Introduction
Globally, colorectal cancer (CRC) accounted for
>1.9 million newly diagnosed cases of cancer in 2020, leading to
almost 935,000 fatalities. This accounted for one-tenth of all
cancer cases and deaths (1). While
chemotherapy is the standard treatment for metastatic CRC, the
median overall survival is 20 months (2), and the 5-year survival rate is 12%
(3). These statistics reveal the
need to unravel the molecular basis of treatment resistance and
develop targeted therapies that induce novel forms of programmed
cell death (PCD). Although oncogene activation, a hallmark of tumor
growth, provides cancer cells with benefits, including resistance
to apoptosis and promotion of invasion and metastasis, it also
induces replication stress (RS) (4). Cells have evolved multiple mechanisms
to manage RS, preventing excess activation that could lead to
genomic instability and DNA damage. One such mechanism is
specialized translesion synthesis (TLS), which allows DNA
replication to proceed past damaged sites, thereby avoiding
replication fork failure (5).
In cancer cells, the TLS core factor REV1 (REV1 DNA
directed polymerase (REV1) promotes chemotherapy resistance and
confers a growth advantage by dynamically recruiting Polζ (B-family
DNA polymerase ζ) in an ‘on the fly’ or ‘gap-filling’ process. The
on the fly process is initiated at platinum-induced DNA damage
sites. In this mechanism, the high-fidelity DNA Polδ is
functionally replaced by the error-prone TLS polymerase REV1. REV1
inserts a cytosine opposite a lesion or abasic sites, allowing Polζ
to continue DNA synthesis before switching back to Polδ. This
mechanism enables cancer cells to bypass the DNA damage and
tolerate the toxicity of platinum-based therapy (6–9). The
gap-filling process involves temporary single-stranded DNA (ssDNA)
gaps resulting from the intermittent synthesis of the lagging
strand during DNA replication. In cancer cells, active oncogenes
and RS create numerous ssDNA gaps that often require REV1-Polζ for
repair, given the fast growth rate of these cells (10,11).
Therefore, targeting the REV1-Polζ complex may counteract the
development of resistance to platinum-based chemotherapy. Secondly,
inhibiting TLS without requiring additional chemotherapy drugs may
facilitate the accumulation of ssDNA gaps. These gaps impede the
ability of cancer cells to replicate and proliferate (12), ultimately leading to cell death.
REV1 mRNA levels are upregulated 10.3-fold in CRC
when DNA mismatch repair is inactivated or p53 is lost (13). The transgenic expression of REV1 in
mice accelerates the development of intestinal adenoma, with the
rate of development being directly proportional to the levels of
REV1 expression (14). A pan-cancer
genomic analysis demonstrated an association between low expression
of REV1 and a more favorable prognosis for CRC (15). Furthermore, high REV1 expression is
associated with a poor prognosis in various types of tumor,
including CRC (15). Therefore,
small molecule drugs that specifically target REV1 hold promise for
CRC treatment.
Research by Wojtaszek et al led to the
discovery of JH-RE-06, a novel compound targeting REV1 (16). Combining JH-RE-06 with cisplatin
arrests tumor growth and significantly prolongs survival in mouse
models (16–18), while decreasing cisplatin-induced
mutations and enhancing tumor cell sensitivity. Although these
findings suggest clinical potential, whether REV1 inhibitors exert
unique mechanisms in CRC compared with other cancers remains
unexplored. Unlike the senescence-inducing effects of REV1
inhibitors reported in melanoma and ovarian cancer cells (17), JH-RE-06 uniquely triggers
NCOA4-mediated ferritinophagy in CRC, which results in increased
levels of the labile iron pool (LIP), inducing ferroptosis.
N-acetylcysteine (NAC), a cysteine prodrug, effectively rescues
this ferroptotic process. While JH-RE-06 does not increase the
sensitivity of CRC cells to oxaliplatin (OXA), it effectively
suppresses clonal proliferation of OXA-resistant (OXR) cell lines
in vitro and inhibits the growth of OXR xenograft tumors
in vivo. As high REV1 expression in CRC tissues is
significantly associated with poor patient prognosis (15), the selective inhibition of REV1 by
JH-RE-06 to induce ferroptosis offers a potential therapeutic
strategy to overcome apoptosis resistance.
Materials and methods
Cell culture
The human CRC cell lines HCT116, SW620 (Wuhan
Servicebio Technology Co., Ltd.), and oxaliplatin-resistant HCT116
(OXR-HCT116) (Xiamen Immocell Biotechnology Co., Ltd.) cells were
maintained under standard physiological conditions (37°C, 5%
CO2) using basal DMEM (cat. no. G4511) enriched with 10%
FBS (cat. no. G8002) and antibiotic-antimycotic solution (1% v/v;
cat. no. G4003; all Wuhan Servicebio Technology Co., Ltd.).
Expression analysis of REV1 in
CRC
The paired-end RNA sequencing files from The Cancer
Genome Atlas (TCGA, portal.gdc.cancer.gov, accession no.
phs000178.v11.p8) for 649 colorectal tumors [colon adenocarcinoma
(COAD) n=456, READ n=193; COAD: colon adenocarcinoma; READ: rectal
adenocarcinoma) were subjected to quality control. Reads were
aligned using STAR (v2.7.10b (19)). Gene expression differences between
paired and unpaired datasets were analyzed using R (version 4.2.1,
R Foundation for Statistical Computing, r-project.org/). Data
visualization was performed with ggplot2 (version 3.4.4,
ggplot2.tidyverse.org/), and receiver operating characteristic
(ROC) curves were generated using pROC (version 1.18.4, http://cran.r-project.org/package=pROC).
Prognostic analysis of gene expression in CRC (GSE17536 and
GSE17537) was conducted with PrognoScan (https://dna00.bio.kyutech.ac.jp/PrognoScan/). CRC
tissue microarrays (93 tissue cores, 37 paired adjacent
non-cancerous and cancer tissue samples, 19 metastatic samples)
were obtained from Shanghai Outdo Biotech Co., Ltd (cat. no.
T21-1063). Antigen retrieval was performed with 3%
H2O2 for 10 min at room temperature.
Following blocking with 5% normal donkey serum (cat. no. G1217-5ML,
Wuhan Servicebio Technology Co., Ltd.) at room temperature for 30
min, tissue sections were probed with REV1-specific primary
antibody (cat. no. AB5088; Abcam, 1:250) at 4°C overnight.
Following thorough PBS washing, they were treated with
HRP-conjugated donkey anti-goat IgG (cat. no. a0181, Beyotime
Institute of Biotechnology; 1:250; room temperature, 45 min)
followed by DAB staining (5 min, room temperature). Counterstaining
with 0.5% Mayer's hematoxylin was then performed (room temperature,
5 min), after which the sections were dehydrated through graded
ethanol, cleared in xylene, and mounted with resin. Stained
sections were observed via light microscope (Nikon Eclipse E100).
REV1 positive rates were analyzed using ImageJ 1.49v (National
Institutes of Health, NIH).
Inhibitors
The following inhibitors were used at 37°C under 5%
CO2 JH-RE-06 (cat. no. HY-12621; 0.5–5.0 µM, 24 or 72 h
treatment; deferoxamine (DFO; cat. no. HY-B1625; 100 or 200 µM, 24
h; Z-VAD-FMK (cat. no. HY-16658B): 5 or 10 µM, 24 h; necrostatin-1
(cat. no. HY-15760): 5 or 10 µM, 24 h; chloroquine (cat. no.
HY-17589A): 10 or 20 µM, 24 h; N-acetylcysteine (cat. no. HY-B0215;
all MedChemExpress): 15 or 20 mM, 24 h; L-penicillamine (cat. no.
196312; 100 mM, 24 h treatment; and 2-mercaptoethanol (cat. no.
M3148; both Sigma-Aldrich; Merck KGaA): 2 mM, 24 h treatment.
RNA interference
Transfection was performed when 2×105
HCT116 cells in the six-well plate reached 60% confluency. For
liposomal transfection complex formation, 15 µl Lipofectamine™ 3000
(cat. no. L3000001, Thermo Fisher Scientific, Inc.) was mixed with
250 µl Opti-MEM™ reduced serum medium (cat. no. 31985070, Gibco)
and incubated at room temperature for 5 min. A total of 15 µl small
interfering (si) RNA [ATG7 (Autophagy-related gene 7) RNA I (cat.
no. 6604, Cell Signaling Technology, Inc., sequences not
available), NCOA4-targeting and scramble siRNA (sequences provided
in Table I; Tsingke Biotechnology
Co., Ltd.)] at a final concentration of 50 nM was separately
diluted in 250 µl Opti-MEM reduced serum medium. Each siRNA diluent
was mixed with the Lipofectamine™ 3000-Opti-MEM™ mixture, and the
transfection complexes were incubated at room temperature for 20
min. The cells were cultured at 37°C under 5% CO2 for 48
h. followed by western blot analysis to determine the knockdown
efficiency of ATG7 and NCOA4.
 | Table I.RNA interference sequences. |
Table I.
