Introduction
According to the International Agency for Cancer
Research, the incidence of colorectal cancer has been steadily
increasing over the past two decades (1). In 2020, colorectal cancer ranked as
the third most common cancer among men and the second most common
among women, with its mortality rate ranking second among all
malignancies (2,3).
5′-Fluorouracil (5-FU) is a key chemotherapeutic
agent that has been used to treat colorectal cancer, exerting its
effects primarily through inhibiting thymidylate synthase (TS);
this inhibition subsequently disrupts RNA and DNA synthesis,
thereby suppressing tumor cell proliferation (4). Although 5-FU has demonstrated
significant efficacy against various types of solid tumor,
especially those of the gastrointestinal tract, its therapeutic
success in advanced colorectal cancer has been shown to be limited,
with response rates of 10–15% (5).
The emergence of 5-FU resistance substantially diminishes its
clinical effectiveness, posing a major challenge in the management
of colorectal cancer; therefore, strategies to enhance the
sensitivity of colorectal cancer cells to 5-FU, and to mitigate
chemotherapy resistance, remain urgent priorities for improving
patient outcomes.
Loperamide (LOP) is a United States Food and Drug
Administration-approved antidiarrheal medicine (6) with a well-established safety profile,
which has been shown to induce autophagy and reverse multidrug
resistance (MDR) in several types of cancer, including glioblastoma
and breast cancer (7,8). The safety profile of LOP, combined
with its efficacy at therapeutic dose (2–8 mg/day) makes it a
promising candidate for drug repurposing in oncology. Its potential
to overcome 5-FU resistance in colorectal cancer is supported by
its ability to modulate cellular stress pathways and enhance
chemosensitivity. These effects have been shown to be mediated
through the induction of autophagy and the downregulation of MDR1
gene expression (7–9). However, despite these promising
findings, the impact of LOP on colorectal cancer, in particular its
potential to overcome 5-FU resistance and the associated underlying
molecular mechanisms, remains poorly understood.
Autophagy is a highly regulated cellular process in
which eukaryotic cells utilize lysosomes to degrade cytoplasmic
proteins and damaged organelles, and it can be characterized into
basal physiological autophagy and stress-induced autophagy
(9). Basal autophagy serves as a
protective mechanism, maintaining cellular homeostasis and
protecting cells from stress or oxidative damage induced by
external stimuli, whereas excessive autophagy can lead to metabolic
stress, extensive degradation of essential cellular components,
and, ultimately, cell death.
In the context of colorectal cancer drug resistance,
numerous studies have suggested that autophagy serves a critical
role in enhancing resistance to various chemotherapeutic agents
(10,11). Consequently, targeting autophagy
represents a potential strategy for modulating the sensitivity of
gastrointestinal tumors to 5-FU treatment.
In the present study, the 5-FU-sensitive colorectal
cancer cell line HCT8 and its 5-FU-resistant counterpart, HCT8R,
were employed in a series of in vitro and in vivo
experiments. The current study investigated whether LOP potentiates
5-FU sensitivity in colorectal cancer cells and elucidated the
underlying molecular mechanisms.
Materials and methods
Cell lines and culture conditions
The human colorectal cancer cell line HCT8 (cat. no.
1101HUM-PUMC000104; Cell Resource Center, Institute of Basic
Medical Sciences, Chinese Academy of Medical Sciences & Peking
Union Medical College) and its 5-FU-resistant derivative, HCT8R
(cat. no. MXC149; ShangHai MEIXUAN Biological Science and
Technology Ltd.), were kindly provided by the research group of
Professor Xi (Hubei University of Medicine, Shiyan, China). Both
cell lines were cultured in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml
streptomycin. Cultures were maintained in a humidified incubator at
37°C containing 5% CO2 to support optimal cell growth.
To inhibit autophagic activity, the HCT8R cells were co-incubated
with an autophagy inhibitor, 3-methyladenine (3-MA; 5 mM), 5-FU (20
µg/ml) and LOP (20 µM) for 24 h at 37°C.
RPMI-1640 medium was purchased as a powder from
Gibco; Thermo Fisher Scientific, Inc., FBS was obtained from
Zhejiang Tianhang Biotechnology Co., Ltd., the antibiotics were
sourced from Beyotime Institute of Biotechnology, and LOP, 5-FU and
3-MA were purchased from MedChemExpress.
Cell counting kit-8 (CCK-8) viability
assay
HCT8 and HCT8R cell metabolic activity was assessed
using the CCK-8 kit (cat. no. C0038; Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Briefly,
cells in the logarithmic growth phase were seeded at
1×103 cells/well in 96-well plates (100 µl/well). After
24 h of adhesion, the medium was replaced with fresh medium
containing 5-FU (0, 10, 20, 40 and 80 µg/ml) and LOP (0, 10, 20, 40
and 80 µM). To evaluate the combined effects of 5-FU and LOP on
cell viability, HCT8R cells were treated with 5-FU (20 µg/ml),
dimethyl sulfoxide (DMSO, 0.2% v/v) or LOP (10 and 20 µM). The
cells were then incubated for 24 h, followed by the addition of 10
µl CCK-8 reagent/well and further incubation for 2 h at 37°C.
Culture medium without cells served as the blank control.
Absorbance at 450 nm was measured using a microplate reader (BioTek
Synergy H1; Biotek; Agilent Technologies, Inc.).
5-Ethynyl-2′-deoxyuridine (EdU)
proliferation assay
Exponentially growing HCT8R cells were counted and
seeded into 48-well plates at a density of 10,000 cells/well. After
24 h of adherence to the plates, the cells were treated with 5-FU
(20 µg/ml), DMSO (0.2% v/v) or LOP (20 µM) for 48 h at 37°C. To
assess cell proliferation, the cells were incubated with EdU
diluted in culture medium at a 100:1 ratio (200 µl/well) for 6 h at
37°C. Following EdU labeling, the medium was subsequently removed
and the cells were fixed with 4% paraformaldehyde (Biosharp Life
Sciences) for 30 min at room temperature. Fixed cells were then
incubated with 2 mg/ml glycine for 5 min to neutralize residual
paraformaldehyde, followed by permeabilization with 0.5% Triton
X-100 for 20 min at room temperature. The EdU reaction solution was
prepared according to the manufacturer's protocol (Beyotime
Institute of Biotechnology) and applied to the cells (100 µl/well)
for 30 min in the dark to ensure precise labeling at room
temperature. Subsequently, the cells were counterstained with DAPI
(Beyotime Institute of Biotechnology) for 10 min to visualize the
nuclei at room temperature. Finally, fluorescent images were
acquired using an inverted fluorescence microscope.
