Non-small cell lung cancer (NSCLC) is one of the
leading causes of cancer-related mortalities globally and accounts
for ~85% of all lung cancer cases (1). Among the molecular subtypes of NSCLC,
Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations are one
of the most commonly identified oncogenic drivers. These mutations
are commonly prevalent in lung adenocarcinoma (LUAD) and are
strongly linked with smoking and environmental carcinogens
(2). KRAS is a member of the rat
sarcoma viral (RAS) oncogene family and accounts for the majority
of RAS-related mutations encountered in human cancer. KRAS
alterations account for ~85% of all RAS mutations (3). The prevalence of KRAS mutations varies
across cancer types, with the highest frequencies found in
pancreatic (88%), colorectal (45–50%) and lung cancer (31–35%)
(4). KRAS mutations are present in
~30% of LUAD cases in Western populations and ~10% of in Asian
populations (5,6).
The KRAS proto-oncogene encodes a small guanine
triphosphate (GTPase) that acts as a binary switch in signal
transductions of multiple receptor tyrosine kinases, such as
mesenchymal-epithelial transition (MET), epidermal growth factor
receptor (EGFR) and anaplastic lymphoma kinase (ALK), thus
regulating cellular functions such as proliferation,
differentiation and survival. KRAS is a clonal transforming
oncogene and is a key oncogenic driver across multiple solid tumors
(7–9).
Structurally, KRAS is a 21-kDa GTPase composed of a
conserved G-domain and two dynamic regulatory regions, Switch I and
Switch II, which control its active (GTP-bound) and inactive
(GDP-bound) states. The Switch II pocket (SII-P) forms the
allosteric site targeted by KRAS G12C inhibitors, while C-terminal
farnesylation enables its anchoring to the plasma membrane
(10,11).
The prevailing outlook on the poor prognosis and
limited treatment options for NSCLC has significantly shifted over
the last 2 decades due to the development of targeted therapy,
molecular profiling and immunotherapy (12,13).
Although several preclinical and clinical studies have investigated
RAS inhibitors, no authorized treatments directly inhibit mutant
variants of KRAS or its downstream signaling (14,15).
These novel KRAS inhibitors are currently being investigated in
combination with other therapies, which may significantly improve
the treatment of KRAS-mutated NSCLC. Targeted therapy and
immunotherapy have shown promising results in treating NSCLC,
therefore the present review explored novel treatment strategies
and their efficacy in targeting the KRAS mutation in NSCLC.
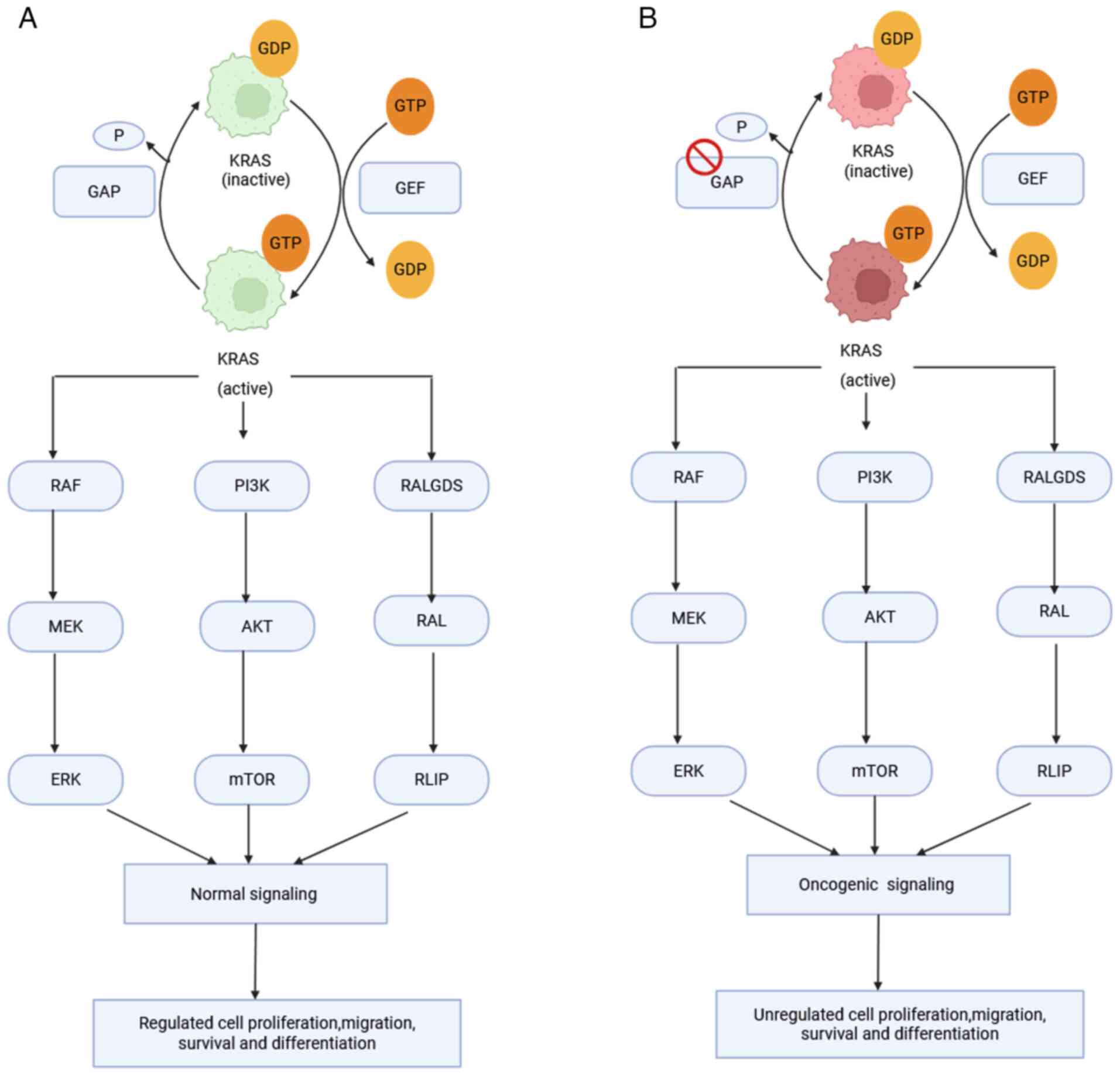
KRAS is a key proto-oncogene that encodes a small
GTPase that controls cell survival, differentiation and
proliferation by acting as a molecular switch in important cellular
signaling cascades (3). The GTPase
cycles between an inactive GDP-bound and an active GTP-bound state.
Activation is promoted by guanine nucleotide exchange factors
(GEFs), which stimulate GDP-GTP exchange, such as SOS1 and the
GTPase-activating proteins (GAPs); for example, neurofibromin 1 is
a GAP that accelerates GTP hydrolysis to inactivate KRAS (14,16).
Multiple downstream effectors of KRAS, primarily,
are part of the RAF-MEK-ERK signaling pathway. Activated KRAS-GTP
recruits RAF kinases to the membrane, which promotes RAF
dimerization and activation. RAF phosphorylates MEK1/2, leading to
activation of ERK1/2. Upon activation, nuclear translocation of ERK
occurs and targets transcription factors, including E26
transformation-specific family, serum response factor and ribosomal
S6 kinase, to influence cell proliferation, differentiation and
migration. Furthermore, KRAS stimulates phosphoinositide 3-kinase
(PI3K), leading to AKT activation that promotes cell survival,
metabolism and growth. KRAS also activates RalGEFs, which regulate
cytoskeleton remodeling, membrane dynamics and vesicle trafficking
(Fig. 1) (14,16,17).
In its wild-type state, KRAS participates in normal cell signaling
but does not directly suppress immune responses. KRAS mutations;
however, it inhibits T-cell activity by stimulating an
immunosuppressive tumor microenvironment (TME) through the
induction of cytokine-(such as IL-6) and chemokine-(such as IL-8)
driven inflammation and immune evasion, and increase programmed
cell death-1 ligand (PD-L1) expression levels via activation of the
MAPK and PI3K-AKT pathways, resulting in immune escape (14,18).
KRAS mutations are clinically significant in NSCLC
due to their frequency, prognostic implications and impact on
treatment decisions (19,20). Point mutations commonly deregulate
the KRAS gene, resulting in an inherently active GTP-bound phase
and activating downstream oncogenic signaling pathways (21–23).
KRAS mutation frequency is lower (~5%) in squamous NSCLC and higher
(20–40%) in LUAD (24).
Furthermore, these mutations are more prevalent in Western
populations compared with Asian populations (26 vs. 11%) and in
smokers compared with non-smokers (30 vs. 11%); the presence of
KRAS mutations in NSCLC is closely associated with smoking
(25). Furthermore, KRAS mutations
are more common in female and younger patients, according to a
pooled study of resected NSCLC tumors; however, only the
association with younger patients was significant in the
multivariate analysis (P=0.044) conducted (26,27).
The previous study by Shepherd et al (27) on the frequency of KRAS mutations did
not include any analysis specific to histology or race. Smoking is
suggested to leave a particular molecular mark on KRAS gene since
point mutations (G12C and G12V) are often detected in former or
current smokers; by contrast, transitional mutations (G12D) are
more common in non-smokers (28,29).
Furthermore, compared with individuals who have never smoked,
smokers often have substantially more complicated KRAS-mutant
tumors with a more considerable mutational burden and a higher
probability of significant co-occurring mutations in tumor protein
P53 (TP53) or serine/threonine kinase 11 (STK11) (30).
Co-mutations, including STK11/LKB1, may influence
the prognosis of KRAS-mutant NSCLC. For example, ERK1/2
phosphorylation is more significant in tumors with KRAS G12C
mutations compared with those with KRAS G12D mutations, and this
enhanced MAPK signaling can be further amplified in the presence of
STK11/LKB1 loss, contributing to more aggressive tumor biology.
Supporting this, a previous study employing animal models driven by
KRAS mutations showed that the MEK inhibitor selumetinib was more
effective in KRAS G12C tumors compared with KRAS G12D tumors
(31). As a result, various KRAS
mutations may induce signal transduction cascades differently,
resulting in unique drug sensitivity profiles (32). Regarding co-occurring mutations,
LUAD is typically associated with KRAS single-driver mutations,
accounting for 95–99% of KRAS-mutant cases, while double mutants
(EGFR/ALK/BRAF and KRAS) are rare, occurring in <1% of cases
(33–35).