RNA interference sequences.
| Target Gene | Primer Name | Sense Sequence
(5′→3′) | Antisense Sequence
(5′→3′) |
|---|
| NCOA4 | NCOA4-01 |
GACCUUAUUUAUCAGCUUA |
UAAGCUGAUAAAUAAGGUC |
|
| NCOA4-02 |
GGAGAACAGUCAGACUUCU |
AGAAGUCUGACUGUUCUCC |
|
| NCOA4-03 |
CCAGGAAGUAUUACUUAAU |
AUUAAGUAAUACUUCCUGG |
| Negative
control | Scramble |
CUAGAUACUAGACUGAUAA |
UUAUCAUCUAGUUAUCUAG |
Tandem mass tags (TMT) proteomic
sequencing
A total of 5×105 HCT116 cells underwent
treatment with DMSO or 3 µM JH-RE-06 for 24 h prior to washing with
PBS and cell harvesting at room temperature. Following
centrifugation at 15,000 g, 4°C for 30 h, the soluble protein
fraction was isolated, and underwent BCA protein quantification
assay. Following concentration normalization (BCA assay: 2 µl
diluted sample/BSA standard in 96-well plate, 200 µl 50:1 Buffer
A/B at 37°C for 30 min, absorbance 562 nm to calculate
concentration; adjusted to uniform level with PBS), samples
underwent tryptic digestion (37°C, overnight), followed by
lyophilization and storage at −80°C. TMTpro reagents (cat. no.
a52047, Thermo Fisher Scientific, Inc.) were used for peptide
labeling, followed by reverse-phase chromatographic separation.
Agilent 1100 HPLC with Zorbax Extend-C18 column was used: mobile
phases A (2:98 ACN-H2O, pH 10) and B (90:10
ACN-H2O, pH 10), 300 µl/min, 210 nm detection. Eluates
(8–54 min) collected cyclically into tubes, lyophilized, frozen for
MS. The samples were loaded onto an Acclaim PepMap RSLC column (75
µm ×50 cm, RP-C18, Thermo Fisher) for separation at a flow rate of
300 nl/min. Data were analyzed using Proteome discoverer 2.4.1.15
(Thermo Fisher Scientific). Pathway analysis of differentially
expressed proteins was performed using the KEGG (Kyoto Encyclopedia
of Genes and Genomes) database (integrated with KEGG annotation
results). The hypergeometric distribution test was applied to
calculate the significance of differential protein enrichment in
each pathway entry, which was represented by the P-value.
Western blotting
A total of 5×105 HCT116 or SW620 cellular
proteins were extracted using RIPA buffer (cat. no. p0013b,
Beyotime Institute of Biotechnology). Protein concentration was
determined using the BCA assay. A total of 30 µg of protein per
lane was resolved via 12% SDS-PAGE and electroblotted onto PVDF
membranes (200 mA, 90 min). Following blocking with 5% skimmed milk
at room temperature for 1 h, membranes were probed with primary
antibodies (4°C, overnight) and corresponding secondary antibodies
(1:1,000 dilution, cat. no. zb-2301 for anti-rabbit; cat. no.
zb-2305 for anti-mouse, Beijing Zhongshan Jinqiao Biotechnology
Co., Ltd.) conjugated to horseradish peroxidase at room temperature
for 90 min, followed visualization using an ECL kit (cat. no.
s6009l, Ue landy). Densitometric analysis was performed using
ImageJ 1.49v (National Institutes of Health). The primary
antibodies were as follows: Glutathione peroxidase 4 (GPX4; cat.
no. 52455), NCOA4 (Nuclear receptor coactivator 4, cat. no. 66849),
FTH1 (Ferritin heavy chain 1, cat. no. 3998), γH2AX (Phosphorylated
histone H2AX, cat. no. 2577), SOD2 (Superoxide dismutase 2, cat.
no. 13141), H2AX (cat. no. 2595), and ATG7 (cat. no. 2631; all
1:1,000, Cell Signaling Technology, Inc.). FTL1 (ferritin light
chain 1, cat. no. 84731-7-RR), GCLC (Glutamate-cysteine ligase
catalytic subunit, cat. no. 12601-1-AP; both Proteintech Group,
Inc.), LC3I/II (Microtubule-associated protein 1 light chain 3
I/II, cat. no. 14600-1-AP), p62 (Sequestosome 1, cat. no.
66184-1-Ig), Tubulin (cat. no. 11224-1-AP), and GAPDH (all 1:2,000,
cat. no. 60004-1-Ig, Proteintech Group, Inc.).
Transmission electron microscopy
(TEM)
5×105 HCT116 or SW620 cells treated with
JH-RE-06 (1.5 and 3.0 µM for HCT116 cells, and 1.0 and 1.5 µM for
SW620 cells), or 9 µl DMSO at 37°C for 24 h, then collected and
fixed overnight at 4°C in a fixation solution (cat. no. G1102;
Wuhan Servicebio Technology Co., Ltd.). This was followed by 2 h
fixation at room temperature using 1% osmium tetroxide in PBS.
Following washing by PBS, samples underwent gradient dehydration
using ethanol and acetone. Samples were embedded in 812 embedding
agent (cat. no. GP2001, Wuhan Servicebio Technology Co., Ltd.) and
acetone. Embedding was performed overnight at 37°C, followed by
polymerization at 60°C for 48 h. Ultrathin sections (60 nm) were
dual-stained: uranyl acetate (5% aqueous solution) at 37°C for 15
min, followed by lead citrate (0.5% aqueous solution) at room
temperature for 15 min, then air-dried and imaged using a Hitachi
HT7700 TE microscope. Mitochondrial numbers were counted, and
autophagic vacuoles with engulfed mitochondria were analyzed using
ImageJ 1.49v (National Institutes of Health).
Immunofluorescence assay
HCT116 cells were fixed with 4% PFA for 15 min at
room temperature. Subsequently, 0.5% Triton X-100 was applied for
20 min for permeabilization at room temperature. Finally,
non-specific binding sites were blocked with 10% goat serum (cat.
no. C0265, Beyotime Institute of Biotechnology) for 30 min at room
temperature. Following blocking, cells were incubated with γH2AX
primary antibody (cat. no. 2577, Cell Signaling Technology, 1:500)
at 4°C for 16 h, then exposed to fluorescent secondary antibody
(Alexa-Fluor 488; cat. no. ab150077, Abcam; 1:1,000) for 1 h at
room temperature in the dark and finally stained with DAPI using a
1 µg/ml aqueous solution at room temperature for 5 min for nuclear
visualization. Samples were mounted with antifade medium (cat. no.
P0128S, Beyotime) and imaged by confocal microscopy, and image
analysis was performed using NIS-elements viewer 5.21 (Nikon
Instruments, Inc.).
Cell Counting Kit (CCK)-8 assay
HCT116 or SW620 cells in the logarithmic growth
phase were trypsinized, resuspended in complete DMEM
(5×103 cells/100 µl) and seeded into 96-well plates (100
µl/well). Following 24 h culture at 37°C, the medium was replaced
with medium containing JH-RE-06 (0.0, 0.5, 1.0, 1.5, 3.0, 5.0 µM),
and cells were cultured at 37°C for 24 or 72 h. CCK-8 reagent (cat.
no. C6005, Us Everbright Inc.) was added (1 h, 37°C in the dark),
and the absorbance was measured at 450 nm.
Clone formation assay
Following 50 µM OXA or 3.0 µM JH-RE-06 treatment (24
h, 37°C), OXR-HCT116 cells were resuspended in DMEM following
trypsin digestion to prepare a cell suspension; subsequent cell
counting was performed, and the cells were then diluted to the same
density using DMEM. After being plated (1,000, 2,000 and 5,000
cells/well) in 6-well plates and maintained at 37°C for 14 days,
the cultured cells were fixed with 4% paraformaldehyde solution for
20 min at room temperature, with subsequent PBS washing. Cells were
stained with 0.2% crystal violet for 5 min at room temperature.
Under a light microscope, positive clones (defined as cell
aggregates containing >50 cells) were identified and counted
manually. Then the cloning rate was determined by dividing the
clone count by the initial number of seeded cells.
Ferroptosis assay
5×105 HCT116 cells were treated with 3.0
µM JH-RE-06, and 5×105 SW620 cells were treated with 1.5
µM JH-RE-06 at 37°C for 6, 12 and 24 h, respectively. A assay kits
were used to measure Fe2+ content (Cell Ferrous Iron
Colorimetric Assay kit; cat. no. E-BC-K881-M, Wuhan Elabscience
Biotechnology Co., Ltd.), malondialdehyde (MDA) levels (MDA
Detection kit; cat. no. KGT003) and glutathione (GSH) content (GSH
Detection kit; cat. no. KGT006, both Jiangsu Kaiji Biotechnology
Co., Ltd.) according to the manufacturers' instructions. Following
3.0 µM JH-RE-06 treatment (24 h, 37°C), 5×105 HCT116
cells were washed with PBS and incubated with FerroOrange
fluorescent probe (cat. no. F374, Dojindo Laboratories Inc.; 1
µmol/l working concentration) at 37°C for 30 min. Cells were washed
again with PBS and imaged using laser confocal microscopy and image
analysis was performed using NIS-Elements Viewer 5.21.
