Introduction
Lysosomes, often referred to as the digestive units
of the cells, are membrane-bound organelles essential for recycling
damaged intracellular components and organelles. In addition to
their degradative functions, lysosomes play key roles in multiple
physiological and pathological processes (1), including nutrient sensing, metabolic
regulation and aging. They have emerged as critical mediators of
drug resistance, particularly in cancer treatment with tyrosine
kinase inhibitors (TKIs) (2,3). The
acidic environment within lysosomes, coupled with the cytosolic pH
gradient, facilitates the passive uptake of hydrophobic, weakly
basic drugs such as TKIs. This pH-dependent sequestration reduces
TKI efficacy by trapping within lysosomes (4–6).
Cancer cells with increased drug sequestration have been shown to
exhibit heightened resistance to therapy, suggesting that lysosomal
trapping is a major therapeutic barrier to treatment success
(3,6). However, the mechanisms by which cells
tolerate high TKI concentrations within lysosomes remain poorly
understood.
Lysosomes are as crucial intracellular
Ca2+ stores, releasing Ca2+ to regulate
signaling pathways essential for organelle homeostasis and
acidification (7). Ca2+
channels, including transient receptor potential mucolipins
(TRPMLs) and two-pore channels located on the endolysosomal
membrane, are essential for proliferation, migration and calcium
homeostasis, all of which are significant both normal physiology
and disease (8). The TRPML family,
also known as mucolipins, consists of three isoforms, TRPML1, −2
and −3, each functioning as inward-rectifying cation channels with
physiological and pathological roles (9). TRPML1, the first identified isoform,
has been linked to type IV mucolipidosis (MLIV), a disorder caused
by loss-of-function mutations (10). It is activated by
phosphatidylinositol-3,5-bisphosphate, voltage changes and low
luminal pH (11). As a
non-selective cation channel, it facilitates lysosomal degradation.
Its deficiency leads to excessively acidic lysosomes due to
impaired H+ efflux (12). Fibroblasts from patients with MLIV
exhibit defective lysosomal degradation of macromolecules (13). TRPML1 regulates autophagy by
releasing Ca2+, which is necessary for mTORC1 modulation
and autophagosome-lysosome fusion (14). Finally, it promotes lysosomal
biogenesis by releasing Ca2+, which triggers
transcription factor EB (TFEB) dephosphorylation and nuclear
translocation (15).
TRPML3 also plays a key role in endocytic and
autophagic pathways (16). As a
phosphatidylinositol-3-phosphate effector, it facilitates
Ca2+ release into the cytoplasm, a process essential for
membrane fusion and autophagosome formation (17). Despite their functional overlap,
TRPML1 and TRPML3 differ in pH sensitivity: The latter is fully
active at a higher pH (~6.3), whereas the former operates optimally
at a lower pH (~4-5.5) (18).
Neutralizing lysosomal pH with uropathogenic E. coli has
been shown to activate TRPML3, triggering Ca2+ release
and lysosomal exocytosis (19).
Reduced luminal Ca2+ in early endosomes has been linked
to increased acidification (20),
while Ca2+ release through TRPML3 supports H+
uptake via the vacuolar H+ pump, helping maintain
lysosomal acidity (21). These
findings suggest the hypothesis that TRPML3 enhances lysosomal TKI
sequestration, contributing to acquired drug resistance. This
hypothesis warrants further investigation.
In the present study, TRPML3 expression in non-small
cell lung cancer (NSCLC) tissues during the development of drug
resistance was initially determined. Using two- and
three-dimensional (2D and 3D, respectively) cell-culture models,
TRPML3 function in the regulation of lysosomal pH, biogenesis and
drug resistance was investigated. It was found that TRPML3 enhances
lysosomal sequestration of osimertinib and compensates for TRPML1
in lysosomal biogenesis during treatment, significantly
contributing to drug resistance.
Materials and methods
Cell culture, reagents and
transfection
Human PC9 and HCC827 NSCLC cell lines were
generously provided by Dr Jin Kyung Rho (Asan Medical Center, Ulsan
University). The cells were cultured in RPMI-1640 medium (HyClone;
Cytiva) supplemented with 10% fetal bovine serum (FBS) and
antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin) in a
humidified incubator with 5% CO2. All cell lines used in
the present study were authenticated by short tandem repeat
profiling (KOMA Biotech, Inc.) and were routinely screened for
microbial contamination as part of standard quality control
procedures. The TRPML3-GCaMP6 construct, generated by inserting the
full-length GCaMP6 sequence into the p3XFLAG-CMV-7.1-TRPML3 vector
(17), was kindly provided by Dr
Hyun Jin Kim (Sungkyunkwan University). The pCMV-TRPML3 expression
vector containing wild-type TRPML3 (NM_018298) was obtained from
OriGene Technologies, Inc. Reagents and their sources were as
follows: osimertinib (Selleck Chemicals), Cell Fractionation Kit
(Cell Signaling Technology, Inc.), anti-TFEB (cat. no. 4240s),
anti-Nanog (cat. no. 3580s; both from Cell Signalling Technology,
Inc.), TRPML agonist ML-SA1 (MilliporeSigma; cat. no. SML0627),
TRPML inhibitor ML-SI1 (GlpBio; cat. no. GC19764), anti-Myc (cat.
no. sc-40; Santa Cruz Biotechnology, Inc.), anti-CD44 (cat. no.
MA5-13890; Thermo Fisher Scientific, Inc.), anti-CD133 (cat. no.
ab16048), anti-lamin-B1 (cat. no. ab19898; both from Abcam),
anti-TRPML1 (cat. no. MBS9205163; MyBioSource, Inc.), anti-TRPML3
(cat. no. 13879-1-AP; Proteintech Group, Inc.) and anti-GAPDH (cat.
no. bs2188R; BIOSS). For transient gene silencing, synthetic small
interfering RNAs (siRNAs) targeting TRPML3 and scrambled
control siRNAs were purchased from Integrated DNA Technologies,
Inc. Their sequences were as follows: TRPML3 siRNA sense,
5′-GUAGAAUUUACCUCUACUCAUUCAT-3′ and antisense,
3′-AUCAUCUUAAAUGGAGAUGAGUAAGUA-5′; scrambled siRNA sense,
5′-CGUUAAUCGCGUAUAAUACGCUAT-3′ and antisense,
3′-AUACGCGUAUUAUACGCGAUUAACGAC-5′. The siRNAs and expression
vectors were introduced using Lipofectamine RNAiMAX and
Lipofectamine 3000, respectively, according to the manufacturer's
protocols (Thermo Fisher Scientific, Inc.). In brief, each mixture
of siRNA (30 nM per 35-mm dish) with RNAiMAX and expression vectors
(2.5 µg per 35-mm dish) with Lipofectamine 3000 was incubated for 5
min and 15 min, respectively, at 24°C. Each sample was then
subjected to subsequent experiment following an additional
incubation of 72 and 24 h, respectively. To generate a stable
TRPML3 knockout cell line, an sgRNA targeting TRPML3 (RefSeq
NM_018298.11) was designed and synthesized by Macrogen, Inc.
(5′-CAGCTACTCACAACTACCTCAGG-3′). It was inserted into the
pSp-U6-Cas9-2A-Puro plasmid; cells were transfected with either the
TRPML3-specific construct or the empty vector control. After
puromycin selection (2–10 µg/ml) for two weeks, the TRPML3 knockout
was confirmed using western blotting.
Osimertinib-resistant NSCLC sub-cell
lines
Resistant sub-lines of PC9 and HCC827, designated as
PC9/OR and HCC827/OR, respectively, were generated by gradually
increasing osimertinib concentrations (1×10−4,
1×10−3, 1×10−2, and 1×10−1 µM)
over five months. Surviving cells were cultured in
1×10−1 µM osimertinib for an additional month to confirm
the stability of resistance. Viability at each subculture step was
assessed using 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl
tetrazolium bromide (MTT) assays.
Collection and analysis of publicly
available RNA sequencing (RNA-Seq) data
To measure gene expression changes in lung
adenocarcinoma (LUAD) tissues during the development of osimertinib
resistance, a publicly available RNA-seq dataset (GSE253742) from
the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) was analyzed,
representing paired LUAD tumor samples collected before and after
osimertinib treatment (22). Data
were processed using Expression Differential Expression Gene
Analysis (ExDEGA v5.1.1.1) software (Ebiogen Inc.). Briefly, raw
data were subjected to quality control with FastQC, followed by
adapter trimming and low-quality read removal using Fastp (23). Processed reads were aligned to the
reference genome with STAR (24),
and quantified using Salmon (25).