Approximately one-third of patients with LUAD have a
KRAS mutation; despite significant advancements in the discovery
and development of targeted therapeutics for molecular subtypes of
LUAD (36), no authorized
medication currently targets any KRAS mutation directly (37). For the past 40 years, significant
efforts have been made to develop medicines for KRAS. These
investigations have targeted the KRAS protein and its membrane
location, interactions between proteins, post-translational
modifications, and downstream signaling pathways; however, these
methods have not shown promising outcomes in clinical trials
(38,39).
Since KRAS has a restricted number of active binding
sites, complicated biochemistry (40) and a high affinity for GTP (41), direct targeting of the protein was
thought to be complex until ~5 years ago (42). Developments in crystallography and
computer modeling (43) have
identified small compounds that directly bind to the inactive
GDP-bound conformation of KRAS G12C within the SII-P, thereby
locking the protein in its inactive state and preventing downstream
signaling (44). However, the
binding affinity of these early-stage drugs would need to be
substantially increased before they can be used as effective
treatments. These early compounds demonstrated that RAS is
principally druggable; however, they lacked sufficient potency and
pharmacological properties to achieve clinical efficacy and often
lacked the precision required to target mutant KRAS effectively
(45)
Prior to the advent of direct KRAS inhibitors,
indirect strategies were investigated, such as by blocking KRAS
post-translational modifications, preventing its anchoring to the
cell membrane or disrupting the downstream signaling pathways it
controls (46–48). Early analyses focused on farnesyl
transferase inhibitors, salirasib and tipifarnib being tested to
prevent membrane anchoring of KRAS. Although the preclinical
activity was promising, the clinical efficacy of salirasib and
tipifarnib in NSCLC was limited due to compensatory prenylation
with geranylgeranyl transferase. In a phase II trial of salirasib
in KRAS-mutant LUAD, no objective responses were observed, although
30–40% of patients achieved temporary disease stabilization at 10
weeks (49,50).
Blockage of the downstream KRAS signaling has also
been investigated. Small molecules that target ERK and MEK decrease
effector phosphorylation and block RAF interactions in preclinical
models (45,51). Despite this, MEK inhibitor clinical
trials as monotherapy have been ineffective. In a phase II study of
previously treated patients with KRAS-mutant NSCLC, FAK inhibition
with defactinib alone had limited clinical activity [median
progression-free survival (PFS), 45 days], and activity was not
associated with TP53 or CDKN2A status (52).
These studies highlight the primary limitations of
indirect KRAS targeting: Biochemical redundancy, adaptive feedback
loops and toxicity due to effects on essential signaling pathways
(53). Post-prenylation processing
enzymes such as Ras-converting enzyme 1 and isoprenylcysteine
carboxyl methyltransferase remain potential targets, but inhibitors
lack specificity and risk broad systemic toxicity (54). However, these limitations are being
overcome through the development of pragmatic strategies. Combined
inhibition of farnesyl- and geranylgeranyl transferase has been
reported as a promising approach in models of pancreatic cancer,
although it has not been tested in LUAD (55). Epidemiological studies suggest that
oral bisphosphonate use is associated with a decreased risk of lung
cancer in non-smoker postmenopausal women (56) and a case report described regression
of hepatic metastases in primary lung adenocarcinoma following
zoledronic acid monotherapy (57).
Preclinical models indicate that these vulnerabilities are
subtype-specific: Mesenchymal-like KRAS-mutant NSCLC is sensitive
to combined FGFR-MEK inhibition, whereas epithelial-like tumors
preferentially respond to ERBB-MEK blockade (58). Such biomarker-led combination
strategies provide novel avenues for indirect targeting.
Collectively, these indirect approaches illustrate the innovative
nature of previous efforts to target KRAS and the associated
limitations, and demonstrate the importance of investigations on
direct interventions in the current therapeutic landscape.
The mutant KRAS G12C protein shows decreased
GAP-stimulated GTPase activity from biochemical analyses,
consistent with earlier G12X mutants (excluding G12P). In contrast
with other mutated KRAS proteins, the mutation KRAS G12C maintains
periodic intrinsic GTPase function and alternates among the active
KRAS-GTP and inactive KRAS-GDP states (59,60).
Structural analysis demonstrated that the KRAS Cys12 mutation is
proximal to a newly identified allosteric site, the SII-P, which is
transiently accessible in the GDP-bound state. This finding allowed
the design of covalent inhibitors to bind Cys12 irreversibly,
trapping KRAS G12C in its inactive state and ablating downstream
signaling (61,62). The first therapies to target this
SII-P were sotorasib, adagrasib and, more recently, garsorasib
(D-1553). These KRAS G12C inhibitors all bind covalently to the
SII-P but differ in their binding kinetics and molecular
interactions (63). Adagrasib
exhibits higher conformational mobility, allowing more sustained
binding to KRAS-G12C, while garsorasib has shown promising clinical
activity in NSCLC and colorectal cancer (64,65).
Structural scaffold differences between KRAS G12C inhibitors, such
as sotorasib and adagrasib, are likely responsible for the
different pharmacodynamic profiles and tissue penetration.
Although the SII-P is the most-validated and
extensively targeted allosteric site, it is not the sole structural
determinant of KRAS inhibitor binding; other regions of KRAS also
influence drug design and activity. KRAS contains key structural
regions, including the Switch I and Switch II domains and a
hypervariable region that regulates membrane attachment. The switch
I region can be indirectly modulated by inhibiting SOS1-KRAS
interactions using SOS1 inhibitors (such as BI-3406 and
BI-1701963). The nucleotide binding pocket has been the focus of
investigation with nucleotide competitive analogs and membrane
localization disrupted with FTase inhibitors through the
hypervariable region (66–68). These findings underscore that KRAS
contains several structural features beyond the SII-P that may be
exploited therapeutically.
The first KRAS G12C inhibitor studied in a clinical
trial for an advanced solid tumor was sotorasib. The initial
investigation was a CodeBreaK 100 phase I/II basket trial to
determine the safety and beneficial effects of sotorasib in
patients with advanced tumors with a KRAS G12C mutation,
particularly those who previously received standard treatment.
Based on the clinical trial outcome, sotorasib received approval
from the Food and Drug Administration (FDA) in 2021 to treat
advanced-stage NSCLC (69,70). In a cohort of 126 patients with KRAS
G12C mutation NSCLC, the objective response rate (ORR) was 37.1%,
with 3.2% exhibiting a complete response (CR). The median overall
survival (OS) was 12.5 months, median PFS was 6.8 months, and
adverse events were seen in 69.8% of patients (71). Another study found a 42.9% confirmed
ORR for 116 patients with KRAS G12C mutated NSCLC. the median OS
was 12.6 months and the median PFS and response duration were 6.5
and 8.5 months, respectively (72).
In a subsequent phase III trial comparing sotorasib and docetaxel,
sotorasib increased PFS [PFS, 5.6 months vs. 4.5 months for
docetaxel, hazard ratio (HR)=0.66, P=0.0017] in patients with NSCLC
who had previously received chemotherapy (73). The IFCT-2102 Lung KG12Ci study, a
nationwide study conducted in France, included 458 patients with
metastatic or advanced KRAS G12C-mutated non-squamous NSCLC across
76 centers. The median age was 65.8 years and 43.4% of the patients
were female. The majority (95.4%) were current or former smokers
and 38.0% had brain metastases at the initiation of sotorasib
therapy. The key efficacy endpoints were a median real-world PFS of
3.5 months and a median real-world OS of 8.3 months (median
follow-up was 15.8 and 16.4 months, respectively). The real-world
ORR was 35.5% and the disease control rate (DCR) was 63.7%. In the
subgroup of patients with brain metastases, the intracranial
real-world ORR was 20.1% and the intracranial real-world DCR was
66.9%. From a safety perspective, sotorasib was considered to be
tolerable. However, 16.5% of patients discontinued their therapy
because of adverse events, while 5.2% of patients experienced grade
3–4 hepatotoxicity (74).
Sotorasib changed the treatment landscape for KRAS
G12C-mutant NSCLC by providing improved clinical outcomes compared
with standard second-line therapies, such as docetaxel,
docetaxel-ramucirumab and immune checkpoint inhibitor (ICI)
monotherapy. Nevertheless, the intracranial efficacy of sotorasib
is low, particularly in patients with brain metastases, and
survival benefits are limited due to the acquisition of resistance;
thus, there is an urgent need for mechanism-based combination
approaches and next-generation inhibitors. The clinical decision to
use sotorasib should be made with caution, particularly for
patients with KRAS G12C-mutant NSCLC with central nervous system
(CNS) involvement or a high disease burden.
Another KRAS G12C inhibitor with a positive clinical
record is adagrasib. The phase I/IB KRYSTAL-1 study of adagrasib
demonstrated a favorable safety profile and a median PFS of 11.1
months (75). KRYSTAL-12 (trial no.
NCT04685135), a phase III study of adagrasib vs. docetaxel in
patients with previously treated KRAS G12C-mutated NSCLC,
demonstrated that 94.0% of patients who received adagrasib and
86.4% of patients who received chemotherapy had treatment-related
adverse events (TRAEs). Grade ≥3 TRAEs occurred in 47.0% of
patients who received adagrasib and 45.7% of patients who received
docetaxel. These patients would have received immunotherapy before
receiving adagrasib (76).
The KRAS G12C inhibitor garsorasib showed strong
efficacy and tolerable safety in a phase II trial (trial no.
NCT05383898) of 123 Chinese patients with pre-treated KRAS
G12C-mutated NSCLC. All participants (median age, 64; 88% male)
were administered 600 mg garsorasib twice daily and achieved an ORR
of 50% (1 CR and 60 partial responses) and a DCR of 89%. With a
median follow-up of 12.3 months, the median PFS was 9.1 months and
the median OS was 14.1 months, the longest reported in
commercialized KRAS G12C inhibitors. TRAEs were seen in 95% of
patients, with grade ≥3 events (50%) mainly comprising hepatic
enzymes (AST elevation, 17%; ALT, 15%) and gastrointestinal
symptoms (nausea/vomiting, 2%); most toxicities were
dose-modifiable (77,78). Garsorasib has demonstrated promising
clinical activity with high response rates, durable responses and
prolonged OS in patients with previously treated KRAS G12C-mutant
NSCLC, and has shown superior outcomes compared with existing
inhibitors, in certain contexts as aforementioned. However,
widespread use and future studies with a larger sample size are
necessary to collect more data on CNS activity, long-term survival
and comparative effectiveness. Presently, the use of garsorasib may
be considered post-chemotherapy and ICI therapy, particularly in
patients with healthy liver function who can be closely monitored.