Animal experiment
24 Male BALB/c nude mice (age: 4–5 weeks; initial
weight: 18–22 g; Ningxia Medical University Laboratory Animal
Center) were housed at 25°C, relative humidity at 50%, a 12 h
light/12 h dark cycle, and ad libitum access to sterile standard
rodent chow and filtered water. All methods received approval from
the Laboratory Animal Ethical and Welfare Committee at Ningxia
Medical University Laboratory Animal Center (approval no.
IACUC-NYLAC-2022-022). Each mouse was injected with
5×106 OXR-HCT116 cells into the axilla. After 12 days of
post-injection culture, the mice were randomized into three groups
(n=8 per group based on power analysis) as follows: i) Oxaliplatin
(5 mg/kg, i.p.), ii) JH-RE-06 (1.6 mg/kg, intratumoral) and iii)
DMSO (100 µl, intratumoral), all of which were administered every
48 h to monitor therapeutic effects and recovery. The maximum
volume of the tumor was <2,000 mm3 (volume=π/6×
length × width × height). The maximum tumor volume and diameter
were 1,450 mm3 and 15 mm, respectively. Following 20
consecutive administrations, animals were euthanized with
isoflurane, perfused with PBS and 4% paraformaldehyde at room
temperature (15 min) and organs (heart, liver, spleen, lung and
kidney) and tumor samples were collected and measured. Anesthesia
for all surgical and tumor measurement procedures consisted of
isoflurane (5% for induction, 2% for maintenance). Confirmation of
death was established by the absence of a pedal/toe pinch reflex
and the cessation of breathing for ≥1 min.
Immunohistochemistry
Paraffin-embedded tumor specimens were prepared by
fixing tissues with 4% paraformaldehyde (4°C, 12 h), embedding in
paraffin, and sectioning into 5 µm-thick slices. Sections were
deparaffinized (xylene/ethanol) and subjected to EDTA-mediated
antigen retrieval (0.01 M EDTA, pH 9.0; 95°C, 5 min). Endogenous
peroxidase was quenched with 3% H2O2 (room
temperature, 10 min). Non-specific binding was blocked with 10%
goat serum (cat. no. C0265, Beyotime; room temperature, 30 min).
Sections were probed with anti-PCNA (cat. no. GB11010-50, Wuhan
Elabscience Biotechnology Co., Ltd.; 1:500) and Ki-67 (cat. no.
GB111499-100, Wuhan Elabscience Biotechnology Co., Ltd.; 1:200)
antibodies (4°C, overnight). Following PBS-T (0.05% Tween-20)
washes, they were treated with HRP-conjugated goat anti-rabbit IgG
(cat. no. G1213-100UL, Wuhan Elabscience Biotechnology Co., Ltd.;
1:200; room temperature, 45 min), followed by DAB chromogen
staining (5 min). Then counterstaining with 0.5% Mayer's
hematoxylin (room temperature, 5 min) and mounting. Stained
sections were observed via light microscope (50 µm scale bar; Nikon
Eclipse E100). PCNA/Ki-67 positive rates were analyzed using ImageJ
1.49v (National Institutes of Health).
H&E staining
Mouse organ wax blocks were sequentially
deparaffinized in graded xylene (analytical grade) and dehydrated
in a series of ethanol (70%→80%→95%→100%, v/v). Hematoxylin
staining (working concentration: 0.5%, w/v; cat. no. Y269827,
Beyotime) was performed at room temperature for 5 min, followed by
tap water washing. Differentiation and eosin counterstaining
(working concentration: 0.5%, w/v; cat. no. C0109, Beyotime
Institute of Biotechnology) were conducted at room temperature for
3 min. After re-dehydration in the above ethanol gradient and
clearing in xylene (analytical grade, room temperature), sections
were mounted with neutral gum and examined under a light microscope
(Nikon Eclipse E100).
Reactive oxygen species (ROS)
detection
5×105 HCT116 cells were treated with 3.0
µM JH-RE-06 for at 37°C for 24 h. Then, the cells were loaded with
10 µM DCFH-DA (dissolved in serum-free DMEM medium) and incubated
at 37°C for 30 min in the dark. Subsequently, cells were washed 3
times with pre-warmed (37°C) PBS. ROS-derived green fluorescence
(488/525 nm) was imaged using a inverted fluorescence microscope.
Image acquisition and fluorescence intensity quantification were
conducted using NIS-Elements Viewer 5.21. ROS levels were
calculated as the mean fluorescence intensity of cells in the
captured fields.
Statistical analysis
The experiments were repeated in triplicate. All
data are presented as mean ± standard error of the mean (SEM).
Statistical analysis was performed using GraphPad Prism 9.5.1
(Dotmatics, Inc.), employing one-way ANOVA followed by Sidak's post
hoc test. Paired comparisons were performed by Wilcoxon signed-rank
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
REV1 is upregulated in CRC tissue and
associated with a poor prognosis
To delineate the role of REV1 in colorectal
carcinogenesis, the present study assessed transcriptomic profiles
from 644 TCGA colorectal adenocarcinoma samples (COAD/READ).
Ggplot2-based analysis demonstrated significantly elevated
expression of both REV1 and REV7 in tumor vs. adjacent normal
tissue, consistent across unpaired (Fig. 1A) and paired sample analyses
(Fig. 1B). The baseline
characteristics of the patients are provided in Table SI. Expression levels were not
significantly associated with clinicopathological parameters,
including sex, age, TNM stage, or perineural invasion. Conversely,
the presence of colon polyps was associated with REV1. Significant
differences were found in lymphatic invasion with respect to REV1
expression (Table SI). The
PrognoScan database was used to assess the association between REV1
expression and CRC prognosis (20).
Analysis of datasets GSE17536 and GSE17537 showed a significant
association between high REV1 expression and poor prognosis
(Fig. 1C). To detect the expression
of REV1, quantitative IHC analysis was performed on 35 paired
specimens using tissue microarrays (Baseline information for CRC
tissue chips is provided in Table
SII). Consistent with TCGA findings, there was a significant
increase in REV1 protein levels in CRC tumors compared with the
paired normal tissues (Fig. 1D and
E). Overall, these findings established high REV1 expression as
a negative prognostic marker in CRC.
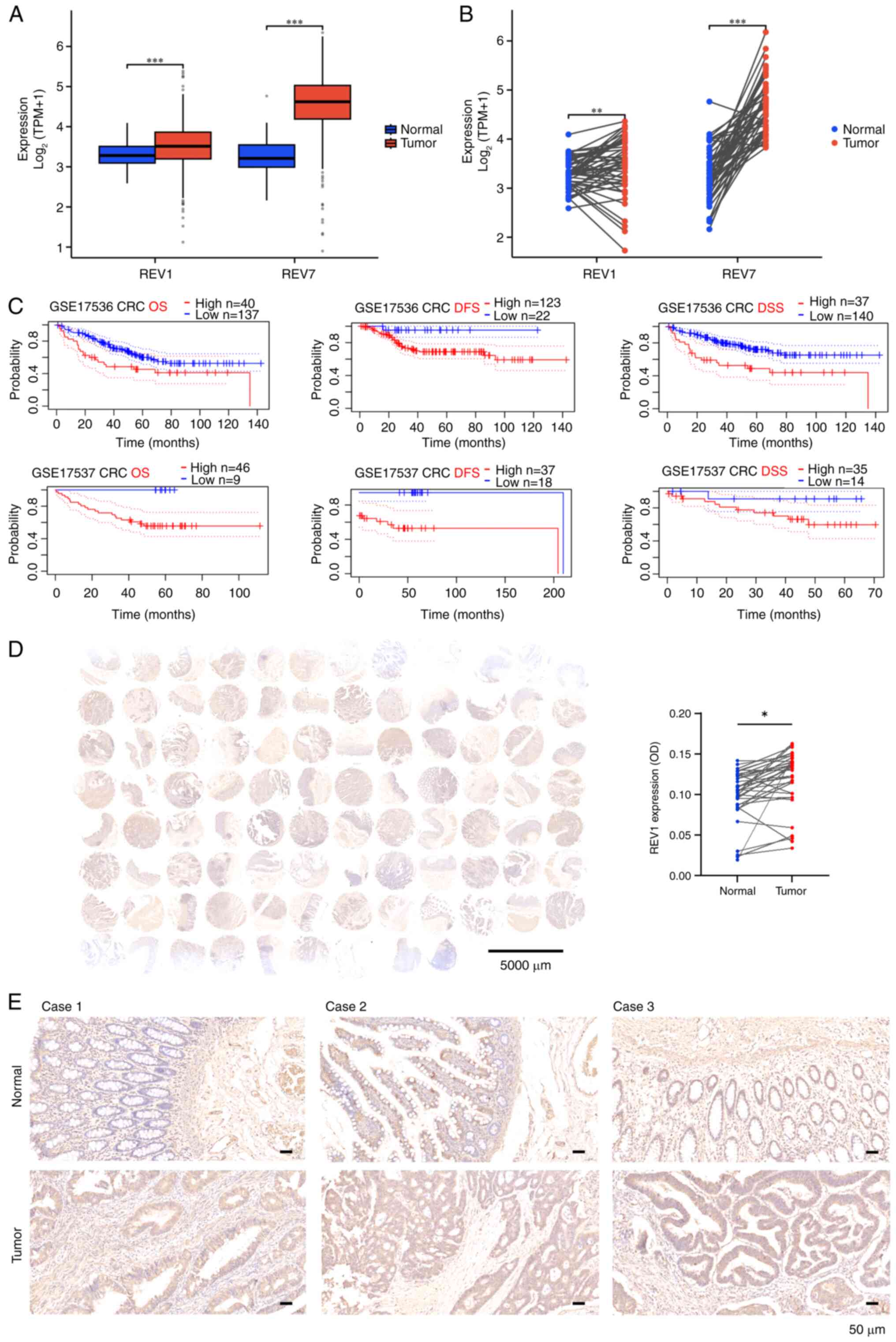 | Figure 1.REV1 expression is upregulated in CRC
tissues and associated with poor prognosis. (A) Analysis of 644
unpaired samples from TCGA database revealed elevated expression of
the REV1 and its binding partner REV7 in clinical CRC samples. (B)
Analysis of 50 paired samples from TCGA database showed that REV1
and REV7 were significantly more highly expressed in cancer
compared with adjacent non-cancerous tissue. (C) Prognostic
analysis of REV1 in CRC was performed using PrognoScan database
datasets (accession no. GSE17536/37). (D) REV1 expression levels
were analyzed in 35 paired CRC tissue samples using a tissue
microarray, which revealed significantly elevated REV1 expression
in CRC vs. adjacent normal tissue. (E) Representative
immunohistochemical REV1 staining in CRC tissue. Scale bar, 50 µm.