Colony formation assay
Exponentially growing HCT8R cells were seeded into
6-well plates at a density of 1,000 cells/well. Once the cells had
formed small colonies, they were treated with 5-FU (20 µg/ml), DMSO
(0.2% v/v) or LOP (20 µM) for 48 h at 37°C. After 6–8 days, colony
formation was assessed under a microscope. When distinct colonies
were visible in the control group, the culture medium was aspirated
and the cells were washed three times with PBS. Cells were
subsequently fixed with 4% paraformaldehyde for 30 min at room
temperature, and colonies were then stained with crystal violet for
30 min at room temperature to enhance visualization. Excess stain
was removed with multiple PBS washes and the plates were left to
air-dry prior to imaging. Crystal violet-stained colonies (>50
cells) were visualized using an inverted light microscope (IX71;
Olympus Corporation). Colony counts per group were manually
quantified in triplicate and averaged.
Wound healing assay
Exponentially growing HCT8R cells were treated with
trypsin until they formed a single-cell suspension, and
~5×105 cells/well were seeded into 6-well plates. The
cells were subsequently cultured at 37°C in a humidified atmosphere
containing 5% CO2 for 24 h to allow them to reach 100%
confluence. A uniform scratch was introduced using a 200-µl pipette
tip and the cells were subsequently washed three times with PBS to
remove any detached cells. Fresh RPMI-1640 medium supplemented with
2% FBS was then added to each well. Initial images were captured to
confirm that the scratch that had been made was uniform, centered
and perpendicular to the plane of the plate. Following treatment
with 5-FU (20 µg/ml), DMSO (0.2% v/v) or LOP (20 µM) for 24 h at
37°C, additional images were acquired under a light microscope.
Cell migration into the scratch area was quantified using ImageJ
(version 4.1; National Institutes of Health), and the wound healing
rate was calculated as: (0 h scratch width −24 h scratch width)/0 h
scratch width ×100 to assess cell motility.
Transwell assay
For the Transwell assay experiments in HCT8 and
HCT8R cells, 600 µl RPMI-1640 medium supplemented with 20% FBS was
added to the lower chamber, with or without 3-MA (5 mM), 5-FU (20
µg/ml) and LOP (10 and 20 µM). Subsequently, 200 µl suspension
containing 105 cells/ml in FBS-free medium was added to
the upper chamber (pore size, 8 µm; Corning, Inc.), which was
placed into a 24-well plate. The cells were cultured for 48 h to
allow for migration. After the incubation period, the insert was
carefully removed and washed once with PBS. The cells that had
migrated to the lower surface of the insert were subsequently fixed
with 4% paraformaldehyde for 30 min at room temperature and washed
three times with PBS. The interior of the insert was gently swabbed
with a soft cotton swab to remove non-adherent cells. Finally,
staining was performed using crystal violet for 20 min at room
temperature, and excess stain was removed by multiple washes with
PBS. Images were captured under a light microscope.
Observation of autophagosomes by
confocal microscopy
Exponentially growing HCT8R cells were plated in
6-well plates, and cultured for 24 h until they reached 70–80%
confluence. Transfection was subsequently performed using
Lipo8000™ reagent (Beyotime Institute of Biotechnology)
in combination with 2 µg pEGFP (cat. no. 165830; Addgene, Inc.) or
pEGFP-microtubule-associated protein 1 light chain 3 (LC3) plasmid
(cat. no. 24920; Addgene, Inc.) and Opti-MEM™ serum-free
medium (Gibco; Thermo Fisher Scientific Inc.). After 36 h of
transfection at 37°C, the cells were subsequently transferred to
confocal dishes, and allowed to continue growing for an additional
24 h. Once cells had reached ~50% confluence, they were incubated
with either 5-FU (20 µg/ml), DMSO (0.2% v/v) or LOP (20 µM) for 24
h at 37°C. Imaging was performed using an Olympus FV3000RS
super-resolution laser confocal microscope (Olympus Corporation).
All samples were analyzed under identical imaging conditions to
ensure consistency and reproducibility.
Western blot analysis
HCT8R cells were harvested into 1.5-ml Eppendorf
tubes containing RIPA lysis buffer (Beyotime Institute of
Biotechnology) for protein extraction at 4°C. The protein
concentration was subsequently quantified using a bicinchoninic
acid protein assay kit, and 20 µg protein samples were separated by
SDS-PAGE on 12% gels and transferred to a PVDF membrane
(MilliporeSigma). Non-specific binding was blocked using PBS
containing 5% nonfat milk for 2 h at room temperature. The membrane
was subsequently incubated overnight at 4°C with primary antibodies
(diluted with PBS) raised against LC3 (1:1,000; cat. no.
18725-1-AP), Beclin (1:1,000; cat. no. 11306-1-AP) (both from
Proteintech Group, Inc.) and β-actin (1:1,000; cat. no. sc-47778;
Santa Cruz Biotechnology, Inc.). Following incubation with the
primary antibodies, the membrane was washed and incubated with
horseradish peroxidase-conjugated anti-rabbit (diluted with PBS,
1:5,000; cat. no. 31460; Invitrogen; Thermo Fisher Scientific,
Inc.) and anti-mouse secondary antibodies (diluted with PBS,
1:5,000; cat. no. 31430; Invitrogen; Thermo Fisher Scientific,
Inc.) for 2 h at room temperature. Finally, the blots were
visualized using an enhanced chemiluminescence detection system
(Bio-Rad Laboratories, Inc.). Images were semi-quantified using
ImageJ (version 4.1; National Institutes of Health).
Apoptosis detection
For apoptosis detection, the HCT8R cells were seeded
in 6-well plates at a density of 4×105 cells/well. Cells
were treated with 5-FU (20 µg/ml) and LOP (10 or 20 µM) for 48 h at
37°C. Both adherent and floating cells were then harvested and
washed with ice-cold PBS. Cells from different experimental groups
were then digested with trypsin (without EDTA), combined with the
culture supernatant and centrifuged at 112 × g for 4 min at 4°C to
pellet the cells. Subsequently, the pellet was gently resuspended
in pre-chilled binding buffer and adjusted to a concentration of
1–5×106 cells/ml. Aliquots of the cell suspension (100
µl) were then mixed with 5 µl FITC-Annexin V and 5 µl PI, gently
mixed, and incubated in the dark at room temperature for 8–10 min.
Subsequently, 400 µl binding buffer was added, and the mixture was
gently mixed on a vortex mixer. Finally, apoptosis was assessed
using a flow cytometer (SA3800; Sony Biotechnology Inc.) within 1
h, with an Annexin V-PI reagent kit purchased from Invitrogen;
Thermo Fisher Scientific, Inc.
Xenograft tumor study in nude
mice
HCT8R cells (5×106 cells/100 µl PBS) were
subcutaneously injected into the lateral abdominal region of female
nude BALB/c mice (age, 4 weeks; weight, 20–24 g; n=3/group; total
n=9) that were housed under the following specific pathogen-free
conditions: Temperature, 23±1°C; humidity, 55±5%; 12-h light/dark
cycle, and with free access to standard chow and drinking water.