Further to the aforementioned studies, numerous other clinical
trials (Table I) are currently
under investigation to identify therapies with increased efficacy
of KRAS G12C inhibitors.
Clinical resistance continues to present a notable
barrier and commonly involves allosteric sites such as the SII-P.
Second-site mutations on or around the SII-P change pocket geometry
and/or local dynamic structures that directly disrupt inhibitor
binding (79). For example, Y96D
eliminates hydrogen bonding in the pocket, A59T, and G13D disrupts
the conformational state essential for drug interaction (80). Furthermore, bypass mechanisms of
resistance, including upstream mechanisms by amplifying the HER2
gene, can induce KRAS activation and reduce dependence on SII-P
inhibition (81). Taken together,
these findings show that the structural confirmation of the SII-P
influences inhibitor binding affinity, and demonstrates how
mutations in this region can lead to resistance. This close
association between the structure and function of KRAS underscores
the importance of combining strategies or developing
next-generation inhibitors to overcome these challenges, including
resistance.
Combining KRAS G12C inhibitors with ICIs is
supported by synergistic immunogenicity: Blocking KRAS can increase
tumor antigen presentation and T-cell infiltration, while ICIs
counteract immune suppression (62,82).
The KRYSTAL-7 trial (trial no. NCT04613596) is a phase III study
assessing first-line adagrasib in combination with pembrolizumab
compared with monotherapy pembrolizumab in patients with advanced
NSCLC with a KRAS G12C mutation. In patients with a PD-L1 tumor
proportion score ≥50%, the confirmed ORR was 63% [32/51 patients;
95% confidence interval (CI), 48–76%]. The DCR in the same subgroup
was 84% (43/51 patients; 95% CI, 71–93%). The median PFS at a
median follow-up of 10.1 months was not achieved (95% CI, 8.2
months to not evaluable). The TRAEs reported among the 148 patients
evaluated for safety led to permanent discontinuation of adagrasib
in 6% (n=9) and pembrolizumab in 11% (n=16) of patients with TRAEs;
4% (n=6) of patients discontinued both agents due to TRAEs
(83). The phase Ib 100/101
CodeBreaK study tested sotorasib in combination with pembrolizumab
or atezolizumab in patients with metastatic KRAS G12C NSCLC.
Although the combination demonstrated clinical activity (ORR,
~29%), numerous patients experienced severe grade 3–4
hepatotoxicity when starting both drugs concomitantly. A ‘sotorasib
lead-in’ dose introduction strategy markedly ameliorated severe
liver toxicity and reduced discontinuation of treatment,
underscoring the relevance of dosage sequence to enhance
tolerability (84). Presently, the
ongoing Krascendo-170 trial (trial no. NCT05789082) is evaluating
divarasib in combination with pembrolizumab sequentially, to
determine whether the treatment dose can be maximized without
losing an optimal balance between efficacy and toxicity in
frontline KRAS G12C-mutated NSCLC.
Combining KRAS G12C inhibitors with ICIs may
potentially improve efficacy, particularly in patients with
PD-L1-high tumors. Adagrasib has potential as a frontline
combination therapy, requiring clarification of the optimal order
and timing for administration. Future clinical plans should
individualize treatment according to tumor biology and immune
status, guided by direct comparison trials initiated through
biomarker-informed pathways.
The toxicities of KRAS G12C inhibitors (sotorasib
and adagrasib) are clinically relevant, although frequently
manageable with individualized approaches. In a prospective
biomarker study, 75% of patients with a history of immune-related
hepatitis from prior immunotherapy subsequently developed severe
hepatotoxicity with sotorasib (85). Nevertheless, attempts at dose
reduction and rechallenge were made in over half of cases, and
long-term rechallenge was successful in some patients using
corticosteroids. Similarly, gastrointestinal AEs (such as diarrhea,
nausea and vomiting) may be controlled with supportive medications
(such as antiemetics, antidiarrheals, hydration and dietary
changes), and therapy is typically held or dose adjusted for grade
≥3 events in the case of adagrasib. Liver toxicity may be managed
with regular liver enzyme monitoring and prompt dose modification.
These results highlight that while toxicities may restrict dosing,
personalized immunosuppression and dose adjustment interventions
can sustain therapeutic benefit in appropriate patients (86,87).
KRAS G12C inhibitors have presented a new era for
NSCLC treatment through direct targeting of KRAS, as both sotorasib
and adagrasib have shown significant clinical activity, and as well
as the more recent therapy, garsorasib. However, modest survival
benefits, low intracranial activity and a resistance mechanism
remain significant limitations. Continued work with rational
combinations, tailored sequencing with immunotherapy,
toxicity-mitigation approaches and the emergence of next-generation
inhibitors are necessary to sustain long-term responses and provide
further treatment options for patients with KRAS G12C tumors.
The lack of effective targeted treatment for
patients with non-G12C KRAS mutations in NSCLC is a notable
clinical issue, particularly considering the recent FDA approval of
KRAS G12C inhibitors. In contrast to G12C inhibitors, which
covalently trap the G12C-specific cysteine residue, non-G12C
mutations require alternative treatment strategies. Immune
profiling studies show that non-G12C mutations are often associated
with increased rates of PD-L1 negativity and STK11 co-mutations,
which are both predictors of poor response to ICIs (88–90).
By targeting pan-KRAS inhibition, TME modulation and
upstream and downstream signaling pathways, emerging strategies aim
to overcome limitations such as allele-specific resistance, pathway
redundancy, and adaptive signaling escape. Pan-RAS and RAS-GTP
inhibitors are being further developed, as they have demonstrated
activity against other KRAS mutations beyond G12C (88). The non-covalent inhibitors MRTX1133
and HRS-4642 disrupt KRAS G12D interactions with SOS1 or RAF1,
resulting in downstream MEK-ERK inhibition. MRTX1133 (trial no.
NCT05737706) has progressed to phase I clinical trials, and
HRS-4642 is in phase I/II trials (91), showing preclinical tumor growth
inhibition in vitro and in vivo (92).
Although tumor mutational burden (TMB) and PD-L1
expression levels are comparable in G12C and non-G12C subgroups,
clinical responses in non-G12C variants are unsatisfactory
(93). These tumors are frequently
associated with co-mutations such as STK11, which shape an
immune-cold microenvironment, contributing to poor response to ICIs
(90). Despite novel targeted
approaches, the therapeutic landscape for non-G12C mutations of
KRAS remains limited.
Due to the constraints of targeted approaches,
immunotherapy has become an essential alternative and complementary
treatment option, especially for KRAS-mutant NSCLC. Furthermore,
novel approaches such as combination regimens with KRAS pathway
inhibitors and ICIs are being explored, based on data that KRAS
mutations alter the immune microenvironment and response to ICIs
(89). These strategies highlight
the need for novel immunotherapeutic regimens and rational
combinations to improve outcomes for patients with non-G12C KRAS
mutations. Although emerging KRAS G12C inhibitors show potential,
patients with non-G12C KRAS mutations in NSCLC have limited options
for targeted therapy and respond poorly to immunotherapy because of
co-mutations and a cold tumor immune microenvironment. Novel
non-covalent KRAS inhibitors and combination approaches targeting
signaling pathways and immune modulation are emerging as promising
treatment options; however, further development of therapies
targeted at specific molecular and immune mechanisms is necessary
to improve outcomes for this complex cohort.
ICIs, combined with platinum-based chemotherapy or
alone, are the conventional first-line treatment for patients with
advanced KRAS mutation-positive NSCLC (94). Smoking has been associated with
elevated TMB in addition to the immune-related characteristics of
KRAS-mutant tumors. Increased TMB may correlate with an improved
response to ICIs (95,96). Mazieres et al (97) retrospectively analyzed NSCLC cases
with different oncogenic driver mutations and reported that
increased PD-L1 expression levels were observed in KRAS-mutant
tumors, which also showed superior responses to ICIs compared with
that of other oncogenic driver mutations such as EGFR, ALK, ROS1
and RET alterations. However, most phase III ICI trials in NSCLC
did not stratify patients by KRAS subtype, and only a few included
post-hoc analyses (98,99). Although KRAS G12C inhibitors have
shown benefit, other KRAS mutations still lack targeted options.
Table II summarizes key
immunotherapy trials in KRAS-mutant NSCLC.
The KEYNOTE-042 study is a phase III trial comparing
pembrolizumab to platinum-based chemotherapy as a first-line
treatment for patients with PD-L1-positive advanced NSCLC. KRAS
mutations, including NRAS mutations, were found in 22.9% (9.6% KRAS
G12C) of 301 patients; these patients exhibited increased levels of
tissue TMB and PD-L1 expression. In KRAS-mutant cohorts,
pembrolizumab treatment showed a significantly improved outcomes,
with an OS hazard ratio of 0.42 (95% CI, 0.22–0.81) and a PFS
hazard ratio of 0.51 (95% CI, 0.29–0.87) compared with KRAS
wild-type tumors (100). The
KEYNOTE-189 study, which examined the efficacies of first-line
platinum-based chemotherapy alone or combined with pembrolizumab in
advanced-stage NSCLC, reported that 12.8% of the 289 patients (95%
CI, 10.1–15.6%) had a KRAS G12C mutation, while 30.8% (95% CI,
22.9–38.8%) reported any KRAS mutation. Furthermore, tumors with
any KRAS mutation had elevated tissue TMB and PD-L1 levels.
Pembrolizumab-based treatment showed higher clinical outcomes
regardless of KRAS status than chemotherapy alone in PFS, OS and
ORR (101). These trials indicate
that KRAS-mutated NSCLC, particularly KRAS G12C, have an enhanced
response to pembrolizumab and immunotherapy due to high-level
expression of PD-L1 and TMB. Pembrolizumab, alone or combined with
chemotherapy, could result in survival and progression-free
benefits for these patients vs. chemotherapy alone.
A retrospective study conducted in China showed that
immunotherapy-based treatment in patients with KRAS-mutant NSCLC
showed improved results compared with that of chemotherapy alone.
The immunotherapy regimens used in this cohort were PD-1/PD-L1
inhibitors either alone or in combination with chemotherapy.