***P<0.001, **P<0.01, *P<0.05. REV1, REV1 DNA directed
polymerase; CRC, colorectal cancer; TCGA, The Cancer Genome Atlas;
TPM, transcripts per kilobase of exon model per million mapped
reads; DFS, disease-free survival; OS, overall survival; DSS,
disease-specific survival; OD, optical density. |
REV1 inhibitor JH-RE-06 suppresses CRC
tumorigenesis both in vitro and in vivo
To exploit REV1 as a drug target, the present study
evaluated JH-RE-06, a compound that impairs REV1 function by
inducing inactive dimer formation (16). CCK-8 assay revealed that JH-RE-06
decreased the proliferation of CRC cells in a dose- and
time-dependent manner and induced DNA damage, as indicated by
increased γH2AX levels (Fig.
2A). While JH-RE-06 is hypothesized to enhance tumor cell
sensitivity to platinum-based drugs (21), western blot and immunofluorescence
analysis show that JH-RE-06 did not modify the effect of OXA in
HCT116 cells (Fig. 2B and C). To
assess its efficacy in resistant models, OXR-HCT116 cells were used
(Fig. S1A). Crystal violet
staining demonstrated that JH-RE-06 effectively inhibited the
clonogenic capacity of OXR-HCT116 cells (Fig. S1B). Therapeutic efficacy of
JH-RE-06 was assessed in vivo using a xenograft mouse model
developed with OXR-HCT116 cells (Fig.
S1C). The JH-RE-06 group displayed a substantial reduction in
tumor volume, indicating its effectiveness against OXR malignancies
(Figs. 2D and S1D). Safety of JH-RE-06 was assessed by
hematoxylin and eosin staining of the hearts, livers, spleens, lung
and kidney of treated mice. No notable pathological damage was
observed in these major organs (Fig.
S1E). Immunohistochemistry for proliferation markers Ki67 and
PCNA revealed that JH-RE-06 significantly suppressed tumor growth
and blood vessel formation (Fig.
2E). In summary, the REV1 inhibitor JH-RE-06 induced DNA damage
in CRC cells and inhibited tumor growth both in vitro and
in vivo.
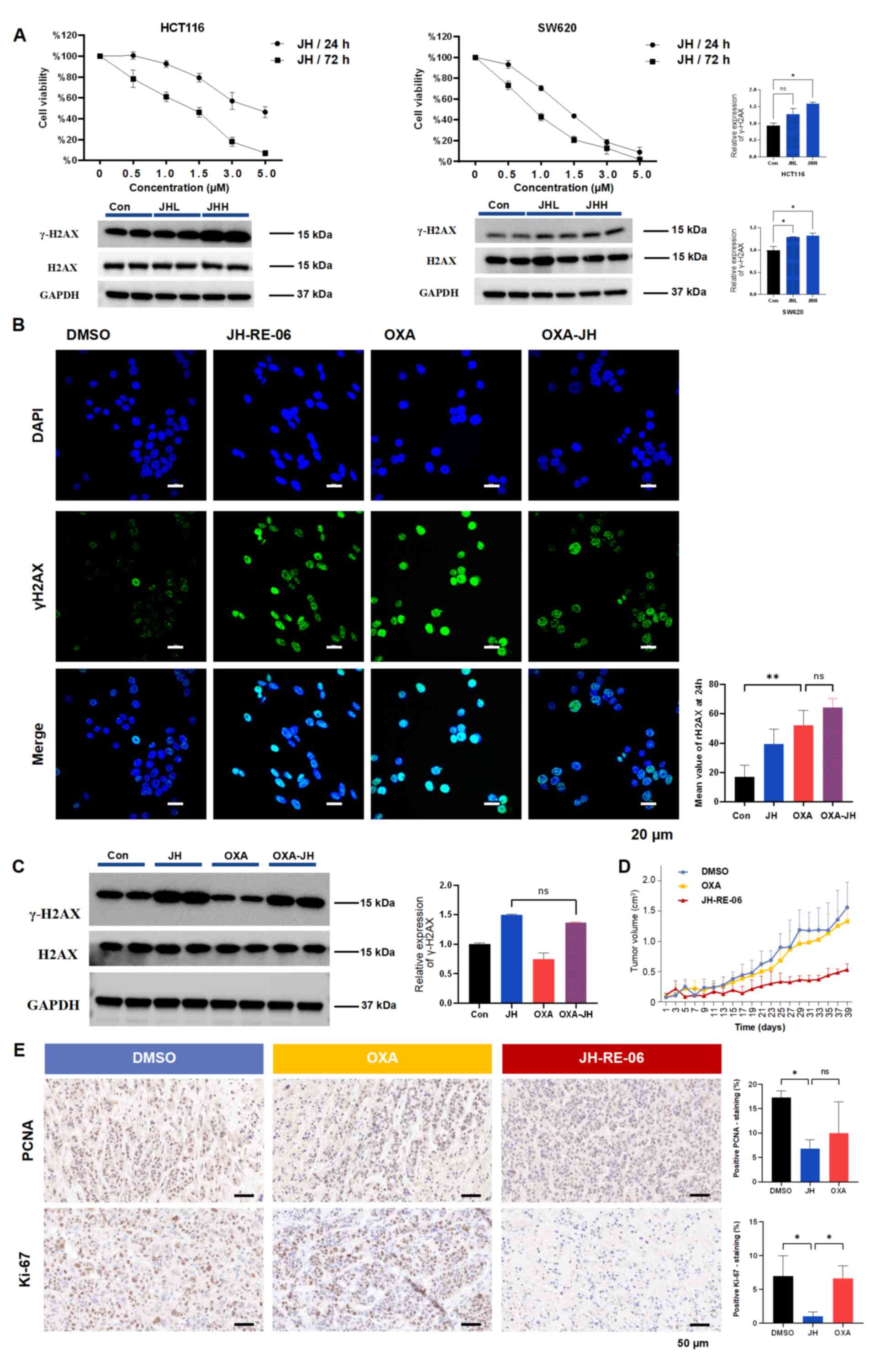 | Figure 2.JH, a REV1 DNA directed polymerase
inhibitor, suppresses CRC tumorigenesis both in vitro and
in vivo. (A) Cell viability assessed by Cell Counting Kit-8
revealed differential sensitivity to JH, with sustained effects
observed at 72 h vs. 24 h in CRC models. WB analysis was used to
detect γH2AX levels in cells treated with JH for 24 h. For HCT116
cells, JHL represents 1.5 and JHH represents 3.0 µM JH. For SW620
cells, JHL represents 1.0 and JHH represents 1.5 µM JH.
Concentrations were selected based on treatments that resulted in
~50% cell viability after 24 or 72 h. GAPDH was used as the
internal control. (B) Immunofluorescence staining of γH2AX in
HCT116 cells treated with 3.0 µM JH and/or 5.0 µM OXA for 12 and 24
h. Scale bar, 20 µm. (C) WB analysis of γH2AX levels after
treatment with JH and/or OXA, with GAPDH as the loading control.
Quantitative analysis revealed no significant differences between
the JH and OXA-JH treatment groups. (D) Tumor growth curves for
OXA-resistant HCT116 cell xenografts in mice. (E) Representative
immunohistochemical images of PCNA and Ki-67 staining in tumor
sections. Scale bar, 50 µm. **P<0.01, *P<0.05. CRC,
colorectal cancer; JHH, JH-RE-06 high; JHL, JH-RE-06 low; WB,
western blot; H2AX, histone H2AX; OXA, oxaliplatin; PCNA,
proliferating cell nuclear antigen; Con, control; ns, not
significant. |
Cell death induced by REV1 inhibition
is associated with oxidative stress and ferroptosis
To investigate the mechanism by which JH-RE-06
causes cell death, CRC cells were pre-treated with inhibitors that
target different PCD pathways, such as apoptosis (Z-VAD-FMK),
necroptosis (necrostatin-1), autophagy (chloroquine), ferroptosis
[deferoxamine, DFO), and antioxidants (L-penicillamine,
2-mercaptoethanol, and N-acetylcysteine). CCK-8 assay showed that
DFO partially alleviated JH-RE-06-induced cell death, while
antioxidants almost completely reversed this in both HCT116 and
SW620 cells (Fig. 3A and B). To
investigate global proteomic changes, TMT proteomics was performed
on HCT116 cells treated with DMSO or 3 µM JH-RE-06 for 24 h.