Read count normalization was performed using the TMM + CPM method
in EdgeR (26). Data mining and
visualization were performed using ExDEGA and Multi Experiment
Viewer (MeV 4.9.0; http://www.tm4.org) (27).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cell lines using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), following the manufacturer's protocol. Subsequently, 1 µg of
RNA was converted into cDNA using the High-Capacity RNA-to-cDNA kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.), following the
manufacturer's protocol. Quantitative PCR (qPCR) was conducted
using VeriQuest SYBR Green qPCR Master Mix (Affymetrix; Thermo
Fisher Scientific, Inc.) on an ABI PRISM 7900 Sequence Detection
System (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermal cycling conditions were as follows: initial denaturation at
95°C for 20 sec, followed by 40 cycles of denaturation at 95°C for
3 sec and annealing/extension at 60°C for 30 sec. Primers were as
follows: TRPML3 forward, 5′-TCCGTTGGGAATCATGCTTAT-3′ and
reverse, 5′-AGTGCTGACAGATTGCCATAGC-3′; GAPDH forward,
5′-ATGGAAATCCCATCACCATCTT-3′ and reverse, 5′-CGCCCCACTTGATTTTGG-3′.
GAPDH expression was used to normalize data and controls for
variations in mRNA concentration. Expression levels were calculated
using the 2−ΔΔCq method (28).
Generation of cancer cell line-derived
spheroid
To generate 3D spheroid cultures, cells were
resuspended in culture medium supplemented with threefold the
standard FBS concentration at a density of 6×104
cells/ml for PC9 and its subline and 4.2×104 cells/ml
for HCC827 and its subline. Cell suspensions were mixed with
VitroGel Hydrogel Matrix (Well Bioscience Inc.; http://www.thewellbio.com) at a 2:1 (v/v) ratio; the
50 µl of the mixture was added per well in U-bottom 96-well plates
(S-bio Sumitomo Bakelite Co., Ltd.). Plates were centrifuged at
1,000 × g for 5 min at 24°C, and 200 µl of regular culture medium
was added per well. Aggregates were incubated in the hydrogel
matrix at 37°C in a 5% CO2 atmosphere for three days to
form multilayered spheroids. These spheroids were treated with
osimertinib or left untreated (DMSO vehicle only) for an additional
9 days, with medium refreshed every 3 days. Spheroids were imaged
using a NIKON Eclipse TS100 microscope equipped with an FL-20BW
CMOS camera (Tucsen Photonics Co., Ltd.). Images were captured with
Mosaic 2.4 software (Tucsen Photonics Co., Ltd.) and analyzed using
ImageJ software (version 1.51k; National Institutes of Health) to
measure spheroid diameters and other parameters. Viability was
measured using the Cell Counting Kit-8 (CCK-8) assay kit (Dojindo
Laboratories, Inc.), according to the manufacturer's protocol. In
brief, 20 µl of CCK-8 solution was added to each well and incubated
at 37°C for 1 h, after which the absorbance was measured at 450 nm
using an iMark microplate reader (Bio-Rad Laboratories, Inc.).
Western blot analysis
Western blotting was performed using established
methods (29). Cell lysates
prepared in RIPA buffer (Invitrogen; Thermo Fisher Scientific,
Inc.) were sonicated for 5 sec at 50% amplitude and incubated on
ice for 10 min. Debris was pelleted at 14,440 × g for 10 min at
4°C. Supernatant protein concentrations were quantified using the
BCA assay. Equal amounts of protein (20 µg per sample) were
separated using 8% SDS-PAGE, followed by transfer to 0.2-µm
polyvinylidene difluoride membranes (Cytiva). The membrane was
blocked with 5% bovine serum albumin (GenDEPOT, LLC) in
Tris-buffered saline with 0.1% Tween 20 at 4°C for 2 h. Primary
antibodies against TRPML1, TRPML3, TFEB, lamin-B1, Myc, CD44,
CD133, Nanog and GAPDH were used at a 1:1,000 dilution. The
membranes were incubated overnight at 4°C with the primary
antibodies, followed by incubation with horseradish peroxidase
(HRP)-linked secondary IgG antibody (1:2,500; Santa Cruz
Biotechnology, Inc.; cat. no. sc-516102 for anti-mouse, and cat.
no. sc-2357 for anti-rabbit) for 2 h at 4°C. Protein bands were
visualized and quantified using the AzureSpot 2.0 software (Azure
Biosystems, Inc.). Whole-cell lysate from spheroids cultured in
VitroGel Hydrogel Matrix (Well Bioscience, Inc.) were extracted
using VitroGel Organoid Recovery Solution (Well Bioscience, Inc.)
according to the manufacturer's protocol. Briefly, spheroids were
transferred to 1.5-ml tubes, washed with PBS, and resuspended in 1
ml of pre-warmed (37°C) VitroGel Organoid Recovery Solution. The
solution was gently pipetted to isolate the spheroids, followed by
a brief incubation at 37°C. After centrifugation at 100 × g for 5
min, pellets were collected and lysed in RIPA buffer. To measure
TFEB translocation from the cytoplasm to the nucleus, cytosolic and
nuclear fractions were isolated using a Cell Fractionation Kit
(Cell Signaling Technology, Inc.), following the manufacturer's
instructions. Briefly, cell pellets were resuspended in Cytoplasm
Isolation Buffer and incubated at 4°C for 5 min. Suspensions were
centrifuged at 500 × g for 5 min at 4°C, and cytosolic supernatants
were collected. Nuclear pellets were resuspended in Membrane
Isolation Buffer, re-pelleted at 8,000 × g for 5 min at 4°C, and
resuspended in Cytoskeleton/Nuclear Isolation Buffer. Both
fractions were analyzed using western blotting.
Cell cycle analysis
Cells were seeded at a density of 7×105
per 60 mm culture dish. Following incubation under the designated
conditions, they were detached using 0.05% Trypsin-EDTA (Gibco;
Thermo Fisher Scientific, Inc.) and fixed in 70% ethanol at −20°C
for 2 h, then stained with 0.5 ml of propidium iodide/RNase
staining buffer (BD Biosciences) for 15 min. Flow cytometry was
performed on at least 20,000 cells using a FACScan analyzer (BD
CellQuestTM Pro version 5.2; BD Biosciences).
MTT assay
Cell viability was assessed using the Viability
Assay Kit (Cellrix; MediFab) following the manufacturer's protocol.
Cells were seeded at a density of 1×104 per well in
96-well plates, treated with 10 µl of assay reagent containing
water-soluble tetrazolium salt, and incubated at 37°C for 30 min.
The production of water-soluble formazan was evaluated by measuring
the absorbance at 450 nm using an iMark microplate reader (Bio-Rad
Laboratories, Inc.).
Imaging of intracellular
Ca2+ (Ca2+i) and green fluorescent
protein (GFP)
Intracellular Ca2+ levels were measured
by incubation with Fura-2/AM (MilliporeSigma) at 37°C for 50 min,
followed by perfusion with a HEPES-buffered solution containing 140
mmol/l NaCl, 5 mmol/l KCl, 1 mmol/l MgCl2, 1 mmol/l
CaCl2, 10 mmol/l HEPES, and 10 mmol/l glucose, adjusted
to pH 7.4 and 310 mOsm. For Ca2+-free conditions, the
CaCl2 was replaced with 1 mmol/l EGTA. Osimertinib and
other compounds, including ML-SI1 and MS-SA1, were diluted in a
Ca2+-free HEPES buffer. Fluorescence excitation was
performed at 340 and 380 nm, with emission at 510 nm-captured using
an sCMOS camera (Andor Technology Ltd.). Ratio-metric data
(F340/F380) were analyzed using Meta-Fluor
Fluorescence Ratio Imaging software (Molecular Devices, LLC).
GCaMP6 fluorescence intensities were measured at an excitation
wavelength of 488 nm, with emission at 510 nm measured using an
sCMOS camera (Andor Technology Ltd.) and analyzed using Meta-Fluor
Fluorescence Ratio Imaging software (Molecular Devices, LLC).
Measurement of lysosomal pH
Lysosomal pH was measured using a LysoSensor
Yellow/Blue DND-160 (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Cells cultured in 35 mm confocal
dishes (SPL Life Sciences) were incubated with the dye for 10 min
at 37°C. After a brief wash with HEPES buffer, cells were
continuously exposed to osimertinib diluted in HEPES buffer.