Overall, the median OS was 22.9 months (95% CI, 14.1–31.7) and PFS
was 9.4 months (95% CI, 6.6–12.1). Immunotherapy regimens had a
median OS of 45.2 vs. 11.3 months (P=1.81E-5) and a median PFS of
10.5 vs. 8.2 months (P=0.706) compared with chemotherapy. The first
line therapy cohort demonstrated a median OS and median PFS of 33.5
vs. 16.1 months (P=0.010) and 32.2 vs. 6.9 months (P=0.00038),
respectively. In the second line therapy cohort, median OS was not
reached vs. 9.23 months (P=0.026) and median PFS was 10.8 vs. 5.5
months (P=0.010). Immunotherapy improved the median OS compared
with that of chemotherapy for both patients with KRAS G12C tumors
and those with non-KRAS G12C tumors, according to subgroup analysis
(G12C, 25.2 vs. 9.1 months, P=0.0037; non-G12C, not reached vs.
25.7 months, P=0.020). In addition, concurrent KRAS/TP53 mutants
also showed an improved median OS with immunotherapy-based regimens
compared to those who received chemotherapy (33.5 vs. 11.8 months;
P=0.036). These findings suggest that immunotherapy is beneficial
for OS relative to chemotherapy in patients with late-stage
KRAS-mutant NSCLC, regardless of the treatment line or KRAS
mutation subtype (102).
Recent studies have provided novel insights into the
role of immunotherapy in KRAS-mutant NSCLC. A retrospective
analysis was conducted in Italy with a cohort of 143 patients with
advanced NSCLC and KRAS mutations received ICIs, with or without
chemotherapy. The most frequent KRAS mutation was G12C (41%),
followed by G12V (23.7%) and G12D (11.8%); ≥50% of the patients
received ICIs as monotherapy, while the rest had it in combination
with chemotherapy, mainly as a first-line therapy. The OS or PFS
among the KRAS subgroups showed no significant differences. By
contrast, patients with KRAS Q61, 13X and G12C mutations had a
relatively longer OS (46.5, 31.8 and 28.7 months, respectively) of
the KRAS subgroups analyzed. The KRAS G12D cohort showed the most
improved response to treatment with an (ORR) of 73%, largely driven
by patients with stable disease (40%), which was greater for
chemoimmunotherapy. The median duration of response (DOR) was 7.4
months overall, with the longest DOR at 10.2 months seen in G12V.
Co-mutations were also assessed: STK11 was present in 24% of cases
and TP53 in 29%. Patients with STK11 co-mutations tended towards
more prolonged OS (39.7 months) compared with those without STK11
co-mutations (26.1 months). By contrast, TP53 co-mutations were
associated with a shorter OS (19.1 months), although the analysis
did not reach statistical significance. Notably, bone metastases
were associated with lower survival and almost doubled the risk of
death (HR, 2.81; P<0.001) (103). However, the analysis was
retrospective, single institution-based, underpowered and the
treatment strategies were heterogeneous, which limited the strength
and external validity of the results. Furthermore, the negative
association between STK11 co-mutations and improved survival is in
contrast to numerous previous findings (104,105) and may be a cohort-specific
artifact. Therefore, the aforementioned study requires further
validation in larger, homogeneously treated cohorts.
A multicenter retrospective study compared
first-line programmed cell death protein 1 (PD-1)/PD-L1 inhibitors
(n=78) vs. platinum-based chemotherapy (n=29) in metastatic
KRAS-mutant NSCLC. The PFS was significantly longer in the
immunotherapy group (7.9 vs. 6.0 months; P=0.030), although there
were no differences in the median OS (16.2 vs. 19.2 months;
P>0.05). Positive PD-L1 status was associated with an improved
PFS, whereas increased CRP, CEA and NLR levels were associated with
poor outcomes. It was also suggested that PD-1/PD-L1 inhibitors may
achieve more robust disease control, particularly in PD-L1-positive
patients, using inflammatory parameters such as C-reactive protein
(CRP), Carcinoembryonic antigen (CEA), and neutrophil-to-lymphocyte
ratio (NLR) as potential prognostic values (106).
Immunotherapy, particularly in combination with
chemotherapy, markedly prolongs survival for advanced KRAS-mutant
NSCLC over chemotherapy alone; this is greater among patients with
PD-L1-positive tumors or high tumor mutational burden. KRAS G12C is
the predominant subtype and exhibits notable responsiveness to
ICIs, providing evidence for ICIs-based chemoimmunotherapy as the
optimal first-line treatment.
Co-mutations in STK11, TP53 and KEAP1 determine the
characteristic phenotype of the TME and clinical prognosis for
KRAS-mutant NSCLC. KRAS/TP53 tumors have an immune-inflamed profile
with high TMB (10–12 mutations/Mb), high PD-L1 expression (≥50%)
and dense CD8+ T-cell infiltration, which supports an
improved response to ICIs (107).
In a pooled analysis of 713 patients, KRAS/TP53 cases had
significantly higher benefit from ICIs compared with chemotherapy,
with an OS HR of 0.71 (95% CI, 0.55–0.92) and interaction HR of
0.53–0.56 favoring immunotherapy (108,109). Thus, KRAS/TP53 cases had
significantly higher benefit from ICIs when compared with that of
chemotherapy.
By contrast, KRAS/STK11 tumors are more consistently
characterized by non-inflamed immune-cold with low PD-L1 expression
(<10%), minimal CD8+ cell infiltration and a TMB of
4–6 mutations/Mb. Poor responses to ICIs and chemoimmunotherapy
suggest the clinical manifestation of this (110). Numerous studies have shown that an
STK11 co-mutation is associated with worse PFS and OS relative to
KRAS-only or KRAS/TP53 disease, which often translates to the level
of benefit derived from chemotherapy alone (111–113). These mutations also significantly
correlated with worse PFS (4.2 months vs. 11.0 months; HR, 2.2; P
< 0.01) and OS (9.8 months vs. NR; HR 2.6; P<0.01). Although
there are conflicting data as to their effects on KRAS G12C
inhibitor efficacy, with some studies showing reduced responses and
others reporting similar outcomes regardless of STK11 status, the
prognosis for patients with KRAS/STK11 co-mutations remains poor
(114–116). This highlights the necessity for
novel single and combination strategies in KRAS/STK11 disease, as
standard chemo-immuno-oncology regimens offer limited benefit.
KRAS/KEAP1 tumors are the most aggressive subtype
and possess infrequent broad resistance to chemotherapy, ICIs and
KRAS-targeting agents. KEAP1/NFE2L2 mutations are associated with
an inferior prognosis to platinum-pemetrexed and ICI regimens, as a
pooled analysis reported an HR of 1.96 (95% CI, 1.33–2.92) for
shorter survival (117). In the
KRYSTAL-1 study, patients with KEAP1 co-mutation had a
significantly shorter median PFS (4.1 months) and OS (5.4 months)
when treated with adagrasib in comparison with their wild-type
counterparts (9.9 and 19.0 months, respectively) (114). Functional validation also
demonstrates that KEAP1 deletion significantly elevates
IC50 of KRAS G12C inhibitors in H358 cells (118). Since KRAS/KEAP1 tumors are
resistant to multiple drugs, KRAS/KEAP1 tumors warrant
prioritization for clinical trials and experimental strategies
beyond current standard therapies.
Together, this emphasizes the need for co-mutation
profiling in treatment algorithms, as patients with KRAS/TP53
tumors may be more responsive to frontline ICI therapy; by
contrast, patients with KRAS/STK11 and KRAS/KEAP1 tumors represent
aggressive, immune-refractory subsets warranting alternative or
combination strategies.
Although targeted medicines provide a notable
response after treatment initiation, a large proportion of tumors
eventually resist targeted medications because of intertumoral
heterogeneity, where pre-existing resistance subclones survive
treatment and expand, driving tumor evolution toward resistance
(119). Secondary mutations in the
KRAS gene also cause resistance to specific inhibitors of the G12C
mutation. Mutations such as Y96D, A59T and G13D result in changes
to the KRAS protein structure that prevent inhibitors, including
sotorasib and adagrasib, from engaging with their target. Such
mutations frequently occur in the SII-P, a key area for drug
binding (120,121).
Concurrent mutations or alterations in driver
oncogenes, including TP53 and STK11/LKB1, are also involved in
resistance. It has been reported that tumors in which KRAS and
STK11 are mutant are less sensitive to immune checkpoint blockade
and KRAS-targeted agents, potentially due to the activation of
compensatory survival signaling pathways (122,123). Second, distant activation of
alternative signaling cascades exists that may mediate escape from
KRAS inhibition. MET amplification is the most extensively known
bypass mechanism, allowing downstream activation of the
PI3K-AKT-mTOR signaling in the presence of KRAS inhibition
(79,124). Other reported bypass routes
include RET fusions and wild-type RAS activation (121,123). These alterations enable tumor
cells to maintain proliferative signaling despite KRAS blockade.
Aside from developing secondary mutations, reactivation of
wild-type KRAS or other RAS family members can resurrect signaling
downstream of the pathway. This reactivation undermines the
efficacy of KRAS G12C inhibitors and leads to therapeutic failure
(121).
Tumor cells can also acquire resistance via
nongenetic means unrelated to new mutations. Cancer cells are kept
alive under drug pressure by cellular plasticity, stress-response
signaling and reprogramming of signal networks. For example,
activation of integrin β4 and paxillin pathways, among others, can
stimulate AKT signaling, contributing to a drug-tolerant persistent
cell phenotype (125,126). Finally, modifications in the TME
are significantly associated with therapy resistance. As such,
hypoxia and TME nutrient deprivation can induce adaptive stress
responses in cancer cells, leading to improved survival and
resistance to targeted therapies (127). Patients receiving KRAS inhibitors
also had worse prognoses when they had oxidative stress
response-related alterations, such as KEAP1 mutations (71). Table
III summarizes the various resistance mechanisms to
KRAS-targeted therapies in NSCLC, including genetic factors,
signaling pathways and TME-associated modifications associated with
treatment resistance.
Multiplex screening via next-generation sequencing
(NGS) is recommended for adequate patient selection in the European
Society for Medical Oncology clinical practice guidelines (4,128).
KRAS G12C inhibitors are a key treatment option for patients who
are not eligible for standard first-line therapies. For example,
co-mutations, including STK11 and KRAS G12C, are associated with
inferior responses to ICIs, indicating that KRAS G12C inhibitors
may be preferential as first-line treatment in those settings.
Another potential strategy is to use chemotherapy or ICIs in
combination with KRAS G12C inhibitors (129–131).