Proteomic analysis revealed 650 significantly altered proteins
following JH-RE-06 treatment (Fig.
3C). Downregulated proteins were predominantly enriched in ‘DNA
replication’, ‘oxidative phosphorylation’, and ‘cell cycle’
(Fig. 3D). Notably, a subset of
these downregulated proteins was also associated with mitochondrial
function, including mitochondrial ribosomal protein small and large
subunits and members of the NADH:ubiquinone oxidoreductase subunit
family (Fig. 3F). Upregulated
proteins were significantly associated with ‘ferroptosis’ and
oxidative stress pathways, including ‘steroid biosynthesis’,
‘cysteine and methionine metabolism’, and ‘citrate cycle (TCA
cycle)’ (Fig. 3E). These findings
suggested that REV1 inhibition modulated oxidative stress,
mitochondrial function and ferroptosis.
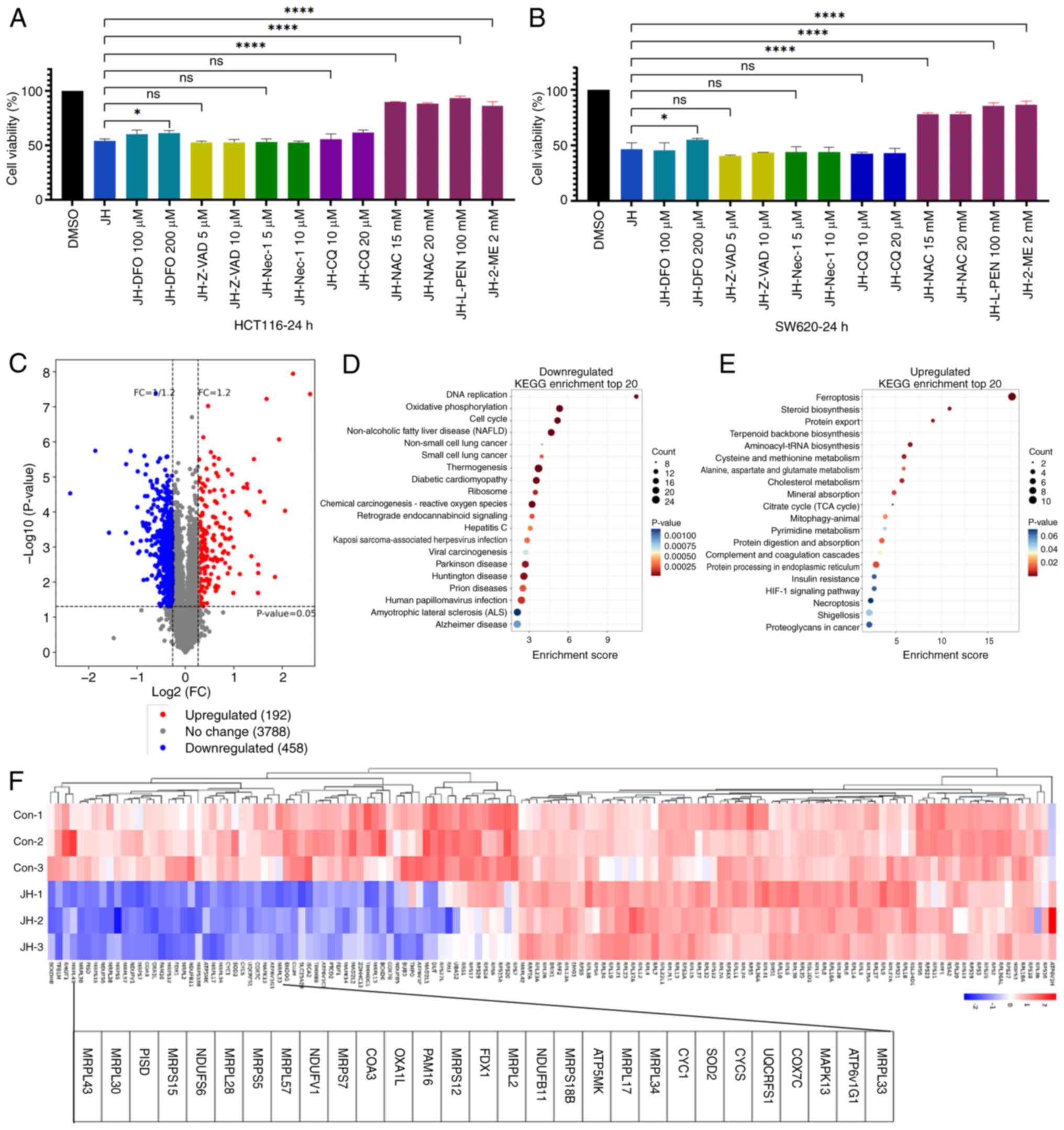 | Figure 3.Cell death induced by REV1 DNA
directed polymerase inhibition is associated with oxidative stress
and ferroptosis. Viability of (A) HCT116 cells treated with 3 µM JH
and (B) SW620 cells treated with 1.5 µM JH in the presence or
absence of inhibitors. (C) Differentially expressed proteins
enriched by proteomics. Compared with the DMSO group, JH
upregulated 192 proteins and downregulated 458 proteins. (D)
Downregulated pathways. The top 20 enriched KEGG pathways are
illustrated. (E) Upregulated pathways. (F) Hierarchical clustering
of differentially expressed mitochondrial-associated genes.
****P<0.0001, *P<0.05. JH, JH-RE-06; KEGG, Kyoto encyclopedia
of genes and genomes; DFO, deferoxamine; Z-VAD, z-vad-fmk; Nec,
necrostatin-1; CQ, chloroquine; NAC, n-acetylcysteine; L-PEN,
l-penicillamine; ME, 2-mercaptoethanol; ns, not significant; Con,
control; FC, fold change; MRPL, mitochondrial ribosomal protein
large subunit; MRPS, mitochondrial ribosomal protein small subunit;
NDUF, NADH:ubiquinone oxidoreductase subunit. |
JH-RE-06 triggers ferroptosis in CRC
cells
Ferroptosis is characterized by iron-catalyzed lipid
peroxidation and is controlled by mitochondrial activity and LIP
concentrations. Therefore, the present study investigated the
subcellular changes in JH-RE-06-treated CRC cells. TEM revealed a
significant reduction in mitochondrial abundance in both HCT116 and
SW620 cells after 24 h JH-RE-06 treatment (Fig. 4A and B). Autophagic vacuoles
containing engulfed mitochondria were observed (Fig. 4A and B). Intracellular free
Fe2+ levels increased following 6 h treatment, peaking
at 24 h in both HCT116 and SW620 cells (Fig. 4C and D). Concurrently, levels of the
lipid oxidation marker MDA and ROS increased, while GSH levels
decreased over time (Figs. 4C and D
and S2A). These results
collectively indicated that JH-RE-06 induced a time-dependent
increase in Fe2+ and MDA levels, alongside a decrease in
GSH levels. Furthermore, western blot analysis confirmed that the
protein expression levels of mitochondrial (SOD2) and
ferritin-(FTL) and glutathione-related proteins (GCLC) decreased
following treatment with 3.0 µM JH-RE-06. This decrease was
consistent with proteomics findings (Fig. 4E). Taken together, these results
suggest that JH-RE-06 triggered ferroptosis in CRC cells.