The mice were randomly divided into the control (vehicle: 0.9% NaCl
+ 0.1% DMSO, i.p. daily), 5-FU monotherapy (25 mg/kg i.p., three
times per week) and combination (5-FU as above + LOP 2 mg/kg/day
i.p., three times per week) groups. All procedures followed
protocols approved by the Hubei University of Medicine Animal
Research Committee (approval no. 2023-034). Treatment was initiated
at day 7 post-inoculation when tumors reached 100±10 mm3
and continued for 21 days. The humane endpoints requiring
euthanasia included ≥20% body weight loss within 48 h, tumor
diameter >1.5 cm, impaired mobility or signs of distress
(labored breathing/vocalization). Animal health and behavior were
assessed twice daily (8:00 a.m./6:00 p.m.) using validated scoring
sheets, and tumor volume (V=LxW2/2) and weight were
measured every 48 h, with the endpoint being day 28 or when tumor
volume exceeded 1,500 mm3. Welfare provisions included:
Individual ventilated cages with environmental enrichment (paper
tunnels, chew blocks), and euthanasia by cervical dislocation under
deep anesthesia (5% isoflurane for induction and 2% for
maintenance). Death was verified by the cessation of respiration,
an absent heartbeat, fixed dilated pupils and bilateral
diaphragmatic transection. All 9 mice were sacrificed, with no
unexpected deaths recorded and no mice excluded from the
analysis.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 9 (Dotmatics). Comparisons between two groups were made using
unpaired Student's t-test, whereas comparisons among multiple
groups were performed by one-way ANOVA followed by Tukey's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
LOP enhances the sensitivity of
colorectal cancer cells to 5-FU
To evaluate the malignant potential of HCT8 and
HCT8R cells, Transwell assays were performed, which revealed that
the migratory capability of the drug-resistant HCT8R cells was
significantly higher compared with that of HCT8 cells (Fig. 1A). The preliminary dose-response
assessments revealed marked insensitivity of HCT8R cells to 5-FU
monotherapy, with conventional therapeutic concentrations (10–80
µg/ml) failing to induce significant inhibition of cell viability
(Fig. S1). Subsequently, the
inhibitory effects of LOP on cell viability were assessed using the
CCK-8 assay. The results demonstrated that LOP inhibited the
viability of both HCT8 and HCT8R cells, with IC50 values
of ~24.74 and 34.29 µM, respectively. These findings suggested that
LOP was able to effectively suppress the viability of both the
parental and 5-FU-resistant colorectal cancer cell lines (Fig. 1B and C).
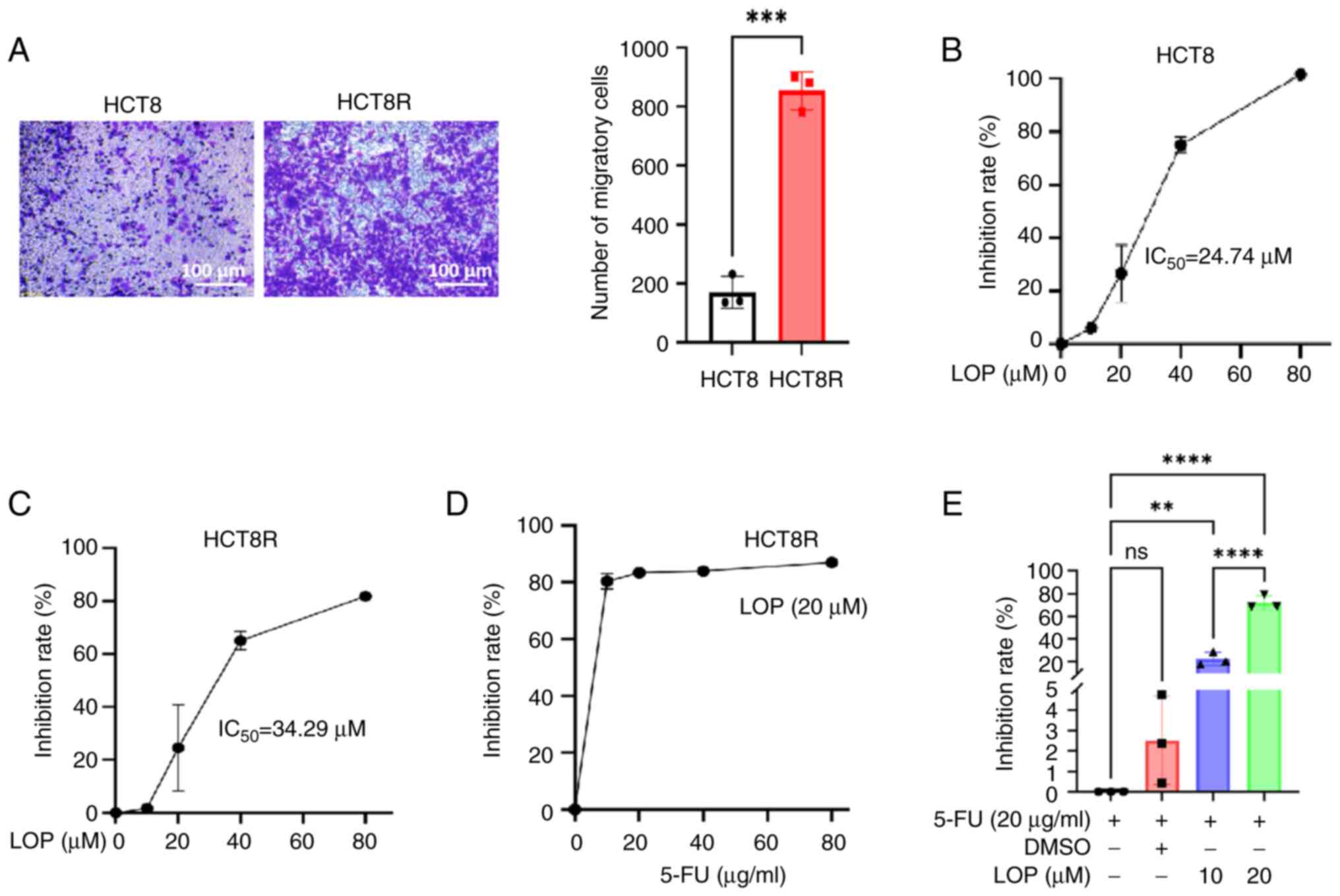 | Figure 1.LOP enhances the sensitivity of
colorectal cancer cells to 5-FU. (A) Transwell assay was performed,
assessing the migratory capability of HCT8 and HCT8R cells. (B)
CCK-8 assay was performed, evaluating the inhibitory effect of LOP
on HCT8 cell proliferation. CCK-8 assay was performed to (C)
evaluate the inhibitory effect of LOP on HCT8R cell proliferation,
(D) examine the effects of LOP combined with various concentrations
of 5-FU on HCT8R cell proliferation, and (E) evaluate the effects
of 5-FU combined with different concentrations of LOP on HCT8R cell
proliferation. **P<0.01, ***P<0.001, ****P<0.0001 (n=3).
