Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
seventh most common malignancy worldwide and accounts for >90%
of all head and neck cancers. According to the GLOBOCAN 2020
estimates, ~890,000 new cases of HNSCC are diagnosed annually
worldwide (1–3). HNSCC arises from the epithelial mucosa
of the larynx, oropharynx, nasopharynx and oral cavity (4). The incidence of HNSCC is primarily
associated with tobacco smoking, alcohol consumption and human
papillomavirus infection (5).
Additionally, the use of smokeless tobacco products and genetic
susceptibility are significant risk factors contributing to its
development (6). The first-line
treatment for HNSCC includes surgery and radiotherapy, often
supplemented with chemotherapy. Among chemotherapeutic agents,
cisplatin is the most commonly used platinum-based drug (7,8).
Cisplatin forms covalent bonds with guanine residues in the DNA,
leading to the crosslinking of the double-stranded helix. This
process inhibits DNA replication, promotes the accumulation of DNA
damage and ultimately induces apoptosis, establishing cisplatin as
an essential chemotherapeutic agent for HNSCC treatment. However,
prolonged cisplatin administration can lead to the development of
resistance mechanisms, necessitating combination therapy with other
chemotherapeutic agents or immunotherapies (9,10).
Despite the availability of established treatment strategies,
patients with HNSCC continue to have a poor prognosis, with a
5-year overall survival rate of ~40–50% worldwide, as documented in
studies published in 2025 (11,12).
Therefore, novel therapeutic strategies are required to improve the
poor prognosis of HNSCC. Although substantial progress has been
made in anticancer research, conventional chemotherapeutic agents
remain associated with significant adverse effects, including
toxicity to normal cells and the development of drug resistance
(13). There is growing evidence
demonstrating the importance of natural product-derived compounds
with potent anticancer activity and minimal side effects. Extensive
research has been conducted to investigate the molecular mechanisms
underlying their effects, including the inhibition of cancer cell
proliferation and angiogenesis. Furthermore, these studies have
demonstrated the induction of cancer cell apoptosis (14,15).
Platycodon grandiflorus (PG), which is widely
used in traditional oriental medicine, is known for its therapeutic
properties in the treatment of respiratory diseases, including
asthma, bronchitis and tonsillitis. Additionally, PG exhibits
immunostimulatory, anti-inflammatory, anti-obesity and
anti-atherosclerotic activities (16,17).
Platycodin D (PD), a triterpenoid saponin abundant in the root of
PG, is the major active natural compound responsible for these
diverse pharmacological effects (18,19).
Notably, numerous studies have demonstrated that PD possesses
anticancer properties, including the inhibition of cell
proliferation, and the regulation of oxidative stress and
autophagy. Furthermore, PD disrupts autophagic flux, thereby
exacerbating cellular stress and promoting cancer cell death
(18–20). However, the anticancer effects of PD
on HNSCC have not yet been adequately investigated.
Autophagy is a highly conserved cellular degradation
and recycling process that is essential for maintaining cellular
homeostasis and enabling adaptation to environmental stressors,
such as nutrient deprivation, oxidative stress and hypoxia. This
process involves the sequestration of damaged organelles and
misfolded proteins within autophagosomes, which subsequently fuse
with lysosomes for degradation (20,21).
Autophagy functions as a cytoprotective mechanism by eliminating
unnecessary or damaged cellular components and thereby promoting
cell survival. However, excessive activation or inhibition of
autophagy can lead to autophagy-mediated cell death (22,23).
Due to this dual role, autophagy has been extensively investigated
as a therapeutic target in cancer (23–25).
The regulation of autophagy is primarily mediated by
autophagy-related genes and key regulatory proteins, such as LC3,
which undergo lipidation to facilitate autophagosome formation.
Following autophagosome formation, fusion with lysosomes enables
the degradation of autophagic cargo (26). In the context of cancer therapy,
autophagy may either promote cell survival through protective
autophagy mechanisms or enhance treatment efficacy by inducing
autophagy-dependent cell death, further underscoring its complex
role in tumor biology (27,28). Oxidative stress, predominantly
mediated by reactive oxygen species (ROS), is known as a key
regulator of autophagy (29).