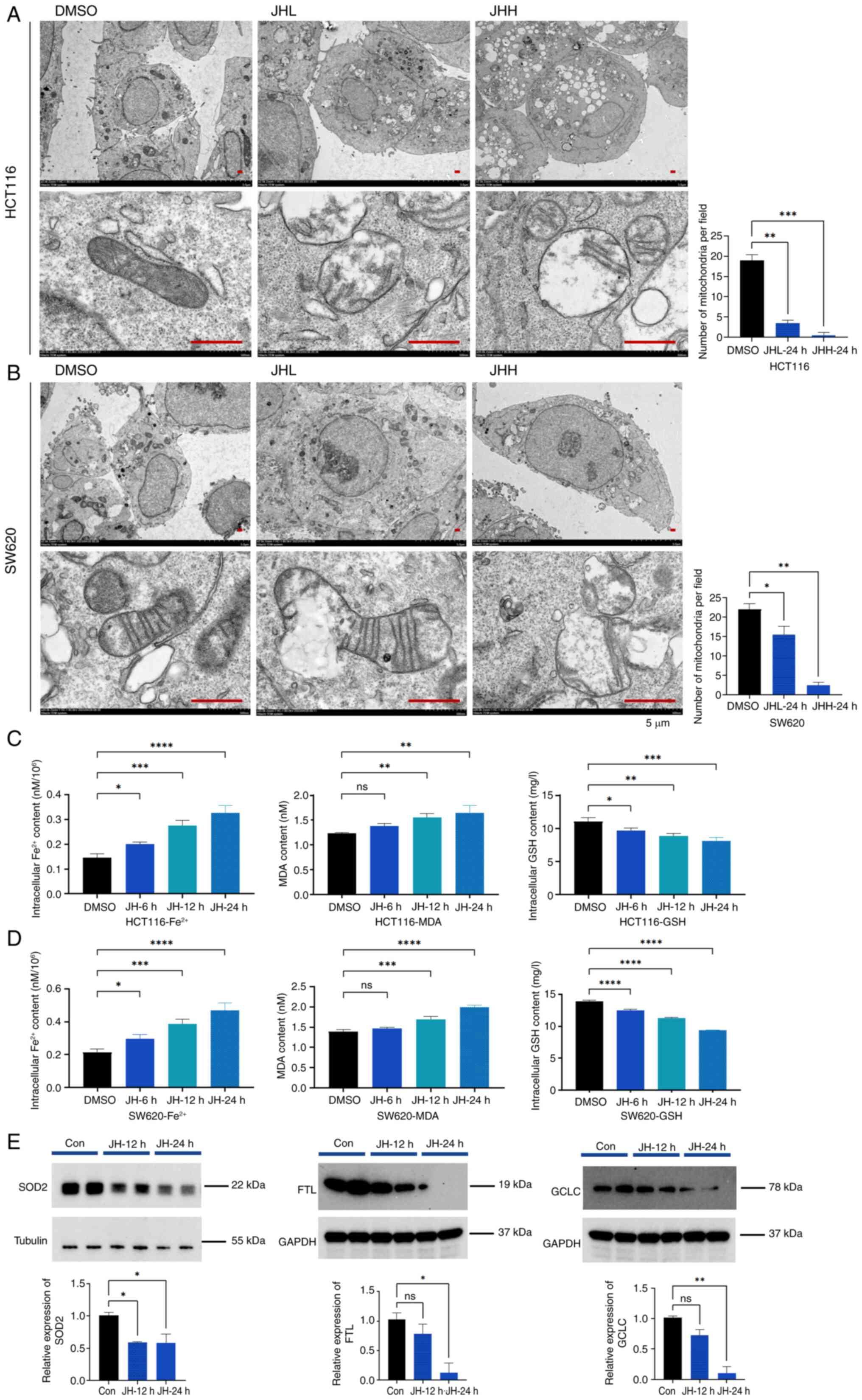 | Figure 4.JH triggers ferroptosis in colorectal
cancer cells. Transmission electron microscopy of (A) HCT116 and
(B) SW620 cells treated with JHL and JHH for 24 h show
intracellular vacuolar changes, with mitochondria-like structures
undergoing digestion inside the vacuoles. Scale bar, 5 µm. Changes
in intracellular Fe2+ concentration, MDA and GSH levels
(normalized to total protein) in (C) HCT116 cells treated with 3.0
µM JH and (D) SW620 cells treated with 1.5 µM JH at 6, 12 and 24 h
post-treatment. (E) Relative protein expression levels of SOD2, FTL
and GCLC following 12 and 24 h JH treatment in HCT116 cells; 24 h
exposure to JH resulted in significant downregulation of
antioxidant proteins SOD2, FTL and GCLC. ****P<0.0001,
***P<0.001, **P<0.01, *P<0.05. JHH, JH-RE-06 high; JHL,
JH-RE-06 low; MDA, malondialdehyde; GSH, glutathione; SOD2,
superoxide dismutase 2; FTL, ferritin light chain; GCLC,
glutamate-cysteine ligase catalytic subunit; ns, not significant;
Con, control. |
JH-RE-06 triggers ferroptosis via
NCOA4-mediated ferritinophagy in CRC cells
Ferroptosis represents an iron-catalyzed, lipid
peroxidation-mediated programmed cell death pathway (22). NCOA4-mediated ferritinophagy plays a
critical role in ferroptosis by regulating cellular iron
homeostasis (23). Ferritinophagy,
the autophagic degradation of ferritin, increases the LIP, which
promotes ferroptosis (24). CRC
cells are both iron-rich and iron-dependent (25), making the regulation of iron
homeostasis a key factor in CRC progression and therapeutic
resistance (26). Proteomics
sequencing revealed elevated NCOA4 expression and a decrease in its
regulatory proteins, suggesting that NCOA4 is key for modulating
cellular iron levels following JH-RE-06 treatment. Therefore, it
was hypothesized that JH-RE-06 increased the LIP in an
NCOA4-dependent manner, thereby inducing ferroptosis via
ferritinophagy. To validate this hypothesis, the present study
assessed changes in the expression of ferritinophagy-associated
proteins following JH-RE-06 treatment in CRC cells using western
blotting. JH-RE-06 increased the expression of NCOA4, p62 and LC3
at both 12 and 24 h (Figs. 5A and B
and S2B and C). FTH1 expression
increased at 12 but decreased by 24 h. To verify the reliability of
the experimental system, DFO was used to chelate Fe2+
and protein levels of LC3II, NCOA4, and p62 were increased
(Figs. 5A and B). Next, the role of
LIP in mediating the effects of JH-RE-06 was explored by
pre-treating cells with DFO to chelate Fe2+, followed by
JH-RE-06 treatment. In the combination group, NCOA4 expression was
sustained for longer period with the DFO group, and FTH1 levels
were also elevated. To investigate the role of ferritinophagy, ATG7
or NCOA4, key proteins involved in ferritinophagy and
Fe2+ recycling, were silenced before treating the cells
with JH-RE-06 (Fig. S2D). In these
cells, the expression of NCOA4, LC3II, and p62 in response to
JH-RE-06 was partially mitigated (Figs.
5C and S2D), and FTH1, a
substrate for ferritinophagy degradation, accumulated due to
impaired autophagy (Fig. 4C and
S2E). LIP was assessed using the
FerroOrange test. In wild-type HCT116 cells, JH-RE-06 treatment
induced a significant increase in LIP at 24 h, indicating iron
overload. However, in siNCOA4 cells, the JH-RE-06-induced increase
in LIP was not observed at 24 h due to the inhibition of ferritin
degradation. This suggested that the iron overload induced by
JH-RE-06 is dependent on functional NCOA4-mediated ferritinophagy
(Fig. 5D). Silencing ATG7 or NCOA4
before JH-RE-06 treatment resulted in decreased MDA levels
(Fig. 5E). Based on these findings,
it was hypothesized that JH-RE-06 induces ferroptosis through
NCOA4-mediated ferritinophagy, disrupting iron homeostasis and
promoting lipid peroxidation.
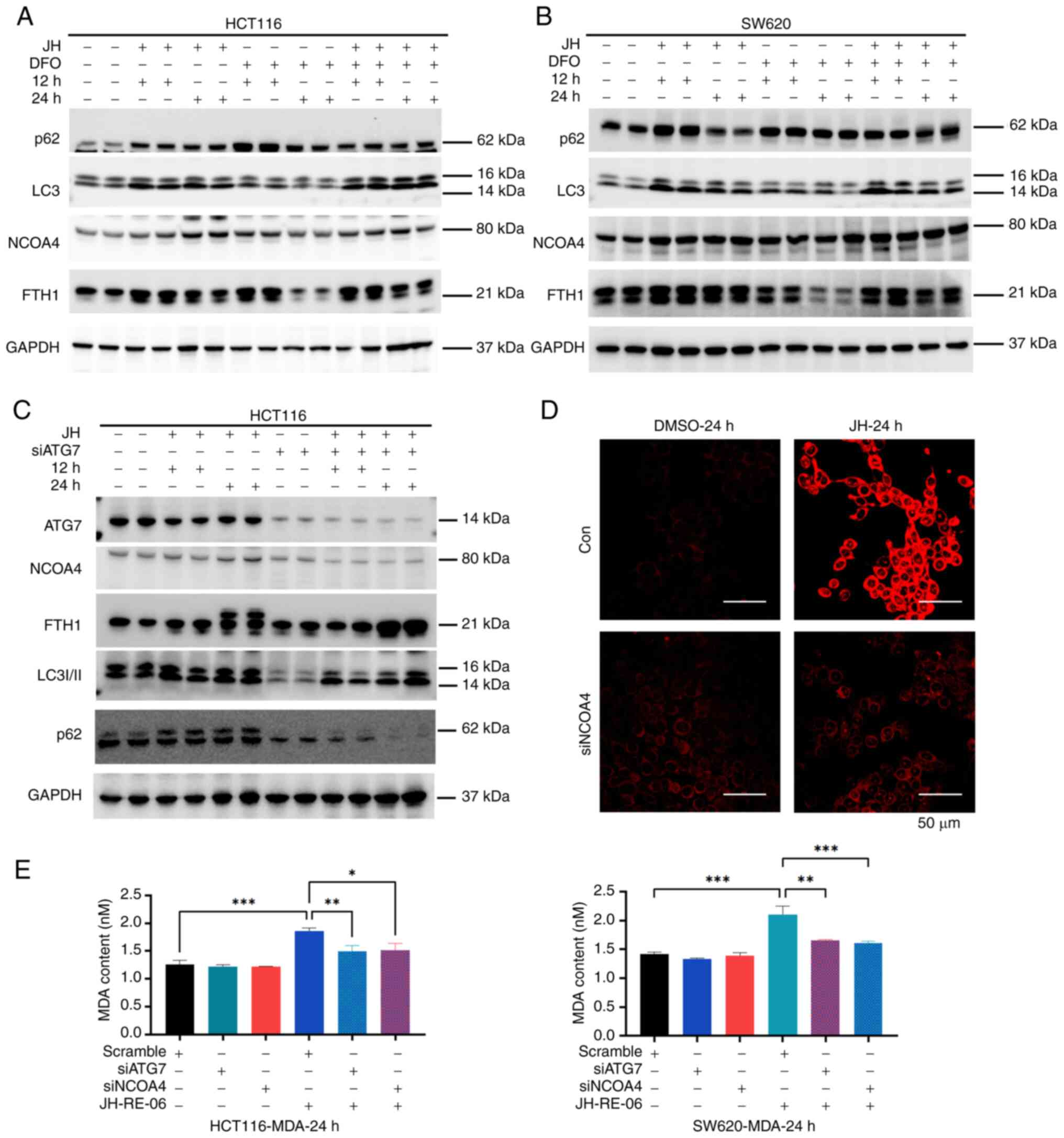 | Figure 5.JH triggers ferroptosis via
NCOA4-mediated ferritinophagy in colorectal cancer cells. (A)
HCT116 cells were treated with 3.0 µM JH and (B) SW620 cells with
1.5 µM JH and/or 50 µM DFO for 12 and 24 h before WB analysis for
p62, LC3, NCOA4 and FTH1, normalized to GAPDH. (C) WB analysis of
HCT116 cells with or without ATG7 knockout, treated with 3.0 µM JH.