5-FU, 5-fluorouracil; CCK-8, Cell Counting Kit-8; DMSO, dimethyl
sulfoxide; LOP, loperamide; ns, not significant. |
To further examine the impact of LOP on the
sensitivity of HCT8R cells to 5-FU, additional CCK-8 assays were
performed. The results showed that the HCT8R cells exhibited
resistance to 5-FU, as increasing concentrations of the drug did
not significantly inhibit cell proliferation; however, treating the
cells with 20 µM LOP in addition to 5-FU led to a marked
enhancement of the inhibitory effect of 5-FU on cell proliferation
(Fig. 1D). Further analysis
revealed that treating the cells with 5-FU (20 µg/ml) alone did not
substantially alter HCT8R cell proliferation; by contrast, compared
with in the 5-FU group, the addition of LOP caused a significant
improvement in the sensitivity of these cells to 5-FU in a
concentration-dependent manner (Fig.
1E).
LOP enhances the 5-FU-induced
inhibition of HCT8R cell proliferation in vitro and in vivo
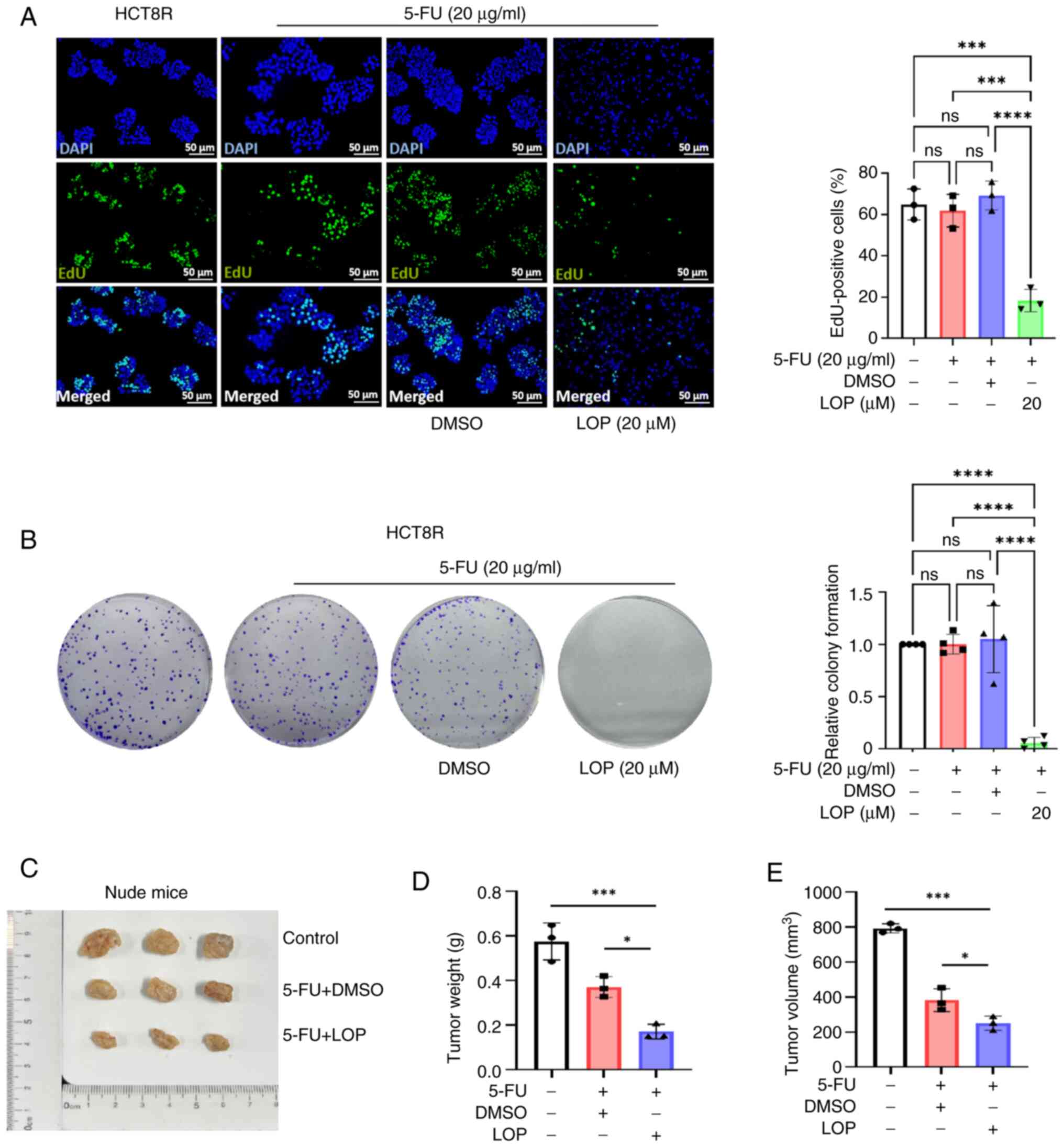
The EdU assay results demonstrated that the
proliferative activity of the HCT8R cells remained unaffected by
the addition of 5-FU or DMSO alone; however, treatment with LOP led
to a significant reduction in the number of proliferating HCT8R
cells (Fig. 2A). Colony formation
assays were subsequently performed, which further demonstrated that
treatment with LOP caused a marked decrease in the number of
colonies formed by HCT8R cells, whereas treatment with 5-FU alone
or DMSO did not lead to any significant inhibitory effects
(Fig. 2B).
To confirm the ability of LOP to enhance the
inhibitory effect of 5-FU on HCT8R cell proliferation in
vivo, a xenograft model was generated using nude mice. HCT8R
cells were implanted into nude mice, which were then randomly
divided into control, DMSO and LOP treatment groups. With the
exception of the control group, the other groups were injected
intraperitoneally with 5-FU (25 mg/kg per week, three times per
week), whereas the LOP treatment group was injected
intraperitoneally with LOP (2 mg/kg per day). The results
demonstrated that treatment with LOP led to a significant
enhancement of the inhibitory effect of 5-FU on tumor growth
(Fig. 2C), as evidenced by a marked
reduction in tumor weight and volume in the LOP treatment group
(Fig. 2D and E). Compared with in
the 5-FU group, the 5-FU and LOP co-treatment group demonstrated a
stronger tumor growth inhibitory effect. Collectively, these
findings suggested that LOP is able to effectively suppress cell
proliferation both in vitro and in vivo.