Fluorescent images were captured using an sCMOS camera (Andor
Technology Ltd.) with excitation at 329 and 384 nm and emission at
440 and 540 nm, respectively. Data were analyzed using MetaMorph
software (Molecular Devices, LLC), and lysosomal pH was determined
by calculating the fluorescence intensity ratio of 384 nm (yellow,
acidic) to 329 nm (blue, less acidic), with fold changes expressed
relative to the control (scr-cells pre-osimertinib treatment).
Measurement of lysosomal area in a
single cell
The lysosomal area in individual cells was measured
using LysoTracker Red DND-99 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions. Cells were plated in
35 mm confocal dishes and transfected with either siML3 or
scrambled RNA as a control. After 72 h, the cells were treated with
0.01 µM osimertinib, incubated for 24 h, and stained with
LysoTracker Red DND-99 for 30 min. Fluorescence was excited at 577
nm and emitted at 590 nm, and images were captured using a confocal
microscope (FV1000; Olympus Corporation). Image analysis was
performed using ImageJ software (version 1.52v; National Institutes
of Health).
Statistical analysis
Data were analyzed using Origin 2020 software
(OriginLab Corporation). Results are presented as the mean ±
standard deviation (SD) based on a minimum of three independent
experiments. Statistical significance was evaluated using one-way
analysis of variance (ANOVA), followed by Tukey's post-hoc test for
multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Endogenous TRPML3 expression is
upregulated in osimertinib-residual LUAD tumor tissues and
osimertinib-resistant NSCLC cell lines
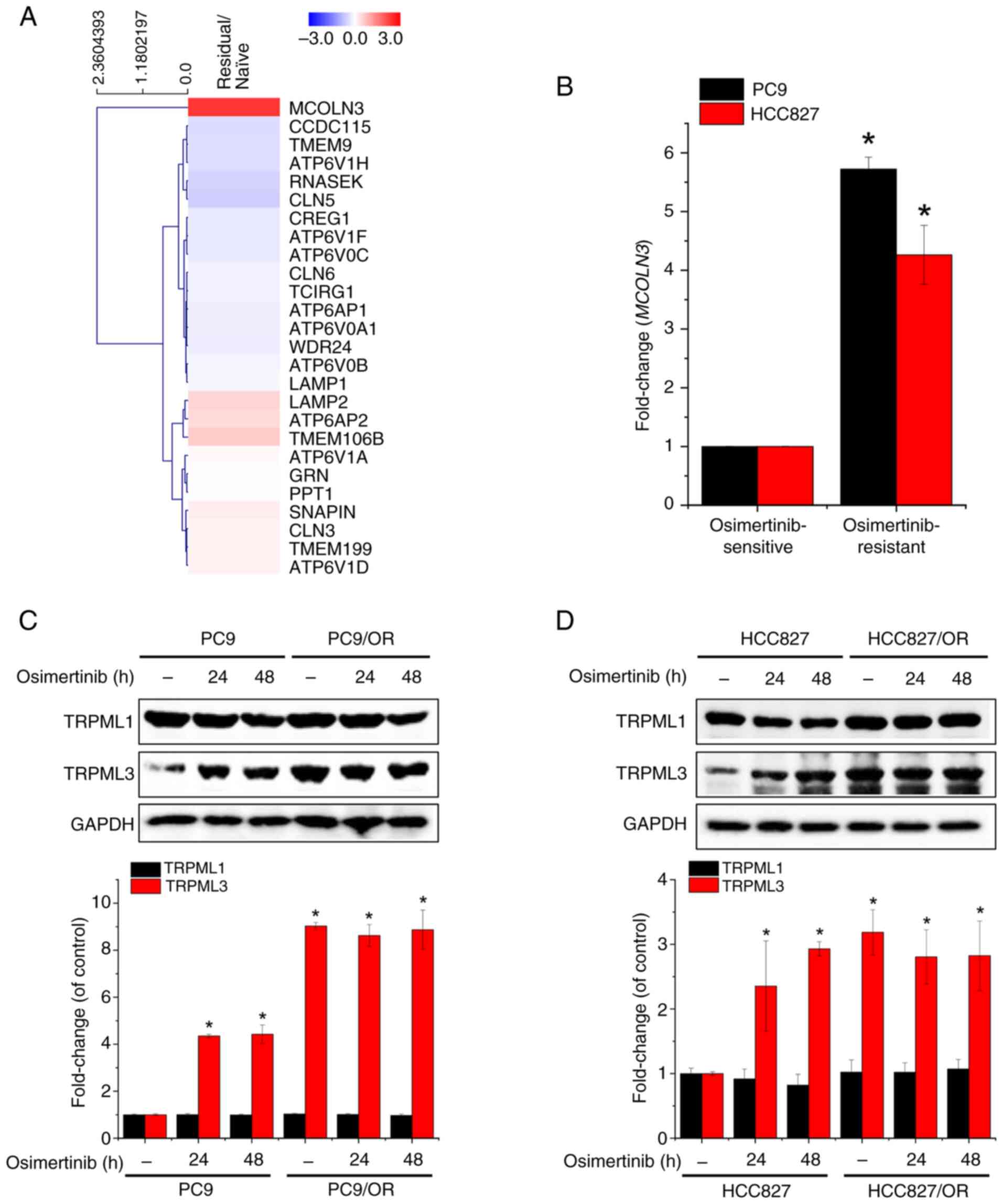
To identify genes involved in osimertinib-induced
lysosomal pH dysregulation, the GEO dataset GSE253742 was analyzed,
which includes RNA-seq data from eight clinical LUAD specimens
(four naive tumors and four osimertinib-residual tumors). Of the
40,929 genes analyzed, TRPML3 was the only gene whose
expression was significantly increased (5.121±0.045; |fold
change|≥2, log2 ≤3, P<0.05) in osimertinib-residual tumors
within a subset of 26 genes related to lysosomal acidification
(GO:0007042) (Fig. 1A). Based on
these findings, the expression was next measured in NSCLC cell
lines, specifically PC9 and HCC827, and in their
osimertinib-resistant sublines. TRPML3 mRNA levels were
significantly higher in osimertinib-resistant cells than in their
parental lines (Fig. 1B).
Additionally, osimertinib exposure and resistance increased TRPML3
protein expression, whereas TRPML1 expression remained unchanged
regardless of treatment or resistance (Fig. 1C and D). These findings suggest that
TRPML3 plays a critical role in the lysosomal response associated
with osimertinib resistance in NSCLC.
TRPML3 knockdown reduces multi-drug
tolerance in osimertinib-resistant PC9 cells
siRNA-mediated TRPML3 knockdown (siML3) was
next performed in PC9 cells and their osimertinib-resistant subline
PC9/OR. TRPML3 expression was efficiently reduced without affecting
TRPML1 expression (Fig. 2A). Next,
the effects of TRPML3 depletion on osimertinib-induced G0/G1
cell-cycle arrest was examined. Cells transfected with siML3 or
scrambled RNA (control) were incubated for 48 h with or without
0.01 µM osimertinib. In PC9 cells, TRPML3 knockdown did not
alter osimertinib-induced G0/G1 arrest but increased the sub-G0
population (Fig. 2B), suggesting
increased cell death. By contrast, TRPML3 knockdown in
PC9/OR cells restored their sensitivity to osimertinib-induced
G0/G1 arrest (Fig. 2C). All tested
TKIs, including osimertinib (Fig.
2D), lapatinib (Fig. 2E),
gefitinib (Fig. 2F) and erlotinib
(Fig. 2G), reduced PC9 viability in
a dose-dependent manner. By contrast, PC9/OR cells displayed
cross-resistance to all TKIs except for lapatinib. Finally,
TRPML3 knockdown restored the sensitivity to osimertinib and
other TKIs in these resistant cells. These findings indicate that
TRPML3 plays a critical role in mediating TKI resistance in NSCLC
cells.