(D) HCT116 cells were transfected with scramble or siNCOA4 for 48
h, then treated with 3.0 µM JH for 24 h. Cells were incubated with
FerroOrange probe and imaged using laser confocal microscopy. Scale
bar, 50 µm. (E) Lipid peroxidation levels were measured in ATG7 and
NCOA4 knockdown cells, as well as wild-type HCT116 and SW620 cells,
treated with JH. ***P<0.001, **P<0.01, *P<0.05. JH,
JH-RE-06; NCOA4, nuclear receptor coactivator 4; WB, western blot;
LC3, microtubule-associated protein 1 light chain 3; FTH, ferritin
heavy chain; ATG, autophagy-related gene; si, small interfering;
DFO, deferoxamine. |
Discussion
REV1 is implicated in cancer development by enabling
cells to bypass DNA damage, thereby accumulating genetic mutations
and increasing cancer risk (14).
Most CRCs originate from polyps, which progress through abnormal
crypts into neoplastic precursor lesions over 10–15 years (27). The present study highlighted the
clinical value of REV1 in CRC. Bioinformatics analysis and
immunohistochemical staining of tissue microarrays revealed that
high tumoral REV1 expression served as a negative prognostic
indicator. Furthermore, bioinformatics analysis identified a
significant association between high REV1 expression and the
presence of colon polyps, indicating its potential role in early
colorectal tumorigenesis. These findings collectively suggested
that REV1 is a promising therapeutic target for both early
intervention and advanced CRC treatment.
Studies have focused on enhancing cancer cell
sensitivity to platinum-based agents by genetically inhibiting REV1
(6,7). Early studies proposed that REV1
ablation may potentiate OXA efficacy by exacerbating DNA damage and
apoptosis (21,28). However, OXA primarily exerts
cytotoxic effects via ribosome biogenesis stress, independent of
DNA damage mechanisms (29).
Similarly, the present study revealed that pretreatment with the
REV1 inhibitor JH-RE-06 enhanced cellular resistance to OXA or
oncogene-induced (cyclin E overexpression) RS can render cancer
cells hypersensitive to TLS inhibition (11,30).
Clinically, while OXA treatment often leads to drug resistance in
CRC, it generates a therapeutic vulnerability that may be exploited
(31). To validate this hypothesis,
the present study established OXR CRC models. Both clonogenic
assays and xenograft experiments revealed that JH-RE-06 selectively
inhibited OXR cell proliferation while exhibiting minimal toxicity,
positioning it as a promising second-line therapeutic candidate for
refractory CRC.
While previous research has explored the effects of
JH-RE-06 in lung and BRCA1/2-deficient cancer (10,18),
the specific PCD mechanisms remain unclear. The present study
established JH-RE-06 as a DNA-damaging agent that inhibited CRC
cell proliferation in a dose-dependent manner. Pharmacological
rescue experiments revealed that JH-RE-06-induced cell death was
mitigated by cysteine regenerators (L-penicillamine,
2-mercaptoethanol and N-acetylcysteine) and the iron chelator DFO,
suggesting that the cell death mechanism was consistent with
ferroptosis rather than conventional PCD pathways such as
apoptosis, necrosis or autophagy. Ferroptosis is a metabolically
regulated cell death process driven by iron-dependent lipid
peroxidation and compromised redox balance (32). In the present study, proteomics
analysis confirmed that JH-RE-06 promoted cell signaling pathways
associated with ‘DNA replication’, ‘ferroptosis’, and ‘oxidative
phosphorylation’.
JH-RE-06 treatment in CRC cells resulted in
decreased mitochondrial abundance and intracellular GSH levels, and
significant elevations in intracellular Fe2+ and MDA
concentrations, which represent key features of ferroptosis.
Consistent with these findings, REV1 deficiency-induced RS causes
metabolic stress, leading to mitochondrial dysfunction in mouse
embryonic fibroblasts (33).
Studies in REV1−/− mice (a replication stress model)
(21) have demonstrated
sex-dependent metabolic disturbances, underscoring the dual role of
REV1 in DNA stability and metabolic control (34).
The association between REV1 inhibition and
ferroptosis remains incompletely understood. However, emerging
evidence indicates NCOA4-mediated ferritinophagy as a key
mechanism, involving iron liberation through ferritin degradation
and RS via minichromosome maintenance protein complex 2–7, helicase
interference (35). JH-RE-06
treatment in wild-type CRC cells significantly upregulated both
autophagy markers (LC3I/II and p62) and ferritinophagy-associated
proteins. This was characterized by increased NCOA4 expression,
decreased FTH1 levels and consequent elevation of LIP. The
experimental system was validated by using DFO to chelate
Fe2+, which demonstrated target protein changes aligned
with prior reports (36,37). By contrast, ATG7 knockout CRC cells
exhibited increased FTH1 levels and no increase in LIP following
JH-RE-06 treatment. Moreover, JH-RE-06-treated autophagy-deficient
CRC cells showed decreased MDA levels, indicating that the
ferroptosis induced by JH-RE-06 may depend on NCOA4-mediated
ferritinophagy. Recent research has highlighted the role of NCOA4
in regulating both DNA replication origins and iron
autophagy-mediated LIP, with potential Fe-S cluster degradation
(38). REV1 protects replication
forks by suppressing their remodeling (11), while NCOA4 regulates DNA replication
origins, preventing RS (39).
However, the precise regulatory association between REV1 inhibition
and NCOA4 remains to be fully elucidated.
Recent studies have indicated the potential of REV1
inhibitors as regulators of cell metabolism for cancer therapy
(40,41). In epithelial ovarian cancer,
fumarate-mediated modulation of TLS may enhance the efficacy of
genotoxic chemotherapy (40). In
pulmonary malignancy, REV1 promotes radioresistance by modulating
amino acid metabolism, specifically via cystathionine γ-lyase
ubiquitination and subsequent disruption of Gly/Ser/Thr metabolic
flux (41). These findings suggest
that the development of more selective REV1 inhibitors may offer
novel therapeutic opportunities, especially when conventional
apoptosis mechanisms are compromised.
The present findings established REV1 as both a
prognostic marker and therapeutic candidate in CRC, with elevated
expression associated with poorer survival. The specific inhibitor
JH-RE-06 induced DNA damage and suppressed tumor cell
proliferation, while also triggering oxidative stress in CRC cells.
JH-RE-06 regulated intracellular iron overload via NCOA4-mediated
ferritinophagy, thereby activating ferroptosis. This led to an
increase in the LIP, contributing to oxidative stress and
initiating ferroptosis, which was reversed by DFO and free radical
scavengers such as NAC. JH-RE-06 effectively inhibited the
proliferation of CRC cells, highlighting its clinical potential as
a therapeutic option for CRC, including both treatment-naive and
chemotherapy-resistant forms.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Ningxia Hui Autonomous
Region Science and Technology Support Program (grant no.
2021BEG03084) and the Ningxia Education Department's Scientific
Research Program (grant no. NYG2024117).
Availability of data and materials
The data generated in the present study may be found
in the Open Archive for Miscellaneous Data, China National Center
for Bioinformation under accession number OMIX007702 or at the
following URL: ngdc.cncb.ac.cn/omix/release/OMIX007702.
Authors' contributions
JC conceived and designed the study, performed
experiments, analyzed data and wrote the manuscript. XY, WZ and JX
performed experiments. FX conceived the study, revised the
manuscript for important intellectual content, and assisted in the
interpretation of key experimental results. XY revised the
manuscript and participated in data analysis. JC and XY confirm the
authenticity of all the raw data. YH participated in key
experimental operations (optimization of ROS detection protocols,
paraffin sectioning techniques, as applicable) and verification of
experimental protocols. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
All procedures adhered to institutional guidelines
and received approval from the Ethics Committee of Ningxia Medical
University Laboratory Animal Center (approval no.