LOP augments the inhibition of HCT8R
cell migration mediated by 5-FU
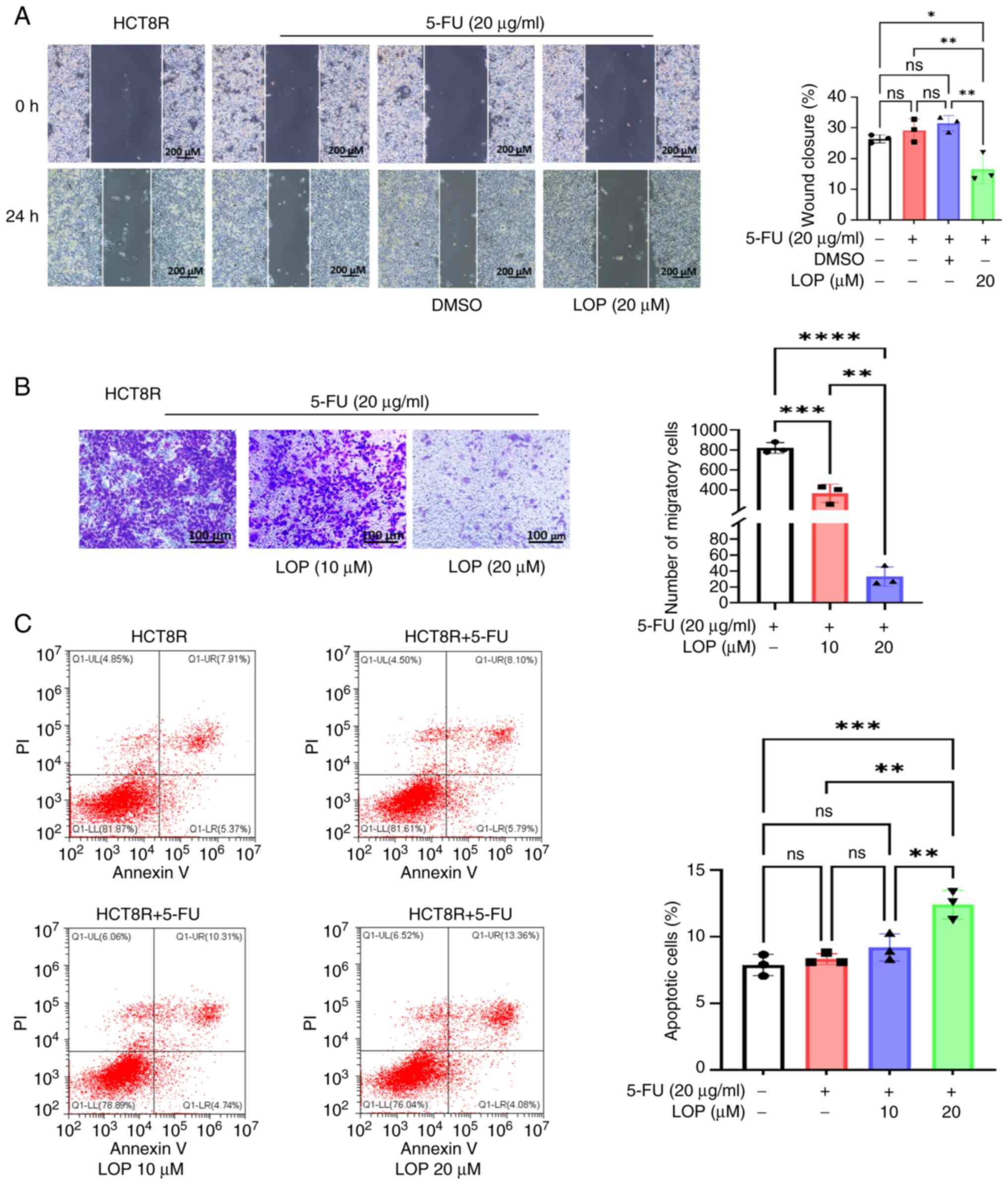
Tumor cell migration is a critical factor for both
tumor invasion into normal tissues and subsequent metastasis. After
performing wound healing assays, it was observed that treatment
with 5-FU or DMSO alone did not significantly affect the migratory
capabilities of drug-resistant HCT8R cells; however, the addition
of 20 µM LOP led to a marked inhibition of cell migration compared
with in the 5-FU group (Fig. 3A).
These findings were further validated by employing Transwell
assays, which demonstrated that LOP was able to effectively
suppress HCT8R cell migration in a dose-dependent manner (Fig. 3B).
LOP enhances 5-FU-induced apoptosis in
HCT8R cells
The balance between tumor cell proliferation and
apoptosis is a crucial determinant of drug sensitivity. The
aforementioned experiments demonstrated that LOP can effectively
inhibit the proliferation, invasion and migration of 5-FU-resistant
cells; however, its impact on apoptosis has not yet been fully
elucidated. To investigate this possibility, Annexin V-PI staining
experiments were performed for apoptosis detection. Flow cytometric
analysis revealed that treatment with 5-FU alone did not cause any
significant induction of apoptosis; by contrast, the addition of
LOP markedly increased the proportions of late apoptotic cells
compared with in the 5-FU group (Fig.
3C).
LOP reverses 5-FU resistance through
the activation of autophagy
To elucidate the molecular mechanism underlying the
ability of LOP to reverse 5-FU resistance, the expression levels of
the autophagy markers LC3 and Beclin were assessed using western
blot analysis. The results demonstrated that LOP treatment led to
an upregulation of Beclin expression and an increase in the
LC3-II/I ratio compared with in the 5-FU group (Fig. 4A), indicating the activation of
autophagy.