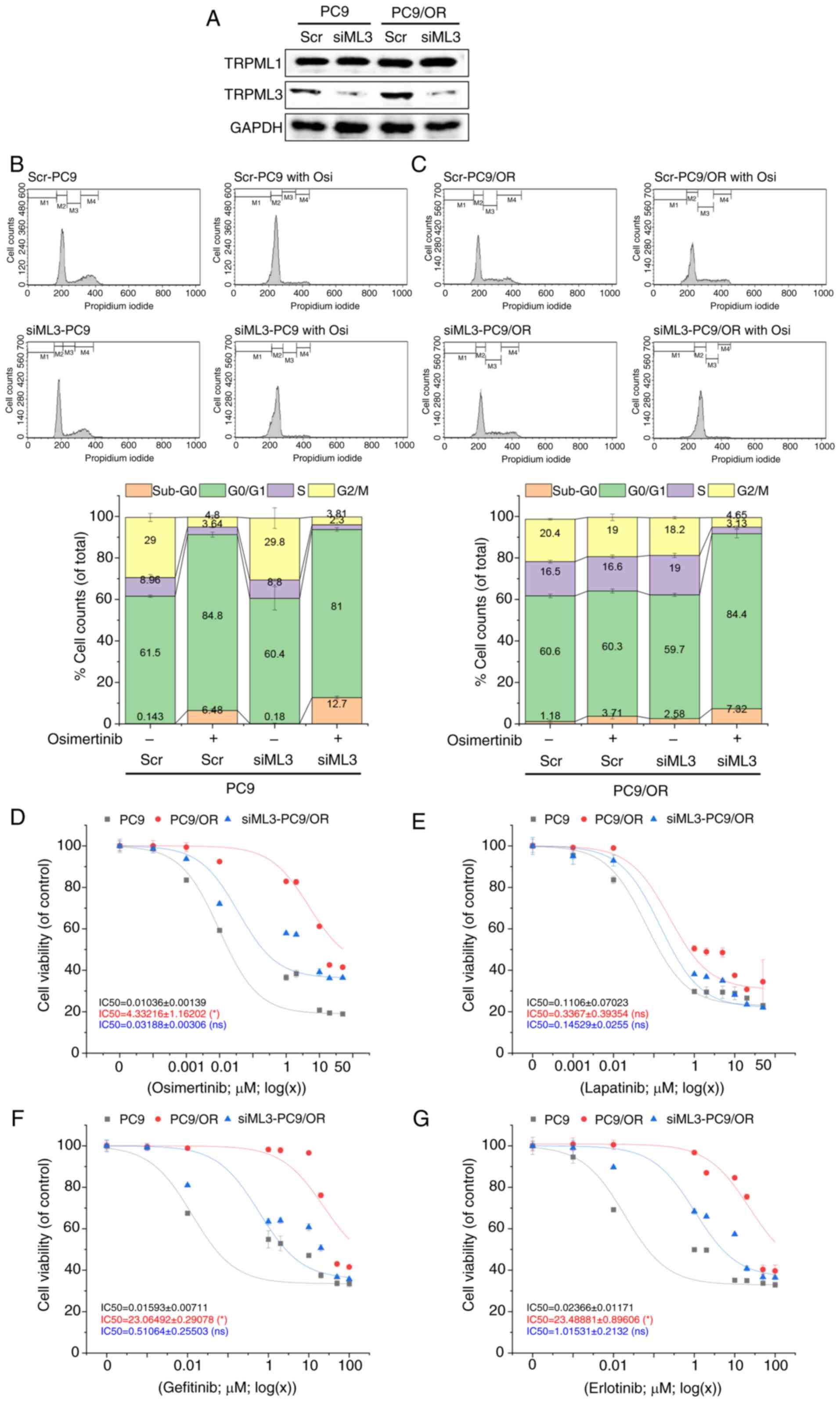 | Figure 2.TRPML3 deletion restores sensitivity
to osimertinib-induced cell-cycle arrest and death in PC9/OR cells.
(A) Confirmation of TRPML3 siRNA knockdown in PC9 and PC9/OR cells,
with no effect on TRPML1 expression. GAPDH was used for
normalization. (B and C) Cell-cycle phase distributions in (B) PC9
and (C) PC9/OR cells transfected with siML3 or scrambled RNA (scr),
then treated with 0.01 µM osimertinib for 48 h. Flow cytometric
data are presented as percentages of cells per phase
(2×104 cells per sample, n=4). (D-G) Viability of PC9,
PC9/OR and siML3-PC9/OR cells treated with increasing
concentrations of (D) osimertinib, (E) lapatinib, (F) gefitinib, or
(G) erlotinib for 72 h, assessed by MTT assay. Data show
percentages relative to DMSO-treated controls (n=3). TRPML,
transient receptor potential mucolipin; si-, small interfering. |
TRPML3 modulates osimertinib
sensitivity and growth in spheroids
To better mimic tumors in vivo, 3D spheroids
derived from TRPML3-overexpressing and -silenced cells
treated with osimertinib treatment for 9 days were employed. was
initiated on day 0 and maintained for 9 days. Spheroid viability
and size were assessed using CCK-8 assays and bright-field imaging,
respectively (Fig. 3A). Western
blotting confirmed changes in TRPML3 expression without affecting
TRPML1 expression (Fig. 3B).
Blotting with antibodies against the myc tag and TRPML3 verified
pCMV-TRPML3 expression in PC9 and HCC827 cells, whereas targeting
TRPML3 with sgRNA reduced endogenous TRPML3 expression in PC9/OR
and HCC827/OR cells. On day 0, increased expression of the spheroid
markers CD44, CD133 and Nanog was observed in spheroids, but not in
parental 2D cultures (Fig. 3C and
D). TRPML3 modulation did not affect spheroid formation. Next,
the effects of TRPML3 overexpression (Fig. 3E and F) and silencing (Fig. 3G and H) on osimertinib-induced
toxicity were assessed in spheroids derived from
osimertinib-sensitive (PC9 and HCC827) and -resistant (PC9/OR and
HCC827/OR) cells. Osimertinib produced a dose-dependent decrease in
spheroid viability in sensitive cell-derived spheroids (Fig. 3E and F), whereas those derived from
resistant cells exhibited greater tolerance (Fig. 3G and H). TRPML3 overexpression in
PC9 (ML3 OE-PC9) and HCC827 (ML3 OE-HCC827) spheroids attenuated
osimertinib-induced viability reductions at 0.1 µM (PC9) and 1 µM
(HCC827). Conversely, TRPML3 silencing significantly reduced
osimertinib tolerance in spheroids derived from PC9/OR and
HCC827/OR cells (Fig. 3G and H).
Consistent with these findings, intracellular TRPML3 levels
influenced osimertinib-induced reductions in spheroid size.
Spheroids were treated with osimertinib at the induced
concentrations; bright-field images were captured every 3 days
until day 9. Those derived from PC9, HCC827 and their sublines
continued to increase in size in the absence of osimertinib
(Fig. 3I-P). By contrast,
osimertinib treatment reduced spheroid size in both PC9 (Fig. 3I) and HCC827 (Fig. 3J) cells in a dose-dependent manner.
Spheroids from TRPML3-overexpressing PC9 (Fig. 3K) and HCC827 (Fig. 3L) cells exhibited increased
resistance to osimertinib, suppressing size reduction, whereas
those derived from PC9/OR (Fig. 3M)
and HCC827/OR (Fig. 3N) cells
maintained their size even at higher osimertinib concentrations
(0.01 and 0.1 µM for PC9/OR; 0.1 µM for HCC827/OR). TRPML3
silencing in PC9/OR (Fig. 3O) and
HCC827/OR (Fig. 3P) cells restored
osimertinib sensitivity, reducing spheroid size at 0.1 µM
osimertinib.