IACUC-NYLAC-2022-022), in compliance with the ARRIVE guidelines and
national regulations for humane treatment of animals. The human
tissue microarrays were commercially sourced from fully
de-identified samples previously collected under ethical oversight.
In accordance with international research standards (Declaration of
Helsinki) and institutional policies for the secondary use of
archival specimens, no additional ethical review or consent
procedures were required.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
2
|
Liu Z, Xu Y, Xu G, Baklaushev VP,
Chekhonin VP, Peltzer K, Ma W, Wang X, Wang G and Zhang C: Nomogram
for predicting overall survival in colorectal cancer with distant
metastasis. BMC Gastroenterol. 21:1032021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Veenstra CM and Krauss JC: Emerging
systemic therapies for colorectal cancer. Clin Colon Rectal Surg.
31:179–191. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Berti M, Cortez D and Lopes M: The
plasticity of DNA replication forks in response to clinically
relevant genotoxic stress. Nat Rev Mol Cell Biol. 21:633–651. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang W and Gao Y: Translesion and repair
DNA polymerases: Diverse structure and mechanism. Annu Rev Biochem.
87:239–261. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hicks JK, Chute CL, Paulsen MT, Ragland
RL, Howlett NG, Gueranger Q, Glover TW and Canman CE: Differential
roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of
intrastrand versus interstrand DNA cross-links. Mol Cell Biol.
30:1217–1230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma S, Hicks JK, Chute CL, Brennan JR,
Ahn JY, Glover TW and Canman CE: REV1 and polymerase ζ facilitate
homologous recombination repair. Nucleic Acids Res. 40:682–691.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baranovskiy AG, Lada AG, Siebler HM, Zhang
Y, Pavlov YI and Tahirov TH: DNA polymerase delta and zeta switch
by sharing accessory subunits of DNA polymerase delta. J Biol Chem.
287:17281–17287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mellor C, Nassar J, Šviković S and Sale
JE: PRIMPOL ensures robust handoff between on-the-fly and
post-replicative DNA lesion bypass. Nucleic Acids Res. 52:243–258.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taglialatela A, Leuzzi G, Sannino V,
Cuella-Martin R, Huang JW, Wu-Baer F, Baer R, Costanzo V and Ciccia
A: REV1-Polzeta maintains the viability of homologous
recombination-deficient cancer cells through mutagenic repair of
PRIMPOL-dependent ssDNA gaps. Mol Cell. 81:4008–4025. e72021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nayak S, Calvo JA, Cong K, Peng M,
Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK and Cantor
SB: Inhibition of the translesion synthesis polymerase REV1
exploits replication gaps as a cancer vulnerability. Sci Adv.
6:eaaz78082020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nayak S, Calvo JA and Cantor SB: Targeting
translesion synthesis (TLS) to expose replication gaps, a unique
cancer vulnerability. Expert Opin Ther Targets. 25:27–36. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin X and Howell SB: DNA mismatch repair
and p53 function are major determinants of the rate of development
of cisplatin resistance. Mol Cancer Ther. 5:1239–1247. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sasatani M, Xi Y, Kajimura J, Kawamura T,
Piao J, Masuda Y, Honda H, Kubo K, Mikamoto T, Watanabe H, et al:
Overexpression of Rev1 promotes the development of
carcinogen-induced intestinal adenomas via accumulation of point
mutation and suppression of apoptosis proportionally to the Rev1
expression level. Carcinogenesis. 38:570–578. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu N, Zhao Y, Mi M, Lu Y, Tan Y, Fang X,
Weng S and Yuan Y: REV1: A novel biomarker and potential
therapeutic target for various cancers. Front Genet. 13:9979702022.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wojtaszek JL, Chatterjee N, Najeeb J,
Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, et al: A
small molecule targeting mutagenic translesion synthesis improves
chemotherapy. Cell. 178:152–159.e11. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chatterjee N, Whitman MA, Harris CA, Min
SM, Jonas O, Lien EC, Luengo A, Vander Heiden MG, Hong J, Zhou P,
et al: REV1 inhibitor JH-RE-06 enhances tumor cell response to
chemotherapy by triggering senescence hallmarks. Proc Natl Acad Sci
USA. 117:28918–28921. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, Jie X, Xing B, Wu Z, Yang X, Rao
X, Xu Y, Zhou D, Dong X, Zhang T, et al: REV1 promotes lung
tumorigenesis by activating the Rad18/SERTAD2 axis. Cell Death Dis.
13:1102022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizuno H, Kitada K, Nakai K and Sarai A:
PrognoScan: A new database for meta-analysis of the prognostic
value of genes. BMC Med Genomics. 2:182009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sharma S, Shah NA, Joiner AM, Roberts KH
and Canman CE: DNA polymerase ζ is a major determinant of
resistance to platinum-based chemotherapeutic agents. Mol
Pharmacol. 81:778–787. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Radulescu S, Brookes MJ, Salgueiro P,
Ridgway RA, McGhee E, Anderson K, Ford SJ, Stones DH, Iqbal TH,
Tselepis C and Sansom OJ: Luminal iron levels govern intestinal
tumorigenesis after Apc loss in vivo. Cell Rep. 2:270–282. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu Q, Wei W, Wu D, Huang F, Li M, Li W,
Yin J, Peng Y, Lu Y, Zhao Q and Liu L: Blockade of GCH1/BH4 axis
activates ferritinophagy to mitigate the resistance of colorectal
cancer to erastin-induced ferroptosis. Front Cell Dev Biol.
10:8103272022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie K, Doles J, Hemann MT and Walker GC:
Error-prone translesion synthesis mediates acquired
chemoresistance. Proc Natl Acad Sci USA. 107:20792–20797. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bruno PM, Liu Y, Park GY, Murai J, Koch
CE, Eisen TJ, Pritchard JR, Pommier Y, Lippard SJ and Hemann MT: A
subset of platinum-containing chemotherapeutic agents kills cells
by inducing ribosome biogenesis stress. Nat Med. 23:461–471. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sekimoto T, Oda T, Kurashima K, Hanaoka F
and Yamashita T: Both high-fidelity replicative and low-fidelity
Y-family polymerases are involved in DNA rereplication. Mol Cell
Biol. 35:699–715. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang G, Wang JJ, Zhi-Min Z, Xu XN, Shi F
and Fu XL: Targeting critical pathways in ferroptosis and enhancing
antitumor therapy of Platinum drugs for colorectal cancer. Sci
Prog. 106:3685042211471732023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fakouri NB, Durhuus JA, Regnell CE,
Angleys M, Desler C, Hasan-Olive MM, Martín-Pardillos A,
Tsaalbi-Shtylik A, Thomsen K, Lauritzen M, et al: Rev1 contributes
to proper mitochondrial function via the PARP-NAD+-SIRT1-PGC1alpha
axis. Sci Rep. 7:124802017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Anugula S, Li Z, Li Y, Hendriksen A,
Christensen PB, Wang L, Monk JM, de Wind N, Bohr VA, Desler C, et
al: Rev1 deficiency induces a metabolic shift in MEFs that can be
manipulated by the NAD(+) precursor nicotinamide riboside. Heliyon.
9:e173922023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Federico G, Carrillo F, Dapporto F,
Chiariello M, Santoro M, Bellelli R and Carlomagno F: NCOA4 links
iron bioavailability to DNA metabolism. Cell Rep. 40:1112072022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu H, Liu Q, Shan X, Gao W and Chen Q: ATM
orchestrates ferritinophagy and ferroptosis by phosphorylating
NCOA4. Autophagy. 19:2062–2077. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang MJ, Song ML, Zhang Y, Yang XM, Lin
HS, Chen WC, Zhong XD, He CY, Li T, Liu Y, et al: SNS alleviates
depression-like behaviors in CUMS mice by regluating dendritic
spines via NCOA4-mediated ferritinophagy. J Ethnopharmacol.
312:1163602023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kuno S and Iwai K: Oxygen modulates iron
homeostasis by switching iron sensing of NCOA4. J Biol Chem.
299:1047012023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bellelli R, Castellone MD, Guida T,
Limongello R, Dathan NA, Merolla F, Cirafici AM, Affuso A, Masai H,
Costanzo V, et al: NCOA4 transcriptional coactivator inhibits
activation of DNA replication origins. Mol Cell. 55:123–137. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li J, Zheng C, Mai Q, Huang X, Pan W, Lu
J, Chen Z, Zhang S, Zhang C, Huang H, et al: Tyrosine catabolism
enhances genotoxic chemotherapy by suppressing translesion DNA
synthesis in epithelial ovarian cancer. Cell Metab.
35:2044–2059.e8. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Y, Feng X, Wu Z, Yang Y, Rao X, Meng
R, Zhang S, Dong X, Xu S, Wu G and Jie X: USP9X-mediated REV1
deubiquitination promotes lung cancer radioresistance via the
action of REV1 as a Rad18 molecular scaffold for cystathionine
γ-lyase. J Biomed Sci. 31:552024. View Article : Google Scholar : PubMed/NCBI
|