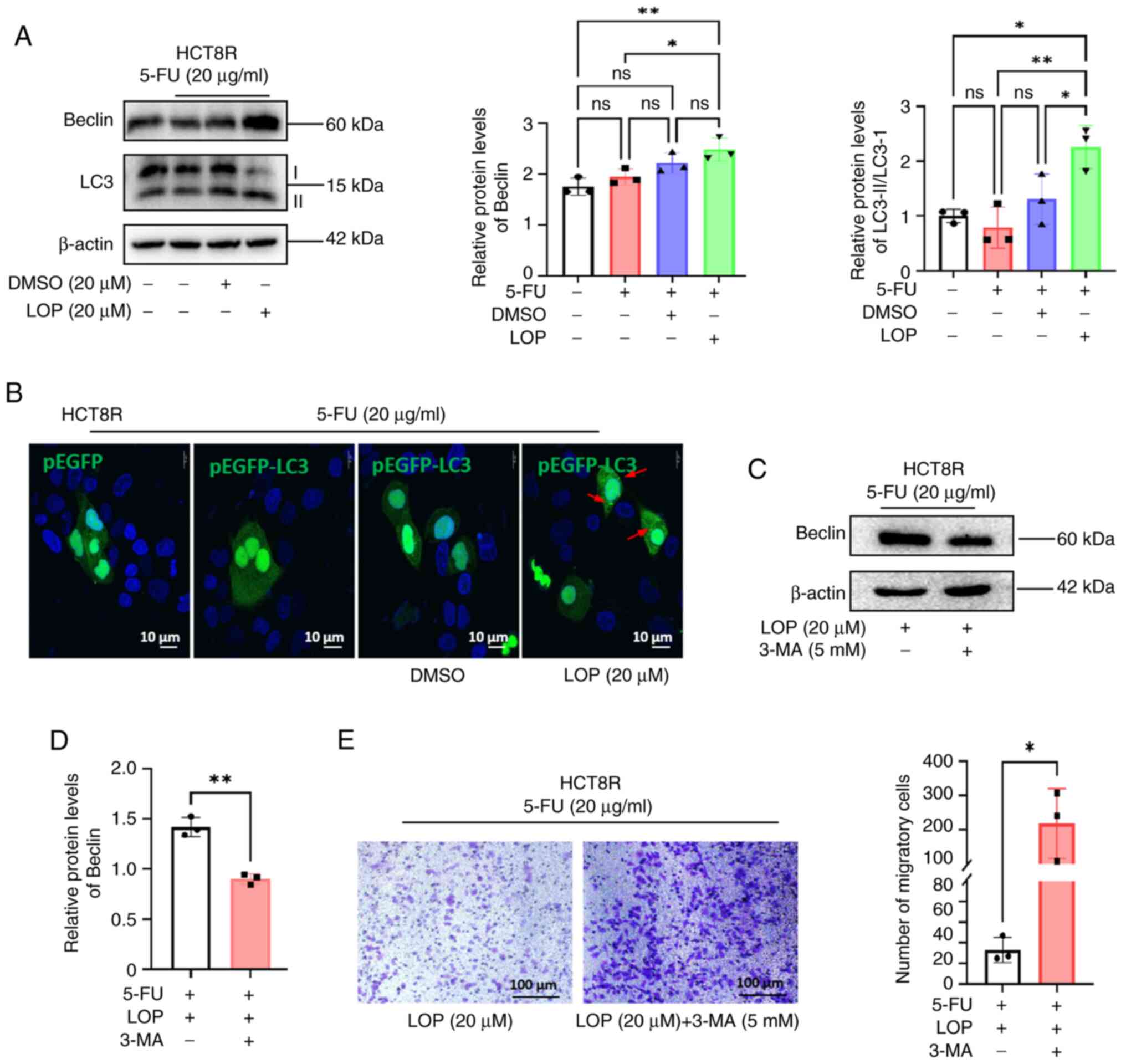 | Figure 4.LOP enhances the inhibitory effects
of 5-FU on HCT8R cells through autophagy activation. (A) Western
blot analysis of changes in LC3 and Beclin protein expression in
HCT8R cells treated with LOP and 5-FU. (B) Confocal microscopy
images, showing the generation of autophagic vesicles in HCT8R
cells transfected with the EGFP-LC3 plasmid, and treated with LOP
and 5-FU. (C) Western blot analysis of Beclin protein expression in
HCT8R cells treated with or without 3-MA. (D) Statistical analysis
of changes in Beclin expression in HCT8R cells treated with or
without 3-MA. (E) Transwell assays were performed, assessing the
migratory capability of HCT8R cells treated with LOP, 5-FU, with or
without 3-MA. *P<0.05, **P<0.01 (n=3). 3-MA, 3-methyladenine;
5-FU, 5-fluorouracil; DMSO, dimethyl sulfoxide; LC3,
microtubule-associated protein 1 light chain 3; LOP, loperamide;
ns, not significant. |
To further investigate this phenomenon, HCT8R cells
were transfected with EGFP or EGFP-LC3 plasmids, and autophagic
vesicles were visualized using confocal microscopy. The results
obtained showed that treatment with LOP in combination with 5-FU
resulted in dispersed, dense fluorescent spots in the cytoplasm of
resistant cells, characteristic of autophagic vesicles; by
contrast, cells treated with 5-FU or DMSO alone exhibited diffuse
fluorescence without the presence of autophagic vesicles (Fig. 4B).
To confirm that the reversal of HCT8R cell
resistance by LOP was associated with autophagy activation, the
autophagy inhibitor 3-MA was used to block autophagy. Western blot
analysis confirmed a significant downregulation of Beclin following
3-MA treatment (Fig. 4C). Moreover,
Transwell assays indicated that treatment with 3-MA led to a
reversal of the inhibition of migration caused by LOP (Fig. 4D) compared with in the group not
treated with 3-MA, highlighting the importance of autophagy as a
target of the action of LOP.
Taken together, these findings suggested that LOP
may enhance autophagy to inhibit cell proliferation, invasion and
migration, while also promoting apoptosis, thereby reversing 5-FU
resistance in HCT8 colorectal cancer cells.
Discussion
Research has highlighted several key mechanisms
contributing to tumor resistance to 5-FU. A notable factor is the
rapid degradation of 5-FU mediated by dihydropyrimidine
dehydrogenase (DPD). The liver, which is rich in DPD, performs a
crucial role in the rapid catabolism of 5-FU, leading to reduced
bioavailability and efficacy (5).
The expression of TS is another critical determinant of 5-FU
sensitivity. In 5-FU-resistant cell lines, gene amplification and
increased protein expression of TS are commonly observed (12,13).
Furthermore, polymorphisms in the TS gene promoter have been shown
to result in elevated TS expression, contributing to decreased
responsiveness to 5-FU (4).
Additionally, microsatellite instability, along with other factors,
including the MDR genes MDR3 and MDR4, the polyamine degradation
enzyme spermidine/spermine N1-acetyltransferase, heat shock protein
co-chaperone Hsp10, chloride transporter MAT8 and Yamaguchi sarcoma
viral oncogene homolog 1, have been associated with 5-FU resistance
(5).
LOP is a widely used antidiarrheal agent that acts
as a peripheral µ-opioid receptor agonist. LOP exhibits minimal
systemic absorption (0.3% bioavailability) due to its charged state
at physiological pH, with >90% excreted unchanged in feces. A
typical dosing regimen starts with 4 mg, followed by 2 mg after
each diarrheal episode (14). Due
to its inability to cross the blood-brain barrier, LOP is
considered a safe drug for clinical use, with minimal central
nervous system side effects (15,16).
Previous studies have explored its potential beyond
gastrointestinal applications, particularly in cancer research,
where LOP has demonstrated notable anti-proliferative and
pro-apoptotic effects on bladder cancer (17), glioma (9), different types of canine cancer
(18), colon cancer (19) and NSCLC cells (20). In vitro studies have
consistently employed LOP at concentrations ranging between 20 and
100 µM to investigate its cellular mechanisms. Specifically, 20, 40
and 60 µM concentrations of LOP have been shown to effectively
inhibit the proliferation of bladder cancer cells (17). These concentrations have been
reported not only to suppress cell proliferation, but also to
promote apoptosis and induce autophagy, suggesting a multifaceted
role of LOP in cancer cell regulation (21,22).