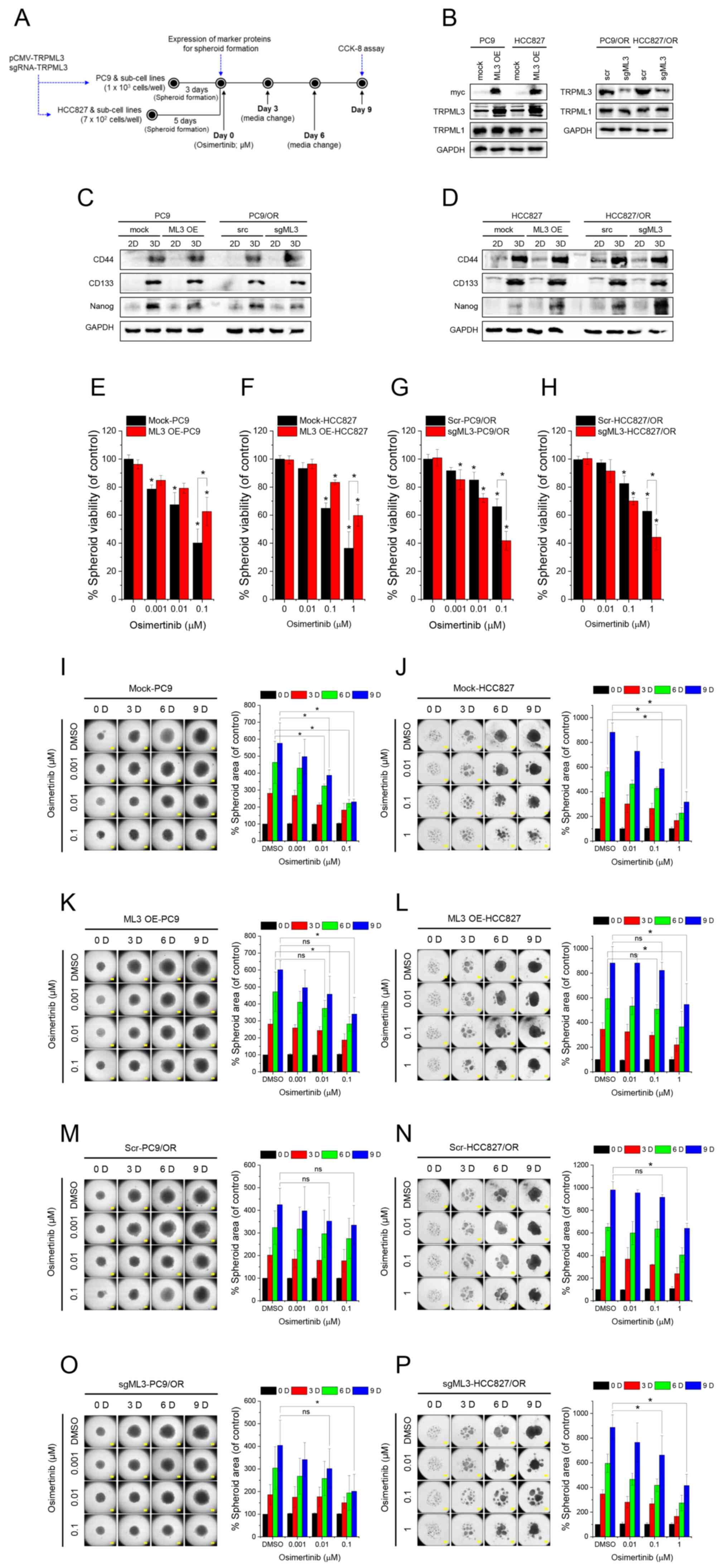 | Figure 3.TRPML3 modulates osimertinib
sensitivity and growth in spheroids. (A) Schematic representation
of the spheroid formation protocol and the subsequent osimertinib
treatment. (B) Expression levels of TRPML3 and −1 in PC9, HCC827
and their sublines (n=3). (C and D) Expression levels of spheroid
formation markers, CD44, CD133 and Nanog in spheroid lysates from
(C) PC9 and PC9/OR, (D) HCC827 and HCC827/OR, and conventional 2D
cultures (2D). (E-H) Spheroid viability assessed using Cell
Counting Kit-8 assays on day 9 of osimertinib treatment. Viability
is expressed as percentage relative to corresponding DMSO-treated
controls: (E) mock-PC9 and ML3 OE-PC9, (F) mock-HCC827 and ML3
OE-HCC827, (G) scr PC9/OR and sgML3-PC9/OR, (H) scr-HCC827/OR and
sgML3-HCC827/OR. Data are shown as the mean ± SD (n=3). (I-P)
Bright-field images of spheroids derived from mock-PC9 (I; n=3),
mock-HCC827 (J; n=4), ML3 OE-PC9 (K; n=3), ML3 OE-HCC827 (L; n=4),
scr-PC9/OR (M; n=3), scr-HCC827/OR (N; n=4), sgML3-PC9/OR (O; n=3),
and sgML3-HCC827/OR (P; n=4) cells, captured at 3-day intervals
following spheroid formation. Osimertinib treatment (DMSO, 0.001,
0.01, 0.1, and 1 µM) was initiated on day 0. Columns adjacent to
each image show overall spheroid size relative to the control
DMSO-treated spheroids on day 0. Data are presented as the mean ±
SD. Scale bar, 400 µm. *P<0.05. TRPML, transient receptor
potential mucolipin; ns, not significant. |
TRPML3-mediated lysosomal
Ca2+ release drives osimertinib-induced intracellular
Ca2+ oscillations
To improve understanding of the TRPML3-mediated TKI
resistance, it was determined whether TRPML3 channel activity
contributed to osimertinib resistance by measuring
[Ca2+]i. Osimertinib treatment induced
intracellular Ca2+ oscillations in PC9 cells,
independently of extracellular Ca2+ (Fig. 4A). These oscillations were more
pronounced in resistant PC9/OR cells (Fig. 4B) and were inhibited by ML-SI1, a
mucolipin channel blocker (Fig.
4C). Ca2+i oscillation frequency was
significantly reduced in TRPML3-deficient cells but not in
TRPML1-deficient cells (Fig. 4D-F).
In TRPML3-deficient PC9 cells, Ca2+ spikes were
significantly reduced, whereas PC9/OR cells with TRPML3
overexpression exhibited enhanced oscillations (Fig. 4G). To determine whether these
oscillations depend on lysosomal Ca2+ release via
TRPML3, the TRPML3-GCaMP6 construct was used. GFP fluorescence
intensity closely mirrored the Fura-2 fluorescence pattern in
osimertinib-treated cells, consistent with TRPML3-mediated
lysosomal Ca2+ release (Fig.
4H and I).
![Osimertinib-induced
Ca2+i oscillations depend on lysosomal
Ca2+ release mediated by TRPML3.
[Ca2+]i measured using Fura-2/AM in cells
treated with 0.01 µM osimertinib in the absence of extracellular
Ca2+. (A and B) Osimertinib-induced
Ca2+i mobilization in (A) PC9 and (B) PC9/OR
cells. (C) Effects of ML-SI1, a TRPML channel blocker, on
osimertinib-induced Ca2+i oscillations. (D-F)
Ca2+i oscillations in PC9 cells transfected
with scrambled RNA (D; scr), TRPML1-targeting siRNA (E; siML1), or
TRPML3-targeting siRNA (F; siML3), following osimertinib treatment.
Each line represents Ca2+i mobilization in a
single cell. (G) Statistical analysis of Ca2+ spike
frequencies (*P<0.05, n=6). (H and I) Lysosomal Ca2+
release in (H) PC9 and (I) HCC827 cells expressing TRPML3-GCaMP6
and treated with 0.01 µM osimertinib in the absence of
extracellular Ca2+. ML-SA1 was used as a positive
control. Lines indicate changes in GFP intensity (F488)
in individual cells, reflecting TRPML3-mediated lysosomal
Ca2+ release. F340/380 and F488
values were normalized to the mean and are presented as
F340/380-norm and F488-norm, respectively.
TRPML, transient receptor potential mucolipin; si-, small
interfering; scr, scrambled.](/article_images/or/54/3/or-54-03-08946-g03.jpg) | Figure 4.Osimertinib-induced
Ca2+i oscillations depend on lysosomal
Ca2+ release mediated by TRPML3.
[Ca2+]i measured using Fura-2/AM in cells
treated with 0.01 µM osimertinib in the absence of extracellular
Ca2+. (A and B) Osimertinib-induced
Ca2+i mobilization in (A) PC9 and (B) PC9/OR
cells. (C) Effects of ML-SI1, a TRPML channel blocker, on
osimertinib-induced Ca2+i oscillations. (D-F)
Ca2+i oscillations in PC9 cells transfected
with scrambled RNA (D; scr), TRPML1-targeting siRNA (E; siML1), or
TRPML3-targeting siRNA (F; siML3), following osimertinib treatment.
Each line represents Ca2+i mobilization in a
single cell. (G) Statistical analysis of Ca2+ spike
frequencies (*P<0.05, n=6). (H and I) Lysosomal Ca2+
release in (H) PC9 and (I) HCC827 cells expressing TRPML3-GCaMP6
and treated with 0.01 µM osimertinib in the absence of
extracellular Ca2+. ML-SA1 was used as a positive
control. Lines indicate changes in GFP intensity (F488)
in individual cells, reflecting TRPML3-mediated lysosomal
Ca2+ release. F340/380 and F488
values were normalized to the mean and are presented as
F340/380-norm and F488-norm, respectively.