The development of resistance to conventional
therapies is a notable problem that has led to a paradigm shift
toward multitargeted therapies and improvements in drug delivery
systems for improved patient prognostic results. KRAS mutations are
considered actionable, particularly for G12C subtype for which
direct inhibitors are now available. Personalization of therapy is
essential because KRAS mutations are heterogeneous and exert
diverse effects on treatment response (132–134). Additional investigation is
warranted for KRAS G12D and G12V mutations, which represents 38% of
KRAS-mutant LUAD cases, as these variants currently lack effective
targeted therapies. Furthermore, emerging pan-KRAS inhibitors
should be further explored as potential treatment strategies
(135).
Emerging strategies for KRAS-mutant NSCLC are
increasingly shaped by advanced bioinformatic and genomic tools
that enable personalized therapy. Artificial intelligence (AI) is
increasingly shaping the drug discovery process, notably with its
rapid expansion induced by drug repurposing strategies. Machine
learning model coupled with molecular docking has demonstrated that
the FDA-approved drugs, including afatinib, neratinib and
zanubrutinib, are likely to be effective against KRAS G12C. This
method demonstrates how AI can identify novel therapeutic
opportunities and is particularly useful for hard-to-target alleles
such as KRAS G12D, where direct inhibitors remain limited (89).
Neoantigen prediction algorithms represent another
avenue of research. Mutant KRAS peptides have demonstrated potent
T-cell responses, which is relevant for STK11 co-mutated or high
TMB tumors. Such subsets are frequently resistant to ICIs
monotherapy, and the options of personalizing a vaccine or
establishing adoptive T-cell therapy constitute a rational approach
for these populations (136,137).
Single-cell RNA sequencing (scRNA-seq) has also been
applied to KRAS-induced tumors, and a tumor-associated epidermal
subpopulation that depended on the oncogene BMI-1 has been
identified, which contributes to tumor growth and treatment
resistance. Treatment approaches targeting this subpopulation with
small-molecule inhibitor PTC596 inhibited tumor growth in
preclinical models, demonstrating how scRNA-seq could detect
resistant subclones and guide targeted therapy (138).
Finally, CRISPR-based therapies are also broadening
treatment choices. High-fidelity Cas9 editing has shown that KRAS
G12C and G12D mutations can be corrected in organoid or xenograft
models with reduced tumor growth (139). In addition, genome-wide CRISPR
screens have identified synthetic lethal relationships that might
be exploited for rational combination strategies or resistance to
KRAS inhibitors by targeting TEA domain transcription factor, CDK4
and Wee1-like protein kinase dependencies (140). Together, these approaches
represent the innovative customized therapy for KRAS-mutation lung
cancer; however, these CRISPR-based and synthetic-lethal strategies
still face key limitations, such as off-target effects and
uncertain clinical translation, additional research is required to
overcome these limitations and improve patient outcomes.
KRAS has emerged as a viable target for therapeutic
inhibition in lung cancer, overcoming the previous challenges
associated with a limited understanding of KRAS biology and protein
biochemistry. Technological progress has deepened understanding of
RAS biology, including the discovery of novel druggable pockets and
the usefulness of molecular subtyping. Among the current
investigational treatments, small-molecule inhibitors targeting the
KRAS G12C mutation have demonstrated promising results. However,
molecular heterogeneity and resistance mechanisms remain concerns,
thus requiring additional translational research. Combination
strategies, such as immuno-targeted therapy, are increasingly
popular to maximize KRAS inhibition. The current challenges and
strategies in KRAS-mutated NSCLC research are overcoming resistance
to ICIs and KRAS inhibitors, discovering predictive biomarkers,
expanding targeted therapies to additional KRAS mutations and
optimizing diagnostic accuracy using NGS analysis of patient plasma
and tissue samples. Furthermore, worldwide accessibility to novel
agents remains essential. These efforts will help to improve the
quality of life and survival of patients with NSCLC.
Not applicable.
Funding: No funding was received.
Not applicable.
US prepared the original draft. HK reviewed the
manuscript. JS, YZ and ZD reviewed and edited the manuscript. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
El Osta B, Behera M, Kim S, Berry LD, Sica
G, Pillai RN, Owonikoko TK, Kris MG, Johnson BE, Kwiatkowski DJ, et
al: Characteristics and outcomes of patients with metastatic
KRAS-mutant lung adenocarcinomas: The lung cancer mutation
consortium experience. J Thorac Oncol. 14:876–889. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moore AR, Rosenberg SC, McCormick F and
Malek S: RAS-targeted therapies: is the undruggable drugged? Nat
Rev Drug Discov. 19:533–552. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Simanshu DK, Nissley DV and McCormick F:
RAS proteins and their regulators in human disease. Cell.
170:17–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Prior IA, Hood FE and Hartley JL: The
frequency of ras mutations in cancer. Cancer Res. 80:2669–2974.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dearden S, Stevens J, Wu YL and Blowers D:
Mutation incidence and coincidence in non small-cell lung cancer:
Meta-analyses by ethnicity and histology (mutMap). Ann Oncol.
24:2371–2376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang H, Liang SQ, Schmid RA and Peng RW:
New horizons in KRAS-mutant lung cancer: Dawn after darkness. Front
Oncol. 9:9532019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Naim MJ, Alam MJ, Ahmad S, Nawaz F,
Shrivastava N, Sahu M and Alam O: Therapeutic journey of
2,4-thiazolidinediones as a versatile scaffold: An insight into
structure activity relationship. Eur J Med Chem. 129:218–250. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bos JL: Ras oncogenes in human cancer: A
review. Cancer Res. 49:4682–4689. 1989.PubMed/NCBI
|
|
9
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yin G, Huang J, Petela J, Jiang H, Zhang
Y, Gong S, Wu J, Liu B, Shi J and Gao Y: Targeting small GTPases:
Emerging grasps on previously untamable targets, pioneered by KRAS.
Signal Transduct Target Ther. 8:2122023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Michalak DJ, Unger B, Lorimer E, Grishaev
A, Williams CL, Heinrich F and Lösche M: Structural and biophysical
properties of farnesylated KRas interacting with the chaperone
SmgGDS-558. Biophys J. 121:3684–3697. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeon H, Wang S, Song J, Gill H and Cheng
H: Update 2025: Management of non-small-cell lung cancer. Lung.
203:532025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang L, Guo Z, Wang F and Fu L: KRAS
mutation: From undruggable to druggable in cancer. Signal Transduct
Target Ther. 6:3862021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang F, Wang B, Wu M, Zhang L and Ji M:
Current status of KRAS G12C inhibitors in NSCLC and the potential
for combination with anti-PD-(L)1 therapy: A systematic review.
Front Immunol. 16:15091732025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uniyal P, Kashyap VK, Behl T, Parashar D
and Rawat R: KRAS mutations in cancer: Understanding signaling
pathways to immune regulation and the potential of immunotherapy.
Cancers (Basel). 17:7852025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parikh K, Banna G, Liu SV, Friedlaender A,
Desai A, Subbiah V and Addeo A: Drugging KRAS: Current perspectives
and state-of-art review. J Hematol Oncol. 15:1522022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hamarsheh S, Groß O, Brummer T and Zeiser
R: Immune modulatory effects of oncogenic KRAS in cancer. Nat
Commun. 11:54392020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghimessy A, Radeczky P, Laszlo V, Hegedus
B, Renyi-Vamos F, Fillinger J, Klepetko W, Lang C, Dome B and
Megyesfalvi Z: Current therapy of KRAS-mutant lung cancer. Cancer
Metastasis Rev. 39:1159–1177. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eser M, Hekimoglu G, Yarar MH, Canbek S
and Ozcelik M: KRAS G12C mutation in NSCLC in a small genetic
center: Insights into sotorasib therapy response potential. Sci
Rep. 14:265812024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bourne HR, Sanders DA and McCormick F: The
GTPase superfamily: A conserved switch for diverse cell functions.
Nature. 348:125–132. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bourne HR, Sanders DA and McCormick F: The
GTPase superfamily: Conserved structure and molecular mechanism.
Nature. 349:117–127. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scheffzek K, Ahmadian MR, Kabsch W,
Wiesmüller L, Lautwein A, Schmitz F and Wittinghofer A: The
Ras-RasGAP complex: Structural basis for GTPase activation and its
loss in oncogenic Ras mutants. Science. 277:333–338. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martin P, Leighl NB, Tsao MS and Shepherd
FA: KRAS mutations as prognostic and predictive markers in
non-small cell lung cancer. J Thorac Oncol. 8:530–542. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adderley H, Blackhall FH and Lindsay CR:
KRAS-mutant non-small cell lung cancer: Converging small molecules
and immune checkpoint inhibition. EBioMedicine. 41:711–716. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matikas A, Mistriotis D, Georgoulias V and
Kotsakis A: Targeting KRAS mutated non-small cell lung cancer: A
history of failures and a future of hope for a diverse entity. Crit
Rev Oncol Hematol. 110:1–12. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shepherd FA, Domerg C, Hainaut P, Jänne
PA, Pignon JP, Graziano S, Douillard JY, Brambilla E, Le Chevalier
T, Seymour L, et al: Pooled analysis of the prognostic and
predictive effects of KRAS mutation status and KRAS mutation
subtype in early-stage resected non-small-cell lung cancer in four
trials of adjuvant chemotherapy. J Clin Oncol. 31:2173–2181. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Riely GJ, Kris MG, Rosenbaum D, Marks J,
Li A, Chitale DA, Nafa K, Riedel ER, Hsu M, Pao W, et al: Frequency
and distinctive spectrum of KRAS mutations in never smokers with
lung adenocarcinoma. Clin Cancer Res. 14:5731–5734. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Redig AJ, Chambers ES, Lydon CA, Dahlberg
SE, Alden RS and Janne PA: Genomic complexity in KRAS mutant
non-small cell lung cancer (NSCLC) from never/light-smokers v
smokers. J Clin Oncol. 34 (15 Suppl):S90872016. View Article : Google Scholar
|
|
30
|
Ferrer I, Zugazagoitia J, Herbertz S, John
W, Paz-Ares L and Schmid-Bindert G: KRAS-mutant non-small cell lung
cancer: From biology to therapy. Lung Cancer. 124:53–64. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li S, Liu S, Deng J, Akbay EA, Hai J,
Ambrogio C, Zhang L, Zhou F, Jenkins RW, Adeegbe DO, et al:
Assessing therapeutic efficacy of MEK inhibition in a
KRASG12C-driven mouse model of lung cancer. Clin Cancer
Res. 24:4854–4864. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garassino MC, Marabese M, Rusconi P, Rulli
E, Martelli O, Farina G, Scanni A and Broggini M: Different types
of K-Ras mutations could affect drug sensitivity and tumour
behaviour in non-small-cell lung cancer. Ann Oncol. 22:235–237.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Imielinski M, Berger AH, Hammerman PS,
Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M,
Sivachenko A, et al: Mapping the hallmarks of lung adenocarcinoma
with massively parallel sequencing. Cell. 150:1107–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee B, Lee T, Lee SH, Choi YL and Han J:
Clinicopathologic characteristics of EGFR, KRAS, and ALK
alterations in 6,595 lung cancers. Oncotarget. 7:23874–23884. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li S, Li L, Zhu Y, Huang C, Qin Y, Liu H,
Ren-Heidenreich L, Shi B, Ren H, Chu X, et al: Coexistence of EGFR
with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A
comprehensive mutation profiling from 5125 Chinese cohorts. Br J
Cancer. 110:2812–2820. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Skoulidis F and Heymach JV: Co-occurring
genomic alterations in non-small-cell lung cancer biology and
therapy. Nat Rev Cancer. 19:495–509. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Christensen JG, Olson P, Briere T, Wiel C
and Bergo MO: Targeting Krasg12c-mutant cancer with a
mutation-specific inhibitor. J Intern Med. 288:183–191. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Haidar M and Jacquemin P: Past and future
strategies to inhibit membrane localization of the KRAS oncogene.