The induction of autophagy, in particular, highlights the potential
of LOP to modulate cellular stress responses, which may be
leveraged in therapeutic strategies targeting cancer cell survival
pathways. However, the impact of LOP on 5-FU-resistant colorectal
cancer has yet to be fully investigated.
In the present study, the colorectal cancer cell
line HCT8R, which is resistant to 5-FU, was observed to exhibit
significantly enhanced invasion and migration compared with the
parental HCT8 cell line, indicating a higher level of malignancy.
It was revealed that LOP effectively inhibited the proliferation of
both HCT8 and HCT8R cells, underscoring its potential as a
therapeutic agent in colorectal cancer. Notably, treatment with LOP
was shown to markedly sensitize 5-FU-resistant HCT8R cells to
5-FU.
Using CCK-8 and EdU assays (to assess cell viability
and cell proliferation, respectively) and clonogenic assays, and an
in vivo nude mouse tumorigenic model the present study
demonstrated that a combination of 5-FU and LOP significantly
suppressed HCT8R cell proliferation, invasion and migration
compared with treatment with 5-FU alone or the control group. This
effect was found to be dose-dependent. Additionally, LOP treatment
was associated with a substantial increase in apoptosis in
5-FU-resistant cells.
The association between autophagy and drug
resistance has garnered notable attention from researchers. The
role of autophagy modulation in cancer treatment, whether via its
inhibition or activation, has been shown to yield different
outcomes, depending on the type of tumor (23–26).
Hu et al (27) demonstrated
that IL-6 promoted chemotherapy resistance in colorectal cancer by
activating autophagy through the IL-6/JAK2/Beclin 1 signaling
pathway. Additionally, Reyes-Castellanos et al (28) identified dysregulated metabolic
pathways in pancreatic ductal adenocarcinoma (PDAC), with autophagy
being a pivotal factor. This process supports tumor survival
through facilitating a continuous supply of intracellular
components via both autonomous and non-autonomous mechanisms. An
elevated level of autophagy in established PDAC tumors may allow
for abnormal proliferation and growth, even under nutrient-limited
conditions.
On the other hand, enhancing cellular autophagy to
induce cell death has been shown to overcome doxorubicin resistance
(29). In the context of colorectal
cancer, numerous studies (30–32)
have suggested that autophagy contributes to resistance against
various chemotherapeutic agents. Moreover, excessive
autophagy-induced cell death has been shown to potentially be an
effective strategy to overcome MDR in tumor cells (33). Therefore, autophagy may serve as a
crucial target for modulating the sensitivity of gastrointestinal
tumors to 5-FU treatment (10,34).
In conclusion, the present study demonstrated that
LOP may activate cellular autophagy, leading to the formation of
autophagosomes. This effect was not observed following treatment
with 5-FU alone or in combination with DMSO, underscoring the
important role of autophagy in enhancing the sensitivity of HCT8R
cells to 5-FU when LOP is present. Additionally, the inhibition of
autophagy reversed the suppressive effect of LOP on the
proliferation of resistant cells, suggesting that LOP could enhance
the sensitivity of 5-FU-resistant colorectal cancer cells through
autophagy activation. This mechanism involved the suppression of
cell proliferation, invasion and migration, as well as the
induction of apoptosis, ultimately overcoming drug resistance.
However, one limitation of the present study is that it lacked
in vivo pharmacokinetic data for LOP, and there is therefore
a need to explore additional mechanisms beyond autophagy, including
apoptosis and metabolic reprogramming. Further studies are also
needed to elucidate the precise molecular mechanisms underlying
these effects, and to explore the potential of LOP in combination
therapies for colorectal cancer and other malignancies.
Supplementary Material
Supporting Data
Acknowledgments
The authors would like to thank Dr Xi (School of
Basic Medical Sciences, Hubei University of Medicine, Shiyan,
China.) for providing the cell lines. The authors would also like
to thank Professor Bei Li (the Biomedical Research Institute, Hubei
University of Medicine) for providing access to the flow cytometer
and chemiluminescence detection system.
Funding
The present study was supported by Joint Funds of the Hubei
Provincial Natural Science Foundation (grant no. 2025AFD176), the
Free Exploration Project of Hubei University of Medicine (grant no.
FDFR201901), the Open Project of Hubei Key Laboratory of Embryonic
Stem Cell Research (grant no. ESOF2024005), the Hubei Provincial
Natural Science Foundation (grant no. 2024AFD284) and the
Innovative Research Program for Graduates of Institute of Hubei
University of Medicine (grant nos. YC2023033 and YC2024007).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JY was responsible for devising the methodology,
choosing the software, performing the investigation, formal
analysis, writing the original draft of the manuscript and funding
acquisition. XA was responsible for devising the methodology,
choosing the software, performing the investigation and funding
acquisition. XQ, JK, HR and YL were responsible for devising the
methodology, choosing the software and performing the
investigation. ZL and DL were responsible for conceptualization of
the study and providing resources. JJ was responsible for
conceptualization of the study, providing resources and supervision
of the project. SL was responsible for conceptualization of the
study, funding acquisition, and writing, reviewing and editing the
manuscript. JY and XA confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal studies were performed according to the
guidelines and with non-retrospective ethics approval from the
Hubei University of Medicine Institutional Animal Care and Use
Committee (approval no. 2023-034).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
5-FU
|
5-fluorouracil
|
|
TS
|
thymidylate synthase
|
|
LOP
|
loperamide
|
|
FBS
|
fetal bovine serum
|
|
EdU
|
5-ethynyl-2′-deoxyuridine
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
DMSO
|
dimethyl sulfoxide
|
|
CCK-8
|
Cell Counting Kit-8
|
|
3-MA
|
3-methyladenine
|
|
DPD
|
dihydropyrimidine dehydrogenase
|
|
MDR
|
multidrug resistance
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108.
2005.PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.PubMed/NCBI
|
|
3
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global CANCER STATISTIcs 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
4
|
Rustum YM: Thymidylate synthase: A
critical target in cancer therapy? Front Biosci. 9:2467–2473. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Longley DB, Harkin DP and Johnston PG:
5-Fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Awouters F, Megens A, Verlinden M,
Schuurkes J, Niemegeers C and Janssen PAJ: Loperamide: Survey of
studies on mechanism of its antidiarrheal activity. Dig Dis Sci.