TRPML, transient receptor potential mucolipin; si-, small
interfering; scr, scrambled. |
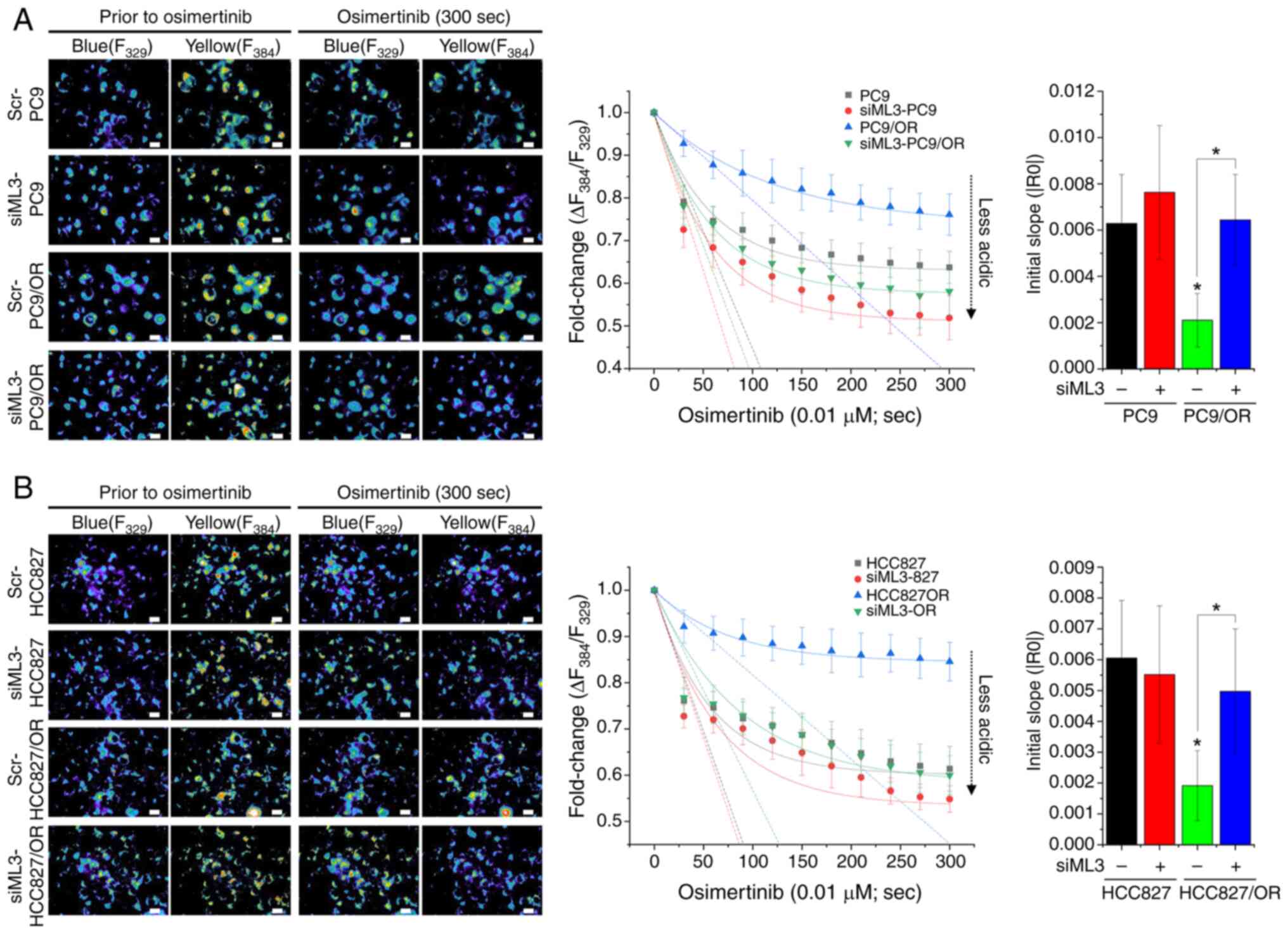
TRPML3 knockdown enhances
osimertinib-induced lysosomal pH increase
Lysosomal pH changes were next monitored using
LysoSensor Yellow/Blue DND-160. The baseline fluorescence
intensities were recorded before treatment, followed by continuous
perfusion with HEPES buffer containing osimertinib. Osimertinib
rapidly increased lysosomal pH in both PC9 (Fig. 5A) and HCC827 cells (Fig. 5B), reaching saturation at 60–65% of
baseline. By contrast, osimertinib-resistant cells (PC9/OR and
HCC827/OR) exhibited both a slower initial increase in lysosomal pH
and a lower saturation level. TRPML3 deletion in
osimertinib-resistant cells accelerated initial lysosomal pH
elevation and increased its saturation level compared with that in
scrambled control cells. These findings indicated that TRPML3 plays
a critical role in the regulation of lysosomal pH changes induced
by osimertinib in PC9 cells. Moreover, osimertinib-resistant NSCLC
cells increased sequestration of osimertinib within lysosomes, a
process that depends on TRPML3 expression. This suggests a link
between TRPML3-mediated lysosomal Ca2+ release and
regulation of lysosomal acidity.
TRPML3 deletion impairs
osimertinib-induced TFEB nuclear translocation and lysosomal
biogenesis
Building on the role of TRPML3 in lysosomal acidity
regulation, it was determined whether it also plays a role in
osimertinib-induced lysosomal biogenesis, which has been primarily
associated with TPRML1. TFEB translocation from the cytosol to the
nucleus was analyzed by fractionating total cellular proteins.
Osimertinib increased the time-dependent nuclear translocation of
TFEB in both PC9 (Fig. 6A) and
HCC827 (Fig. 6B) cells. However,
TRPML3 deletion completely blocked TFEB translocation. LysoTracker
Red DND-99 staining revealed that the osimertinib-induced increase
in lysosomal area was significantly reduced in TRPML3-deficient PC9
(Fig. 6C) and HCC827 (Fig. 6D) cells. In TRPML3-deficient cells,
the lysosomal area was comparable to that in untreated control
cells (scr-PC9 and scr-HCC827). These findings indicate that TRPML3
is essential for the regulation of TFEB nuclear translocation and
lysosomal biogenesis in response to osimertinib-induced lysosomal
pH alteration.
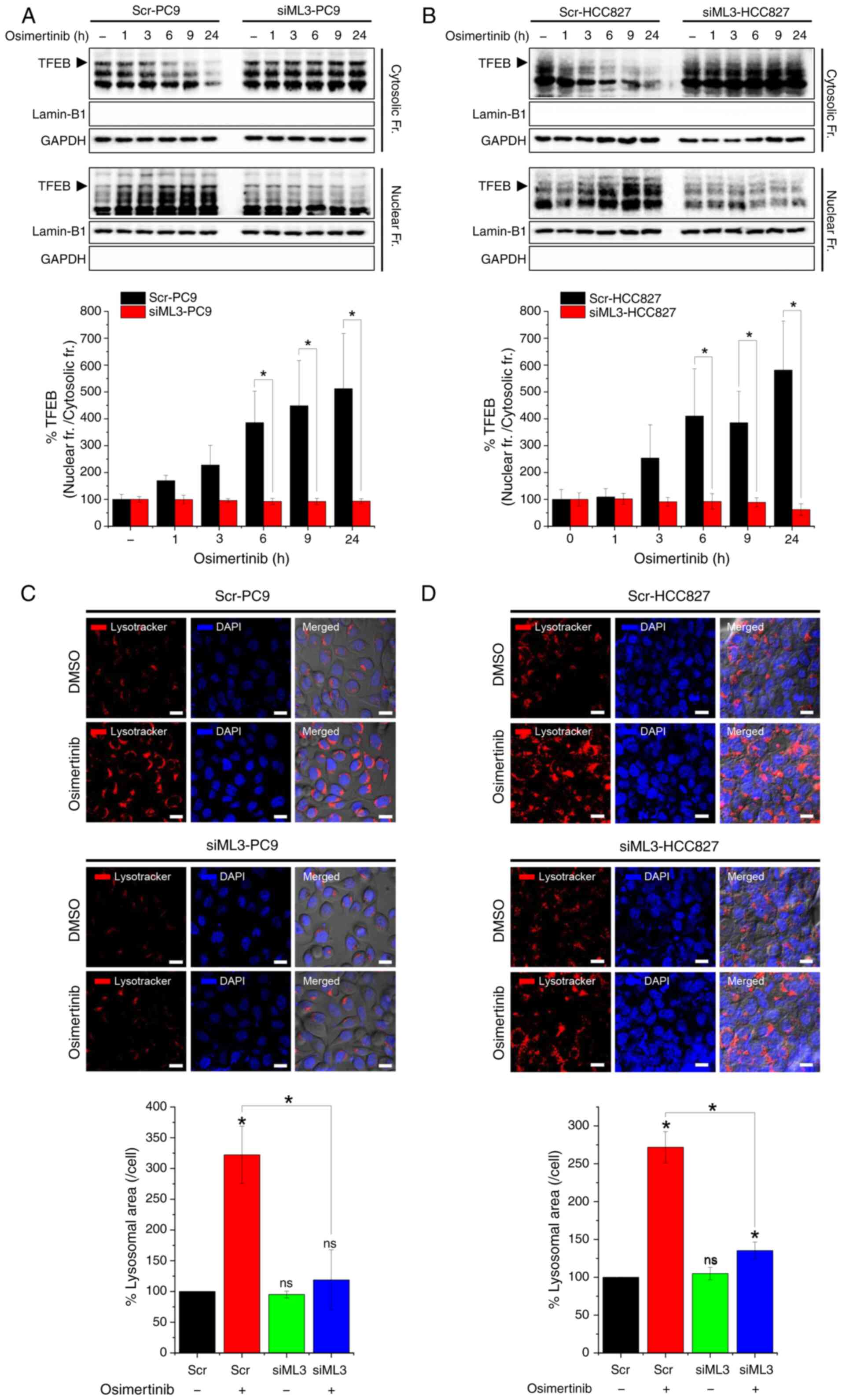 | Figure 6.TRPML3 deficiency impairs
osimertinib-induced lysosomal biogenesis. (A and B) TFEB levels in
cytosolic and nuclear fractions of PC9 and HCC827 cells transfected
with TRPML3-targeting siRNA (siML3) or scrambled RNA (control) and
treated with 0.01 µM osimertinib for 1, 3, 6, 9, and 24 h. GAPDH
and lamin-B1 served as cytosolic and nuclear markers, respectively.