Int J Mol Sci. 22:131932021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tomasini P, Walia P, Labbe C, Jao K and
Leighl NB: Targeting the KRAS pathway in non-small cell lung
cancer. Oncologist. 21:1450–1460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gysin S, Salt M, Young A and McCormick F:
Therapeutic strategies for targeting ras proteins. Genes Cancer.
2:359–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Spoerner M, Herrmann C, Vetter IR,
Kalbitzer HR and Wittinghofer A: Dynamic properties of the Ras
switch I region and its importance for binding to effectors. Proc
Natl Acad Sci USA. 98:4944–4949. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Buhrman G, O'Connor C, Zerbe B, Kearney
BM, Napoleon R, Kovrigina EA, Vajda S, Kozakov D, Kovrigin EL and
Mattos C: Analysis of binding site hot spots on the surface of Ras
GTPase. J Mol Biol. 413:773–789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ostrem JM, Peters U, Sos ML, Wells JA and
Shokat KM: K-Ras(G12C) inhibitors allosterically control GTP
affinity and effector interactions. Nature. 503:548–551. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shima F, Matsumoto S, Yoshikawa Y,
Kawamura T, Isa M and Kataoka T: Current status of the development
of Ras inhibitors. J Biochem. 158:91–99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Berndt N, Hamilton AD and Sebti SM:
Targeting protein prenylation for cancer therapy. Nat Rev Cancer.
11:775–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zimmermann G, Papke B, Ismail S, Vartak N,
Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens
PI and Waldmann H: Small molecule inhibition of the KRAS-PDEδ
interaction impairs oncogenic KRAS signalling. Nature. 497:638–642.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Riely GJ, Johnson ML, Medina C, Rizvi NA,
Miller VA, Kris MG, Pietanza MC, Azzoli CG, Krug LM, Pao W and
Ginsberg MS: A phase II trial of Salirasib in patients with lung
adenocarcinomas with KRAS mutations. J Thorac Oncol. 6:1435–1437.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Whyte DB, Kirschmeier P, Hockenberry TN,
Nunez-Oliva I, James L, Catino JJ, Bishop WR and Pai JK: K- and
N-Ras are geranylgeranylated in cells treated with farnesyl protein
transferase inhibitors. J Biol Chem. 272:14459–14464. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shima F, Yoshikawa Y, Ye M, Araki M,
Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, et al: In
silico discovery of small-molecule Ras inhibitors that display
antitumor activity by blocking the Ras-effector interaction. Proc
Natl Acad Sci USA. 110:8182–8187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gerber DE, Camidge DR, Morgensztern D,
Cetnar J, Kelly RJ, Ramalingam SS, Spigel DR, Jeong W, Scaglioni
PP, Zhang S, et al: Phase 2 study of the focal adhesion kinase
inhibitor defactinib (VS-6063) in previously treated advanced KRAS
mutant non-small cell lung cancer. Lung Cancer. 139:60–67. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cox AD, Fesik SW, Kimmelman AC, Luo J and
Der CJ: Drugging the undruggable RAS: Mission possible? Nat Rev
Drug Discov. 13:828–851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang M and Casey PJ: Protein prenylation:
Unique fats make their mark on biology. Nat Rev Mol Cell Biol.
17:110–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kazi A, Xiang S, Yang H, Chen L, Kennedy
P, Ayaz M, Fletcher S, Cummings C, Lawrence HR, Beato F, et al:
Dual farnesyl and geranylgeranyl transferase inhibitor thwarts
mutant KRAS-driven patient-derived pancreatic tumors. Clin Cancer
Res. 25:5984–5996. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tao MH, Chen S, Freudenheim JL, Cauley JA,
Johnson KC, Mai X, Sarto GE, Wakelee H, Boffetta P and
Wactawski-Wende J: Oral bisphosphonate use and lung cancer
incidence among postmenopausal women. Ann Oncol. 29:1476–1485.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nagao S, Hattori N, Fujitaka K, Iwamoto H,
Ohshimo S, Kanehara M, Ishikawa N, Haruta Y, Murai H and Kohno N:
Regression of a primary pulmonary adenocarcinoma after zoledronic
acid monotherapy. Hiroshima J Med Sci. 60:7–9. 2011.PubMed/NCBI
|
|
58
|
Kitai H, Ebi H, Tomida S, Floros KV,
Kotani H, Adachi Y, Oizumi S, Nishimura M, Faber AC and Yano S:
Epithelial-to-mesenchymal transition defines feedback activation of
receptor tyrosine kinase signaling induced by MEK inhibition in
KRAS-mutant lung cancer. Cancer Discov. 6:754–769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Johnson C, Burkhart DL and Haigis KM:
Classification of KRAS-activating mutations and the implications
for therapeutic intervention. Cancer Discov. 12:913–923. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hunter JC, Manandhar A, Carrasco MA,
Gurbani D, Gondi S and Westover KD: Biochemical and structural
analysis of common cancer-associated KRAS mutations. Mol Cancer
Res. 13:1325–1335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ostrem JML and Shokat KM: Direct
small-molecule inhibitors of KRAS: From structural insights to
mechanism-based design. Nat Rev Drug Discov. 15:771–785. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Canon J, Rex K, Saiki AY, Mohr C, Cooke K,
Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al: The
clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity.
Nature. 575:217–223. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pandey D, Chauhan SC, Kashyap VK and Roy
KK: Structural insights into small-molecule KRAS inhibitors for
targeting KRAS mutant cancers. Eur J Med Chem. 277:1167712024.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Warnecke B and Nagasaka M: Adagrasib in
the treatment of KRAS p.G12C positive advanced NSCLC: Design,
development and place in therapy. Drug Des Devel Ther.
18:5673–5683. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li Z, Song Z, Zhao Y, Wang P, Jiang L,
Gong Y, Zhou J, Jian H, Dong X, Zhuang W, et al: D-1553
(Garsorasib), a potent and selective inhibitor of
KRASG12C in patients with NSCLC: Phase 1 study results.
J Thorac Oncol. 18:940–951. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
D'Alessio-Sands L, Gaynier J,
Michel-Milian V, Agbowuro AA and Brackett CM: Current strategies
and future dimensions in the development of KRAS inhibitors for
targeted anticancer therapy. Drug Dev Res. 86:e700422025.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Deming DA: Development of KRAS inhibitors
and their role for metastatic colorectal cancer. J Natl Compr Canc
Netw. 23:e2470672025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhao T and Liang SH: Novel tricyclic KRAS
inhibitors for the treatment of cancer. ACS Med Chem Lett.
16:178–179. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hong DS, Fakih MG, Strickler JH, Desai J,
Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS,
et al: KRASG12C inhibition with sotorasib in advanced
solid tumors. N Engl J Med. 383:1207–1217. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
No authors listed. FDA approves first KRAS
inhibitor: Sotorasib. Cancer Discov. 11:OF42021. View Article : Google Scholar
|
|
71
|
Skoulidis F, Li BT, Dy GK, Price TJ,
Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F,
et al: Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl
J Med. 384:2371–2381. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jänne PA, Riely GJ, Gadgeel SM, Heist RS,
Ou SI, Pacheco JM, Johnson ML, Sabari JK, Leventakos K, Yau E, et
al: Adagrasib in non-small-cell lung cancer harboring a
KRASG12C mutation. N Engl J Med. 387:120–131. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
de Langen AJ, Johnson ML, Mazieres J,
Dingemans AC, Mountzios G, Pless M, Wolf J, Schuler M, Lena H,
Skoulidis F, et al: Sotorasib versus docetaxel for previously
treated non-small-cell lung cancer with KRASG12C
mutation: A randomised, open-label, phase 3 trial. Lancet.
401:733–746. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wislez M, Mascaux C, Cadranel J, Thomas
QD, Ricordel C, Swalduz A, Pichon E, Veillon R, Gounant V,
Rousseau-Bussac G, et al: Real-world effectiveness and tolerability
of sotorasib in patients with KRAS G12C-mutated metastatic
non-small cell lung cancer: The IFCT-2102 lung KG12Ci study. Eur J
Cancer. 219:1153012025. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ou SHI, Jänne PA, Leal TA, Rybkin II,
Sabari JK, Barve MA, Bazhenova L, Johnson ML, Velastegui KL,
Cilliers C, et al: First-in-human phase I/IB dose-finding study of
adagrasib (MRTX849) in patients with advanced KRASG12C
solid tumors (KRYSTAL-1). J Clin Oncol. 40:2530–2538. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mok TSK, Yao W, Duruisseaux M, Doucet L,
Azkárate Martínez A, Gregorc V, Juan-Vidal O, Lu S, De Bondt C, de
Marinis F, et al: KRYSTAL-12: Phase 3 study of adagrasib versus
docetaxel in patients with previously treated advanced/metastatic
non-small cell lung cancer (NSCLC) harboring a KRASG12C
mutation. J Clin Oncol. 42 (Suppl 17):LBA85092024. View Article : Google Scholar
|
|
77
|
Li Z, Dang X, Huang D, Jin S, Li W, Shi J,
Wang X, Zhang Y, Song Z, Zhang J, et al: Abstract CT246:
Open-label, single-arm, multicenter, phase 2 trial of garsorasib in
KRAS G12C-mutated non-small-cell lung cancer. Cancer Res. 84 (7
Suppl):CT2462024. View Article : Google Scholar
|
|
78
|
Li Z, Dang X, Huang D, Jin S, Li W, Shi J,
Wang X, Zhang Y, Song Z, Zhang J, et al: Garsorasib in patients
with KRASG12C-mutated non-small-cell lung cancer in
China: An open-label, multicentre, single-arm, phase 2 trial.