38:977–995. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Y, Sridhar R, Shan L, Sha W, Gu X and
Sukumar S: Loperamide, an FDA-approved antidiarrhea drug,
effectively reverses the resistance of multidrug resistant
MCF-7/MDR1 human breast cancer cells to doxorubicin-induced
cytotoxicity. Cancer Invest. 30:119–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zielke S, Meyer N, Mari M, Abou-El-Ardat
K, Reggiori F, van Wijk SJL, Kögel D and Fulda S: Loperamide,
pimozide, and STF-62247 trigger autophagy-dependent cell death in
glioblastoma cells. Cell Death Dis. 9:9942018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meyer N, Henkel L, Linder B, Zielke S,
Tascher G, Trautmann S, Geisslinger G, Münch C, Fulda S, Tegeder I
and Kögel D: Autophagy activation, lipotoxicity and lysosomal
membrane permeabilization synergize to promote pimozide- and
loperamide-induced glioma cell death. Autophagy. 17:3424–3443.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang JC, Feng YL, Liang X and Cai XJ:
Autophagy in 5-fluorouracil therapy in gastrointestinal cancer:
Trends and challenges. Chin Med J (Engl). 129:456–563. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang BR, Han JB, Jiang Y, Xu S, Yang R,
Kong YG, Tao ZZ, Hua QQ, Zou Y and Chen SM: CENPN suppresses
autophagy and increases paclitaxel resistance in nasopharyngeal
carcinoma cells by inhibiting the CREB-VAMP8 signaling axis.
Autophagy. 20:329–348. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johnston PG, Drake JC, Trepel J and
Allegra CJ: Immunological quantitation of thymidylate synthase
using the monoclonal antibody TS 106 in 5-fluorouracil-sensitive
and -resistant human cancer cell lines. Cancer Res. 52:4306–4312.
1992.PubMed/NCBI
|
|
13
|
Copur S, Aiba K, Drake JC, Allegra CJ and
Chu E: Thymidylate synthase gene amplification in human colon
cancer cell lines resistant to 5-fluorouracil. Biochem Pharmacol.
49:1419–1426. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baker DE: Loperamide: A pharmacological
review. Rev Gastroenterol Disord. 7 (Suppl 3):S11–S18.
2007.PubMed/NCBI
|
|
15
|
Parkar N, Spencer NJ, Wiklendt L, Olson T,
Young W, Janssen P, McNabb WC and Dalziel JE: Novel insights into
mechanisms of inhibition of colonic motility by loperamide. Front
Neurosci. 18:14249362024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu PE and Juurlink DN: Clinical review:
Loperamide toxicity. Ann Emerg Med. 70:245–252. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu J, Guo Q, Li J, Yuan H, Xiao C, Qiu J,
Wu Q and Wang D: Loperamide induces protective autophagy and
apoptosis through the ROS/JNK signaling pathway in bladder cancer.
Biochem Pharmacol. 218:1158702023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Regan RC, Gogal RM Jr, Barber JP,
Tuckfield RC, Howerth EW and Lawrence JA: Cytotoxic effects of
loperamide hydrochloride on canine cancer cells. J Vet Med Sci.
76:1563–1568. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim IY, Shim MJ, Lee DM, Lee AR, Kim MA,
Yoon MJ, Kwon MR, Lee HI, Seo MJ, Choi YW and Choi KS: Loperamide
overcomes the resistance of colon cancer cells to bortezomib by
inducing CHOP-mediated paraptosis-like cell death. Biochem
Pharmacol. 162:41–54. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tong CWS, Wu MMX, Yan VW, Cho WCS and To
KKW: Repurposing loperamide to overcome gefitinib resistance by
triggering apoptosis independent of autophagy induction in KRAS
mutant NSCLC cells. Cancer Treat Res Commun.
25:1002292020.PubMed/NCBI
|
|
21
|
Yang S, Li Z, Pan M, Ma J, Pan Z, Zhang P
and Cao W: Repurposing of antidiarrheal loperamide for treating
melanoma by inducing cell apoptosis and cell metastasis suppression
in vitro and in vivo. Curr Cancer Drug Targets. 24:1015–1030. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roncuzzi L, Perut F and Baldini N:
Repurposing of loperamide as a new drug with anticancer activity
for human osteosarcoma. Anticancer Res. 44:1063–1070. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang T, Song X, Yang Y, Wan X, Alvarez
AA, Sastry N, Feng H, Hu B and Cheng SY: Autophagy and hallmarks of
cancer. Crit Rev Oncog. 23:247–267. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh SS, Vats S, Chia AYQ, Tan TZ, Deng
S, Ong MS, Arfuso F, Yap CT, Goh BC, Sethi G, et al: Dual role of
autophagy in hallmarks of cancer. Oncogene. 37:1142–1158. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yun CW, Jeon J, Go G, Lee JH and Lee SH:
The dual role of autophagy in cancer development and a therapeutic
strategy for cancer by targeting autophagy. IJMS. 22:1792020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang H and Zou Z: Targeting autophagy to
overcome drug resistance: Further developments. J Hematol Oncol.
13:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu F, Song D, Yan Y, Huang C, Shen C, Lan
J, Chen Y, Liu A, Wu Q, Sun L, et al: IL-6 regulates autophagy and
chemotherapy resistance by promoting BECN1 phosphorylation. Nat
Commun. 12:36512021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reyes-Castellanos G, Abdel Hadi N and
Carrier A: Autophagy contributes to metabolic reprogramming and
therapeutic resistance in pancreatic tumors. Cells. 11:4262022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen C, Lu L, Yan S, Yi H, Yao H, Wu D, He
G, Tao X and Deng X: Autophagy and doxorubicin resistance in
cancer. Anticancer Drugs. 29:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J,
Qian Y, Kryczek I, Sun D, Nagarsheth N, et al: Fusobacterium
nucleatum promotes chemoresistance to colorectal cancer by
modulating autophagy. Cell. 170:548–563.e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng Z, Zhang S, Han Q, Chu T, Wang H, Yu
L, Zhang W, Liu J, Liang W, Xue J, et al: Liensinine sensitizes
colorectal cancer cells to oxaliplatin by targeting HIF-1α to
inhibit autophagy. Phytomedicine. 129:1556472024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Z, Chen H, Huang L, Duan B, Dai S,
Cai W, Sun M, Jiang Z, Lu R, Jiang Y, et al: ATB°,+-targeted
nanoparticles initiate autophagy suppression to overcome
chemoresistance for enhanced colorectal cancer therapy. Int J
Pharm. 641:1230822023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang R, Pan T, Xiang Y, Zhang M, Feng J,
Liu S, Duan T, Chen P, Zhai B, Chen X, et al: β-Elemene reverses
the resistance of p53-deficient colorectal cancer cells to
5-fluorouracil by inducing pro-death autophagy and cyclin
D3-dependent cycle arrest. Front Bioeng Biotechnol. 8:3782020.
View Article : Google Scholar : PubMed/NCBI
|