Quantitative analysis (lower panel) shows nuclear-to-cytosolic TFEB
ratios expressed as percentages relative to control (scr-PC9
without osimertinib treatment; n=3). (C and D) Lysosomes and nuclei
stained with LysoTracker Red DND-99 and DAPI after 24 h osimertinib
treatment of cells transfected with siML3 or scr. Relative
lysosomal area per cell is presented as a percentage of the control
(scr-PC9 without osimertinib treatment; n=3). Scale bar, 20 µm.
*P<0.05. TRPML, transient receptor potential mucolipin; si-,
small interfering; scr, scrambled; ns, not significant. |
Discussion
Osimertinib (AZD9291) is a first-line TKI used to
treat NSCLC with the T790M EGFR allele (30). Although initially effective, it
often fails to prevent the emergence of residual tumors that become
resistant over time (31). A known
mechanism underlying this resistance involves the lysosomal
sequestration of hydrophobic, weakly basic TKIs, such as gefitinib
and osimertinib, which reduces TKI toxicity and promotes multi-drug
resistance (32,33). However, the precise molecular
mechanisms regulating this sequestration remain poorly understood.
In the present study, lysosomal calcium channel TRPML3 (MCOLN3) was
assessed as a potential contributor to TKI resistance. RNA
expression analyses EGFR-mutant LUAD samples before and
after osimertinib treatment, revealed that TRPML3 expression
was specifically increased in osimertinib-residual tumors, unlike
TRPML1 and −2, suggesting a distinct role for TRPML3 in mediating
drug resistance. It was demonstrated that osimertinib-induced
increases in [Ca2+]i levels depend on
TRPML3-mediated lysosomal Ca2+ release, which is
critical for maintaining lysosomal acidity. Furthermore, TRPML3,
rather than TRPML1, was found to be crucial for lysosomal
biogenesis through Ca2+-dependent TFEB activation in
osimertinib-treated NSCLC cells. Experiments using NSCLC-derived
spheroids further confirmed that modulating TRPML3
expression alters osimertinib sensitivity, underscoring TRPML3 as a
potential therapeutic target to overcome drug resistance in
NSCLC.
Several studies have addressed the role of TRPML1 in
lysosomal signaling and autophagy; however, it remains less
characterized in cancer biology. The Cancer Genome Atlas data show
that TRPML3 expression is decreased in multiple cancers, including
LUAD (34). Our data provide the
first evidence, to the best of our knowledge, that increased TRPML3
expression in residual tumors following osimertinib treatment
contributes to drug resistance.
As an inward-rectifying cation channel (35), TRPML3 has been implicated in
autophagy, particularly in autophagosome biogenesis during
endocytosis and phagocytosis (17,36).
Autophagy plays a dual role in cancer, as it prevents tumor
initiation by mitigating cellular stress but supports tumor
progression by sustaining proliferation and metabolic activity
(37). Although the relationship
between TRPML3 and autophagy was not fully explored, they suggest
that TRPML3-mediated enhancement of autophagic flux in
osimertinib-resistant contributes to drug resistance. Key
limitations of the present study include the absence of in
vivo validation and the lack of direct investigation into the
interaction between TRPML3 and autophagy in the context of TKI
resistance. Given the established involvement of TRPML3 in
autophagy (17,36), future studies should determine
whether TRPML3-driven lysosomal Ca2+ signaling enhances
autophagy-mediated drug sequestration. To this end, important
future directions include monitoring autophagic flux in NSCLC cells
in response to changes in TRPML3 expression and evaluating whether
co-targeting TRPML3 and autophagy enhances the efficacy of
osimertinib in vivo. Clarifying the relationship between
TRPML3 and autophagy will deepen our understanding of resistance
mechanisms and guide the development of combination therapies.
Our study underscores the critical role of TRPML3 in
regulating the lysosomal sequestration of TKIs and promoting
lysosomal biogenesis in NSCLC cells. Previously, the authors
identified TRPML3 as a key contributor to drug resistance and
demonstrated that lysosomal Ca2+ release via TRPML3
facilitates lysosomal trafficking to the plasma membrane, enhancing
resistance. This mechanism is influenced by gefitinib-induced
lysosomal pH changes, as gefitinib is a lysosomotropic weak base
(38). Emerging evidence suggests
that lysosomal Ca2+ release mediated by TRPML3 is
essential for maintaining lysosomal acidification, as
Ca2+ efflux is coupled with H+ uptake into
the lumen (20,39–41).
Given that this acidification drives lysosomotropic agent
sequestration, TRPML3 likely enhances sequestration by preserving
the lysosomal proton gradient. Based on these studies, it was
hypothesized that TKI sequestration elevates lysosomal pH, which in
turn triggers lysosomal Ca2+ release via TRPML3 but not
TRPML1. This Ca2+ release is coupled with increased
H+ uptake, further enhancing sequestration. The present
findings support this hypothesis: osimertinib treatment rapidly
increased lysosomal pH; moreover, the rate of pH increase was
directly correlated with TRPML3 expression. Additionally,
osimertinib-induced Ca2+ mobilization depends on
TRPML3-mediated lysosomal Ca2+ release. These findings
suggest that osimertinib resistance is enhanced in NSCLC cells by
increasing the lysosomal sequestration of TKIs. By elucidating the
role of TRPML3 in these processes, the present study provides
valuable insights into drug resistance mechanisms and identifies
potential therapeutic targets.
It was observed that osimertinib-induced lysosomal
pH elevation does not favor TRPML1 activation, as TRPML1 functions
optimally in a more acidic environment. Prolonged TKI treatment
increases both lysosomal number and size (33,42).
While TRPML1 is well known for its role in lysosomal biogenesis
through Ca2+ release and TFEB dephosphorylation
(13), the mechanism by which
weak-base chemotherapeutics such as osimertinib induce lysosomal
Ca2+ release remains unclear. Given that TRPML3 is
activated at higher pH, it was hypothesized that it compensates for
TRPML1 by activating TFEB and promoting lysosomal biogenesis.
Consistent with this, it was observed that TRPML3 depletion
inhibited TFEB nuclear translocation and reduced
osimertinib-induced lysosomal biogenesis. These results provide new
insights into lysosomal regulation in cancer cells treated with
TKIs such as osimertinib. Further studies are needed to elucidate
the molecular pathways involved and evaluate the therapeutic
potential of targeting TRPML channels in cancer treatment.
In conclusion, a novel mechanism was identified, by
which TRPML3 contributes to osimertinib resistance in NSCLC by
regulating lysosomal biogenesis and function. Our results suggest a
compensatory relationship between TRPML3 and TRPML1 in response to
TKI-induced lysosomal changes. By identifying TRPML3 as a key
regulator, the present study provides new insights into the
molecular basis of drug resistance and highlights TRPML3 as a
promising therapeutic target.
Acknowledgements
The authors would like to thank Dr Jin Kyung Rho
(Asan Medical Center, Ulsan University) and Dr Hyun Jin Kim
(Sungkyunkwan University) for providing NSCLC cell lines and
GCaMP-TRPML3 plasmid constructs, respectively.
Funding
The present study was supported by the National Research
Foundation of Korea and funded by the Ministry of Education (grant
no. RS-2023-NR076426).