Lancet Respir Med. 12:589–598. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Awad MM, Liu S, Rybkin II, Arbour KC,
Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al:
Acquired resistance to KRASG12C inhibition in cancer. N
Engl J Med. 384:2382–2393. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Cox AD and Der CJ: ‘Undruggable KRAS’:
Druggable after all. Genes Dev. 39:132–162. 2025.PubMed/NCBI
|
|
81
|
Maruyama K, Shimizu Y, Nomura Y, Oh-Hara
T, Takahashi Y, Nagayama S, Fujita N and Katayama R: Mechanisms of
KRAS inhibitor resistance in KRAS-mutant colorectal cancer
harboring Her2 amplification and aberrant KRAS localization. NPJ
Precis Oncol. 9:42025. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mugarza E, van Maldegem F, Boumelha J,
Moore C, Rana S, Llorian Sopena M, East P, Ambler R, Anastasiou P,
Romero-Clavijo P, et al: Therapeutic KRASG12C inhibition
drives effective interferon-mediated antitumor immunity in
immunogenic lung cancers. Sci Adv. 8:eabm87802022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Garassino MC, Jänne PA, Barlesi F, Spira
AI, Mok TSK, Hochmair M, O'Byrne KJ, Felip E, Kim SW, Cappuzzo F,
et al: 1394TiP KRYSTAL-7: A phase III study of first-line adagrasib
plus pembrolizumab versus pembrolizumab alone in patients with
advanced NSCLC with KRASG12C mutation mutation. Ann Oncol. 35
(Suppl 2):S872–S873. 2024. View Article : Google Scholar
|
|
84
|
Li BT, Falchook GS, Durm GA, Burns TF,
Skoulidis F, Ramalingam SS, Spira A, Bestvina CM, Goldberg SB,
Veluswamy R, et al: OA03.06 CodeBreaK 100/101: First report of
safety/efficacy of sotorasib in combination with pembrolizumab or
atezolizumab in advanced KRAS p.G12C NSCLC. J Thorac Oncol. 17
(Suppl):S10–S11. 2022. View Article : Google Scholar
|
|
85
|
Ernst SM, Hofman MM, van der Horst TE,
Paats MS, Heijboer FWJ, Aerts JGJV, Dumoulin DW, Cornelissen R, von
der Thüsen JH, de Bruijn P, et al: Hepatotoxicity in patients with
non-small cell lung cancer treated with sotorasib after prior
immunotherapy: A comprehensive clinical and pharmacokinetic
analysis. EBioMedicine. 102:1050742024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chour A, Basse C, Lebossé F, Bonte PE,
Girard N and Duruisseaux M: Management of sotorasib-related adverse
events and hepatotoxicities following anti-PD-(L)1 therapy:
Experience with sotorasib in two French anti-cancer centers and
practical guidance proposal. Lung Cancer. 191:1077892024.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang J, Johnson M, Barve M, Bazhenova L,
McCarthy M, Schwartz R, Horvath-Walsh E, Velastegui K, Qian C and
Spira A: Practical guidance for the management of adverse events in
patients with KRASG12C-mutated non-small cell lung cancer receiving
adagrasib. Oncologist. 28:287–296. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mina SA, Shanshal M, Leventakos K and
Parikh K: Emerging targeted therapies in non-small-cell lung cancer
(NSCLC). Cancers (Basel). 17:3532025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Fancelli S, Petroni G, Pillozzi S and
Antonuzzo L: Unconventional strategy could be the future: From
target to KRAS broad range treatment. Heliyon. 10:e297392024.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Masfarre Pinto L, Parreira ASDFM, Castro
Unanua N, Rocha P, Morilla Ruiz I, Teijeira Sanchez L, Clave S,
Navarro Gorro N, Bach Mora R, Sánchez I, et al: 1408P Differences
on immune biomarkers between KRAS G12C and KRAS non-G12C mutated
non-small cell lung cancer. Ann Oncol. 34 (Suppl2):S805–S806. 2023.
View Article : Google Scholar
|
|
91
|
Zhou C, Li C, Luo L, Li X, Jia K, He N,
Mao S, Wang W, Shao C, Liu X, et al: Anti-tumor efficacy of
HRS-4642 and its potential combination with proteasome inhibition
in KRAS G12D-mutant cancer. Cancer Cell. 42:1286–1300.e8. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Li Y, Zhao J and Li Y: New exploration of
KRASG12D inhibitors and the mechanisms of resistance.
Exp Hematol Oncol. 14:392025. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Arbour KC, Rizvi H, Plodkowski AJ,
Hellmann MD, Knezevic A, Heller G, Yu HA, Ladanyi M, Kris MG,
Arcila ME, et al: Treatment outcomes and clinical characteristics
of patients with KRAS-G12C-mutant non-small cell lung cancer. Clin
Cancer Res. 27:2209–2215. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bironzo P, Cani M, Jacobs F, Napoli VM,
Listì A, Passiglia F, Righi L, Di Maio M, Novello S and Scagliotti
GV: Real-world retrospective study of KRAS mutations in advanced
non-small cell lung cancer in the era of immunotherapy. Cancer.
129:1662–1671. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Rodenhuis S, van de Wetering ML, Mooi WJ,
Evers SG, van Zandwijk N and Bos JL: Mutational activation of the
K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of
the lung. N Engl J Med. 317:929–935. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Goodman AM, Kato S, Bazhenova L, Patel SP,
Frampton GM, Miller V, Stephens PJ, Daniels GA and Kurzrock R:
Tumor mutational burden as an independent predictor of response to
immunotherapy in diverse cancers. Mol Cancer Ther. 16:2598–2608.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mazieres J, Drilon A, Lusque A, Mhanna L,
Cortot AB, Mezquita L, Thai AA, Mascaux C, Couraud S, Veillon R, et
al: Immune checkpoint inhibitors for patients with advanced lung
cancer and oncogenic driver alterations: Results from the
IMMUNOTARGET registry. Ann Oncol. 30:1321–1328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Rothschild SI: KRAS and immune checkpoint
inhibitors-serendipity raising expectations. J Thorac Oncol.
14:951–954. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Torralvo J, Friedlaender A, Achard V and
Addeo A: The activity of immune checkpoint inhibition in KRAS
mutated non-small cell lung cancer: A single centre experience.
Cancer Genomics Proteomics. 16:577–582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mok TSK, Lopes G, Cho BC, Kowalski DM,
Kasahara K, Wu YL, de Castro G Jr, Turna HZ, Cristescu R,
Aurora-Garg D, et al: Associations of tissue tumor mutational
burden and mutational status with clinical outcomes in KEYNOTE-042:
Pembrolizumab versus chemotherapy for advanced PD-L1-positive
NSCLC. Ann Oncol. 34:377–388. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Gadgeel S, Rodriguez-Abreu D, Felip E,
Esteban E, Speranza G, Reck M, Hui R, Boyer M, Garon EB, Horinouchi
H, et al: LBA5-KRAS mutational status and efficacy in KEYNOTE-189:
Pembrolizumab (pembro) plus chemotherapy (chemo) vs placebo plus
chemo as first-line therapy for metastatic non-squamous NSCLC. Ann
Oncol. 30 (Suppl 11):xi64–xi65. 2019. View Article : Google Scholar
|
|
102
|
Peng L, Guo J, Kong L, Huang Y, Tang N,
Zhang J, Wang M, He X, Li Z, Peng Y, et al: Efficacy of
immunotherapy in KRAS-mutant advanced NSCLC: A real-world study in
a Chinese population. Front Oncol. 12:10707612023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Urtecho SB, Provenzano L, Spagnoletti A,
Bottiglieri A, Pircher C, Massa G, Sposetti C, Proto C, Brambilla
M, Occhipinti M, et al: Decoding KRAS mutation in non-small cell
lung cancer patients receiving immunotherapy: A retrospective
institutional comparison and literature review. Lung Cancer.
199:1080512025. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Koyama S, Akbay EA, Li YY, Aref AR,
Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM,
Denning WL, et al: STK11/LKB1 deficiency promotes neutrophil
recruitment and proinflammatory cytokine production to suppress
T-cell Activity in the lung tumor microenvironment. Cancer Res.
76:999–1008. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Knetki-Wróblewska M, Wojas-Krawczyk K,
Krawczyk P and Krzakowski M: Emerging insights into STK11, KEAP1
and KRAS mutations: Implications for immunotherapy in patients with
advanced non-small cell lung cancer. Transl Lung Cancer Res.
13:3718–3730. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wei K, Sun T, Feng X, Chen Y, Liu Q and
Tang H: PD-1/L1 immune checkpoint inhibitors for KRAS-mutant
non-small cell lung cancer: A multicenter retrospective real-world
study. BMC Cancer. 25:4442025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Dong ZY and Wu YL: What is the
significance of TP53 and KRAS mutation for immunotherapy in
non-small cell lung cancer? Transl Cancer Res. 6 (Suppl
6):S1115–S1117. 2017. View Article : Google Scholar
|
|
108
|
Budczies J, Romanovsky E, Kirchner M,
Neumann O, Blasi M, Schnorbach J, Shah R, Bozorgmehr F, Savai R,
Stiewe T, et al: KRAS and TP53 co-mutation predicts benefit of
immune checkpoint blockade in lung adenocarcinoma. Br J Cancer.
131:524–533. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Bischoff P, Reck M, Overbeck T,
Christopoulos P, Rittmeyer A, Lüders H, Kollmeier J, Kulhavy J,
Kemper M, Reinmuth N, et al: Outcome of first-line treatment with
pembrolizumab according to KRAS/TP53 mutational status for
nonsquamous programmed death-ligand 1-high (≥50%) NSCLC in the
German national network genomic medicine lung cancer. J Thorac
Oncol. 19:803–817. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Skoulidis F, Goldberg ME, Greenawalt DM,
Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco
SE, Gay L, et al: STK11/LKB1 mutations and PD-1 inhibitor
resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov.
