stimulation by inhibiting Notch1-Jagged1/NF-κB(p65)
signaling. Phytomedicine. 123:1549282023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang R, Zong J, Peng Y, Shi J, Du X, Liu
H, Shen Y, Cao J, Jia B, Liu F and Zhang J: GPR30 knockdown weakens
the capacity of CAF in promoting prostate cancer cell invasion via
reducing macrophage infiltration and M2 polarization. J Cell
Biochem. 3:299382021.
|
|
107
|
Louault K, Li RR and DeClerck YA:
Cancer-Associated fibroblasts: Understanding their heterogeneity.
Cancers (Basel). 12:31082020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Shi L, Zhu W, Huang Y, Zhuo L, Wang S,
Chen S, Zhang B and Ke B: Cancer-associated fibroblast-derived
exosomal microRNA-20a suppresses the PTEN/PI3K-AKT pathway to
promote the progression and chemoresistance of non-small cell lung
cancer. Clin Transl Med. 12:e9892022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
McAndrews KM, Chen Y, Darpolor JK, Zheng
X, Yang S, Carstens JL, Li B, Wang H, Miyake T, Correa de Sampaio
P, et al: Identification of functional heterogeneity of
carcinoma-associated fibroblasts with distinct IL6-mediated therapy
resistance in pancreatic cancer. Cancer Discov. 12:1580–1597. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Galbiati A, Zana A, Bocci M, Millul J,
Elsayed A, Mock J, Neri D and Cazzamalli S: A Dimeric FAP-targeting
small-molecule radioconjugate with high and prolonged tumor uptake.
J Nucl Med. 63:1852–1858. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lee IK, Noguera-Ortega E, Xiao Z, Todd L,
Scholler J, Song D, Liousia M, Lohith K, Xu K, Edwards KJ, et al:
Monitoring therapeutic response to anti-FAP CAR T cells using
[18F]AlF-FAPI-74. Clin Cancer Res. 28:5330–5342. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Van Cutsem E, Tempero MA, Sigal D, Oh DY,
Fazio N, Macarulla T, Hitre E, Hammel P, Hendifar AE, Bates SE, et
al: Randomized phase III trial of pegvorhyaluronidase alfa with
nab-paclitaxel plus gemcitabine for patients with hyaluronan-high
metastatic pancreatic adenocarcinoma. J Clin Oncol. 38:3185–3194.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wei TT, Lin YT, Tang SP, Luo CK, Tsai CT,
Shun CT and Chen CC: Metabolic targeting of HIF-1alpha potentiates
the therapeutic efficacy of oxaliplatin in colorectal cancer.
Oncogene. 39:414–427. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cowman SJ and Koh MY: Revisiting the HIF
switch in the tumor and its immune microenvironment. Trends Cancer.
8:28–42. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Guo J, Zeng H and Chen Y: Emerging nano
drug delivery systems targeting cancer-associated fibroblasts for
improved antitumor effect and tumor drug penetration. Mol Pharm.
17:1028–1048. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhang Y, Fang Z, Pan D, Li Y, Zhou J, Chen
H, Li Z, Zhu M, Li C, Qin L, et al: dendritic polymer-based
nanomedicines remodel the tumor stroma: Improve drug penetration
and enhance antitumor immune response. Adv Mater. 36:e24013042024.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Huang TX, Tan XY, Huang HS, Li YT, Liu BL,
Liu KS, Chen X, Chen Z, Guan XY, Zou C and Fu L: Targeting
cancer-associated fibroblast-secreted WNT2 restores dendritic
cell-mediated antitumour immunity. Gut. 71:333–344. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Edelman MJ, Wang X, Hodgson L, Cheney RT,
Baggstrom MQ, Thomas SP, Gajra A, Bertino E, Reckamp KL, Molina J,
et al: Phase III randomized, placebo-controlled, double-blind trial
of celecoxib in addition to standard chemotherapy for advanced
non-small-cell lung cancer with cyclooxygenase-2 overexpression:
CALGB 30801 (Alliance). J Clin Oncol. 35:2184–2192. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Pelly VS, Moeini A, Roelofsen LM, Bonavita
E, Bell CR, Hutton C, Blanco-Gomez A, Banyard A, Bromley CP,
Flanagan E, et al: Anti-inflammatory drugs remodel the tumor immune
environment to enhance immune checkpoint blockade efficacy. Cancer
Discov. 11:2602–2619. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wang C, Li S, Wang Y, An Y, Shen K, Wang
X, Luan W, Ma F, Ni L, Zhou H, et al: Targeting IRS-1/mPGES-1/NOX2
to inhibit the inflammatory response caused by insulin-like growth
factor-I-induced activation of NF-κB and NLRP3 in cancer cells. Vet
Comp Oncol. 18:689–698. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Guo B, Fu S, Zhang J, Liu B and Li Z:
Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci
Rep. 6:361072016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Dorst DN, Smeets EMM, Klein C, Frielink C,
Geijs D, Trajkovic-Arsic M, Cheung PFY, Stommel MWJ, Gotthardt M,
Siveke JT, et al: Fibroblast activation protein-targeted
photodynamic therapy of cancer-associated fibroblasts in murine
models for pancreatic ductal adenocarcinoma. Mol Pharm.
20:4319–4330. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kato T, Furusawa A, Okada R, Inagaki F,
Wakiyama H, Furumoto H, Fukushima H, Okuyama S, Choyke PL and
Kobayashi H: Near-infrared photoimmunotherapy targeting
podoplanin-expressing cancer cells and cancer-associated
fibroblasts. Mol Cancer Ther. 22:75–88. 2023. View Article : Google Scholar : PubMed/NCBI
|