Availability of data and materials
The data generated in the present study are included
in the figures of this article. The data generated in the present
study may be requested from the corresponding author.
Authors' contributions
MSeoK contributed to data curation, formal analysis,
investigation, methodology, software, and wrote the initial draft.
MSeuK was responsible for conceptualization, formal analysis,
funding acquisition, investigation, project administration,
supervision, validation, and editing the manuscript. Both authors
read and approved the final version of the manuscript and confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TKI
|
tyrosine kinase inhibitor
|
|
NSCLC
|
non-small cell lung cancer
|
|
LUAD
|
lung adenocarcinoma
|
|
TFEB
|
transcription factor EB
|
|
FBS
|
fetal bovine serum
|
|
TRPMLs
|
transient receptor potential
mucolipins
|
|
MLIV
|
type IV mucolipidosis
|
|
RNA-seq
|
RNA-sequencing
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
References
|
1
|
Zhang Z, Yue P, Lu T, Wang Y, Wei Y and
Wei X: Role of lysosomes in physiological activities, diseases, and
therapy. J Hematol Oncol. 14:792021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Savini M, Zhao Q and Wang MC: Lysosomes:
Signaling hubs for metabolic sensing and longevity. Trends Cell
Biol. 29:876–887. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hraběta J, Belhajová M, Šubrtová H, Merlos
Rodrigo MA, Heger Z and Eckschlager T: Drug sequestration in
lysosomes as one of the mechanisms of chemoresistance of cancer
cells and the possibilities of its inhibition. Int J Mol Sci.
21:43922020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yamagishi T, Sahni S, Sharp DM, Arvind A,
Jansson PJ and Richardson DR: P-glycoprotein mediates drug
resistance via a novel mechanism involving lysosomal sequestration.
J Biol Chem. 288:31761–31771. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seebacher N, Lane DJ, Richardson DR and
Jansson PJ: Turning the gun on cancer: Utilizing lysosomal
P-glycoprotein as a new strategy to overcome multi-drug resistance.
Free Radic Biol Med. 96:432–445. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhitomirsky B and Assaraf YG: Lysosomes as
mediators of drug resistance in cancer. Drug Resist Updat.
24:23–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu H and Ren D: Lysosomal physiology. Annu
Rev Physiol. 77:57–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kendall RL and Holian A: The role of
lysosomal ion channels in lysosome dysfunction. Inhal Toxicol.
33:41–54. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu M and Dong XP: Endolysosomal TRPMLs in
cancer. Biomolecules. 11:652021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bargal R, Avidan N, Ben-Asher E, Olender
Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D and
Bach G: Identification of the gene causing mucolipidosis type IV.
Nat Genet. 26:118–123. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santoni G, Maggi F, Amantini C, Marinelli
O, Nabissi M and Morelli MB: Pathophysiological role of transient
receptor potential mucolipin channel 1 in calcium-mediated
stress-induced neurodegenerative diseases. Front Physiol.
11:2512020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y,
Bashllari E, Bisceglia J, Muallem S and Kiselyov K: TRP-ML1
regulates lysosomal pH and acidic lysosomal lipid hydrolytic
activity. J Biol Chem. 281:7294–7301. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Di Paola S, Scotto-Rosato A and Medina DL:
TRPML1: The Ca(2+)retaker of the lysosome. Cell Calcium.
69:112–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi J, Li Q, Xin T, Lu Q, Lin J, Zhang Y,
Luo H, Zhang F, Xing Y, Wang W, et al: MCOLN1/TRPML1 in the
lysosome: A promising target for autophagy modulation in diverse
diseases. Autophagy. 20:1712–1722. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Medina DL, Di Paola S, Peluso I, Armani A,
De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso
C, Forrester A, et al: Lysosomal calcium signalling regulates
autophagy through calcineurin and TFEB. Nat Cell Biol. 17:288–299.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SW, Kim DH, Park KS, Kim MK, Park YM,
Muallem S, So I and Kim HJ: Palmitoylation controls trafficking of
the intracellular Ca2+ channel MCOLN3/TRPML3 to regulate
autophagy. Autophagy. 15:327–340. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim SW, Kim MK, Hong S, Choi A, Choi JH,
Muallem S, So I, Yang D and Kim HJ: The intracellular
Ca2+ channel TRPML3 is a PI3P effector that regulates
autophagosome biogenesis. Proc Natl Acad Sci USA.
119:e22000851192022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paroutis P, Touret N and Grinstein S: The
pH of the secretory pathway: Measurement, determinants, and
regulation. Physiology (Bethesda). 19:207–215. 2004.PubMed/NCBI
|
|
19
|
Miao Y, Li G, Zhang X, Xu H and Abraham
SN: A TRP channel senses lysosome neutralization by pathogens to
trigger their expulsion. Cell. 161:1306–1319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gerasimenko JV, Tepikin AV, Petersen OH
and Gerasimenko OV: Calcium uptake via endocytosis with rapid
release from acidifying endosomes. Curr Biol. 8:1335–1338. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Martina JA, Lelouvier B and Puertollano R:
The calcium channel mucolipin-3 is a novel regulator of trafficking
along the endosomal pathway. Traffic. 10:1143–1156. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang J, Bi G, Sui Q, Zhao G, Zhang H,
Bian Y, Chen Z, Huang Y, Xi J, Shi Y, et al: Transcription factor
ZNF263 enhances EGFR-targeted therapeutic response and reduces
residual disease in lung adenocarcinoma. Cell Rep. 43:1137712024.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen S, Zhou Y, Chen Y and Gu J: fastp: An
ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Patro R, Duggal G, Love MI, Irizarry RA
and Kingsford C: Salmon provides fast and bias-aware quantification
of transcript expression. Nat Methods. 14:417–419. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chu VT, Gottardo R, Raftery AE, Bumgarner
RE and Yeung KY: MeV+R: Using MeV as a graphical user interface for
Bioconductor applications in microarray analysis. Genome Biol.
9:R1182008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kurien BT and Scofield RH: Western
blotting: An introduction. Methods Mol Biol. 1312:17–30. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ramalingam SS, Vansteenkiste J, Planchard
D, Cho BC, Gray JE, Ohe Y, Zhou C, Reungwetwattana T, Cheng Y,
Chewaskulyong B, et al: Overall survival with osimertinib in
untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 382:41–50.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ohashi K, Maruvka YE, Michor F and Pao W:
Epidermal growth factor receptor tyrosine kinase
inhibitor-resistant disease. J Clin Oncol. 31:1070–1080. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Halaby R: Influence of lysosomal
sequestration on multidrug resistance in cancer cells. Cancer Drug
Resist. 2:31–42. 2019.PubMed/NCBI
|
|
33
|
Zhitomirsky B and Assaraf YG: Lysosomal
sequestration of hydrophobic weak base chemotherapeutics triggers
lysosomal biogenesis and lysosome-dependent cancer multidrug
resistance. Oncotarget. 6:1143–1156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu M, Li X, Zhang T, Liu Z and Zhao Y:
Identification of a nine-gene signature and establishment of a
prognostic nomogram predicting overall survival of pancreatic
cancer. Front Oncol. 9:9962019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim HJ, Li Q, Tjon-Kon-Sang S, So I,
Kiselyov K and Muallem S: Gain-of-function mutation in TRPML3
causes the mouse Varitint-Waddler phenotype. J Biol Chem.
282:36138–36142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim HJ, Soyombo AA, Tjon-Kon-Sang S, So I
and Muallem S: The Ca(2+) channel TRPML3 regulates membrane
trafficking and autophagy. Traffic. 10:1157–1167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim MS, Yang SH and Kim MS: TRPML3
enhances drug resistance in non-small cell lung cancer cells by
promoting Ca2+-mediated lysosomal trafficking. Biochem
Biophys Res Commun. 627:152–159. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Christensen KA, Myers JT and Swanson JA:
pH-dependent regulation of lysosomal calcium in macrophages. J Cell
Sci. 115:599–607. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Morgan AJ, Platt FM, Lloyd-Evans E and
Galione A: Molecular mechanisms of endolysosomal Ca2+ signalling in
health and disease. Biochem J. 439:349–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen D, Wang X, Li X, Zhang X, Yao Z,
Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD and Xu H: Lipid
storage disorders block lysosomal trafficking by inhibiting a TRP
channel and lysosomal calcium release. Nat Commun. 3:7312012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
de Araujo MEG, Liebscher G, Hess MW and
Huber LA: Lysosomal size matters. Traffic. 21:60–75. 2020.
View Article : Google Scholar : PubMed/NCBI
|