8:822–835. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Sun L, Handorf EA, Zhou Y, Borghaei H,
Aggarwal C and Bauman J: Outcomes in patients treated with
frontline immune checkpoint inhibition (ICI) for advanced NSCLC
with KRAS mutations and STK11/KEAP1 comutations across PD-L1
levels. Lung Cancer. 190:1075102024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ricciuti B and Garassino MC: Precision
immunotherapy for STK11/KEAP1-mutant NSCLC. J Thorac Oncol.
19:877–882. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
De Giglio A, De Biase D, Favorito V,
Maloberti T, Di Federico A, Zacchini F, Venturi G, Parisi C,
Gustavo Dall'Olio F, Ricciotti I, et al: STK11 mutations correlate
with poor prognosis for advanced NSCLC treated with first-line
immunotherapy or chemo-immunotherapy according to KRAS, TP53,
KEAP1, and SMARCA4 status. Lung Cancer. 199:1080582025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Negrao MV, Paula AG, Molkentine D, Hover
L, Nilsson M, Vokes N, Engstrom L, Calinisan A, Briere DM, Waters
L, et al: Impact of co-mutations and transcriptional signatures in
non-small cell lung cancer patients treated with adagrasib in the
KRYSTAL-1 trial. Clin Cancer Res. 31:1069–1081. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Olsen A, Lebedeva A, Nosova P, Nikulin V,
Sharova M, Ignatova E, Mileyko V and Ivanov M: Impact of the
STK11/KRAS co-mutation on the response to immunotherapy in a
real-world pan-cancer cohort. Tumori. 110:146–152. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Sisca L, Cascetta P, Aijaz A, Catania C,
Facchinetti F, Naqash AR, Ricciuti B and Cortellini A: KRAS and
STK11 co-mutations in resectable non-small cell lung cancer:
Enduring prognostic value and impaired immunotherapy response.
Transl Lung Cancer Res. 14:2374–2382. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Hellyer JA, Padda SK, Diehn M and Wakelee
HA: Clinical implications of KEAP1-NFE2L2 mutations in NSCLC. J
Thorac Oncol. 16:395–403. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tian L, Liu C, Zheng S, Shi H, Wei F,
Jiang W, Dong Y, Xu H, Yin E, Sun N, et al: KEAP1 mutations as key
crucial prognostic biomarkers for resistance to KRAS-G12C
inhibitors. J Transl Med. 23:822025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jänne PA, van den Heuvel MM, Barlesi F,
Cobo M, Mazieres J, Crinò L, Orlov S, Blackhall F, Wolf J, Garrido
P, et al: Selumetinib plus docetaxel compared with docetaxel alone
and progression-free survival in patients with KRAS-mutant advanced
non-small cell lung cancer. JAMA. 317:1844–1853. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Sreter KB, Catarata MJ, von Laffert M and
Frille A: Resistance to KRAS inhibition in advanced non-small cell
lung cancer. Front Oncol. 14:13578982024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Blaquier JB, Cardona AF and Recondo G:
Resistance to KRASG12C inhibitors in non-small cell lung
cancer. Front Oncol. 11:7875852021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Cascetta P, Marinello A, Lazzari C,
Gregorc V, Planchard D, Bianco R, Normanno N and Morabito A: KRAS
in NSCLC: State of the art and future perspectives. Cancers
(Basel). 14:54302022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Chour A, Toffart AC, Berton E and
Duruisseaux M: Mechanisms of resistance to KRASG12C inhibitors in
KRASG12C-mutated non-small cell lung cancer. Front Oncol.
14:13287282024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J,
Kiedrowski LA, Michel AG, Syed MU, Fella KA, Sakhi M, et al:
Clinical acquired resistance to KRASG12C inhibition
through a novel KRAS switch-II pocket mutation and polyclonal
alterations converging on RAS-MAPK reactivation. Cancer Discov.
11:1913–1922. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Mohanty A, Nam A, Srivastava S, Jones J,
Lomenick B, Singhal SS, Guo L, Cho H, Li A, Behal A, et al:
Acquired resistance to KRAS G12C small-molecule inhibitors via
genetic/nongenetic mechanisms in lung cancer. Sci Adv.
9:eade38162023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Akhave NS, Biter AB and Hong DS:
Mechanisms of resistance to KRASG12C-targeted therapy.
Cancer Discov. 11:1345–1352. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Labrie M, Brugge JS, Mills GB and
Zervantonakis IK: Therapy resistance: Opportunities created by
adaptive responses to targeted therapies in cancer. Nat Rev Cancer.
22:323–339. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Planchard D, Popat S, Kerr K, Novello S,
Smit EF, Faivre-Finn C, Mok TS, Reck M, Van Schil PE, Hellmann MD,
et al: Metastatic non-small cell lung cancer: ESMO clinical
practice guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 29 (Suppl 4):iv192–iv237. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Seo KY, Jelinsky SA and Loechler EL:
Factors that influence the mutagenic patterns of DNA adducts from
chemical carcinogens. Mutat Res. 463:215–246. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Goulding RE, Chenoweth M, Carter GC, Boye
ME, Sheffield KM, John WJ, Leusch MS, Muehlenbein CE, Li L, Jen MH,
et al: KRAS mutation as a prognostic factor and predictive factor
in advanced/metastatic non-small cell lung cancer: A systematic
literature review and meta-analysis. Cancer Treat Res Commun.
24:1002002020.PubMed/NCBI
|
|
131
|
Awad MM, Gadgeel SM, Borghaei H, Patnaik
A, Yang JC, Powell SF, Gentzler RD, Martins RG, Stevenson JP, Altan
M, et al: Long-term overall survival from KEYNOTE-021 cohort G:
Pemetrexed and carboplatin with or without pembrolizumab as
first-line therapy for advanced nonsquamous NSCLC. J Thorac Oncol.
16:162–168. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Feng W: Advancements in targeted therapies
based on the EGFR, and KRAS genetic mutations in lung cancer. Theor
Nat Sci. 67:22–29. 2024. View Article : Google Scholar
|
|
133
|
Harris E and Thawani R: Current
perspectives of KRAS in non-small cell lung cancer. Curr Probl
Cancer. 51:1011062024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Jiang Z: A review of KRAS inhibitors and
potential improvement in the future. Transact Mater Biotechnol Life
Sci. 7:322–327. 2024.
|
|
135
|
Ricciuti B, Leonardi GC, Metro G, Grignani
F, Paglialunga L, Bellezza G, Baglivo S, Mencaroni C, Baldi A,
Zicari D and Crinò L: Targeting the KRAS variant for treatment of
non-small cell lung cancer: Potential therapeutic applications.
Expert Rev Respir Med. 10:53–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Rosner S, Connor S, Sanber K, Zahurak M,
Zhang T, Gurumurthy I, Zeng Z, Presson B, Singh D, Rayes R, et al:
Divergent clinical and immunologic outcomes based on STK11
co-mutation status in resectable KRAS-mutant lung cancers following
neoadjuvant immune checkpoint blockade. Clin Cancer Res.
31:339–351. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Choi J, Goulding SP, Conn BP, McGann CD,
Dietze JL, Kohler J, Lenkala D, Boudot A, Rothenberg DA, Turcott
PJ, et al: Systematic discovery and validation of T cell targets
directed against oncogenic KRAS mutations. Cell Rep Methods.
1:1000842021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Gondal MN, Cieslik M and Chinnaiyan AM:
Integrated cancer cell-specific single-cell RNA-seq datasets of
immune checkpoint blockade-treated patients. Sci Data. 12:1392025.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Álvarez-Pérez JC, Sanjuán-Hidalgo J,
Arenas AM, Hernández-Navas I, Benitez-Cantos MS, Andrades A,
Calabuig-Fariñas S, Jantus-Lewintre E, Paz-Ares L, Ferrer I and
Medina PP: High-fidelity Cas9-mediated targeting of KRAS driver
mutations restrains lung cancer in preclinical models. Nat Commun.
16:70802025. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Wang Y, Bui TA, Yang X, Hutvagner G and
Deng W: Advancements in gene therapies targeting mutant KRAS in
cancers. Cancer Metastasis Rev. 44:242025. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho
BC, Turna HZ, Castro G Jr, Srimuninnimit V, Laktionov KK,
Bondarenko I, et al: Pembrolizumab versus chemotherapy for
previously untreated, PD-L1-expressing, locally advanced or
metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised,
open-label, controlled, phase 3 trial. Lancet. 393:1819–1830. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Borghaei H, Paz-Ares L, Horn L, Spigel DR,
Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al:
Nivolumab versus docetaxel in advanced nonsquamous non-small-cell
lung cancer. N Engl J Med. 373:1627–1639. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Rodríguez-Abreu D, Powell SF, Hochmair MJ,
Gadgeel S, Esteban E, Felip E, Speranza G, De Angelis F, Dómine M,
Cheng SY, et al: Pemetrexed plus platinum with or without
pembrolizumab in patients with previously untreated metastatic
nonsquamous NSCLC: Protocol-specified final analysis from
KEYNOTE-189. Ann Oncol. 32:881–895. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Socinski MA, Jotte RM, Cappuzzo F, Orlandi
F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D,
Thomas CA, Barlesi F, et al: Atezolizumab for first-line treatment
of metastatic nonsquamous NSCLC. N Engl J Med. 378:2288–2301. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Garassino MC, Theelen WSME, Jänne PA, Boom
LN, De Marinis F, Dómine Gómez M, Nadal E, Paz-Ares L, Gu W, Felip
E, et al: 5MO: First-line adagrasib (ADA) with pembrolizumab
(PEMBRO) in patients (pts) with advanced/metastatic
KRASG12C-mutated non-small cell lung cancer (NSCLC) and PD-L1 ≥50%
from the phase II portion of KRYSTAL-7. J Thorac Oncol. 20 (Suppl
1):S8–S10. 2025. View Article : Google Scholar
|
|
146
|
Neal J, Pavlakis N, Kim SW, Goto Y, Lim
SM, Mountzios G, Fountzilas E, Mochalova A, Christoph DC, Bearz A,
et al: CONTACT-01: A randomized phase III trial of atezolizumab +
cabozantinib versus docetaxel for metastatic non-small cell lung
cancer after a checkpoint inhibitor and chemotherapy. J Clin Oncol.
42:2393–2403. 2024. View Article : Google Scholar : PubMed/NCBI
|