Understanding the interaction between ROS, autophagy and cell death
is crucial for improving anticancer treatment strategies. Based on
these considerations, it was hypothesized that cisplatin-induced
ROS accumulation may initially activate autophagy as a cellular
stress response, whereas the known autophagic flux-blocking
activity of PD could further exacerbate cellular stress by
impairing autophagic degradation. Furthermore, the combined effect
of excessive ROS production and autophagic flux inhibition was
proposed to synergistically promote cytotoxic stress and cell death
in HNSCC cells.
Therefore, the present study aimed to investigate
whether PD enhanced cisplatin chemosensitivity in HNSCC cells and
to elucidate the underlying mechanism, with a particular focus on
ROS accumulation and autophagic flux regulation.
Materials and methods
Reagents
PD powder was purchased from MilliporeSigma and
dissolved in DMSO at a stock concentration of 20 mM. Cisplatin was
purchased from MilliporeSigma and dissolved according to the
manufacturer's instructions.
Cell culture
The human HNSCC cell line HSC3 was purchased from
the Japanese Collection of Research Bioresources Cell Bank. The
FaDu cell line was obtained from the American Type Culture
Collection. The HaCaT cell line was kindly provided by Dr Jin-Woo
Lee (Kyung Hee Medical Center, Seoul, South Korea) and the original
supplier of the cell line was Cell Lines Service GmbH. All cell
lines were maintained at 37°C in a humidified incubator containing
5% CO2. HSC3 cells were cultured in Roswell Park
Memorial Institute-1640 medium (RPMI-1640; HyClone; Cytiva) and
FaDu cells were cultured in Minimum Essential Medium (MEM; HyClone;
Cytiva), both supplemented with 10% heat-inactivated FBS (HyClone;
Cytiva) and 1% penicillin-streptomycin (Corning, Inc.). HaCaT cells
were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone;
Cytiva) supplemented with 10% heat-inactivated FBS and 1%
penicillin-streptomycin. When cell confluency reached ~80% in a
100-mm dish, cells were washed with Dulbecco's PBS (DPBS; pH
7.0–7.6) and detached using trypsin-EDTA for subculturing. Cell
line authentication was conducted by short tandem repeat profiling
using the Korean Cell Line Bank (Korean Cell Line Research
Foundation) within the past year. During the initial thawing of
cell cultures, all cell lines were treated with a mycoplasma
removal agent (cat. no. 093050044; MP Biomedicals, LLC) at 37°C for
24 h in a humidified incubator to eliminate potential mycoplasma
contamination.
Cell viability assay
HaCaT, HSC3 and FaDu cells were seeded into 96-well
plates at a density of 3×103 cells/well and allowed to
attach by incubating them at 37°C for 24 h. Subsequently, HSC3 and
FaDu cells were treated with PD (0, 5, 10 and 15 µM) and cisplatin
(0, 5 and 10 µM) at 37°C for 48 h, either as a single treatment or
in combination, whereas HaCaT cells were treated with PD alone (0,
0.5, 1, 3, 5 and 10 µM) at 37°C for 48 h. Control cells were
treated with an equivalent volume of vehicle (DMSO) under the same
experimental conditions. Thereafter, cell viability was assessed
using a WST-8-based cell viability assay according to the
manufacturer's protocol (EZ-Cytox; DoGenBio), and 10 µl of the
reagent was added to each well. After incubation for 1 h, the
formation of chromogenic formazan was quantified by measuring the
absorbance at 450 nm using a SPARK® multimode microplate
reader (Tecan Group, Ltd.). Cell viability (%) was calculated as
[mean optical density (OD) of the sample/mean OD of the control]
×100%.
Colony formation assay
HSC3 and FaDu cells were seeded into 6-well plates
at a density of 300 cells per well and maintained at 37°C. After 24
h of incubation, the cells were treated at 37°C with 5 µM PD and 5
µM cisplatin at 48-h intervals. HSC3 and FaDu cells were divided
into four groups: Control, PD-treated, cisplatin-treated and
combination-treated. HSC3 cells were cultured for 7 days and FaDu
cells were cultured for 10 days. The colonies that developed from
surviving cells were stained with crystal violet solution (0.5%
crystal violet in 50% methanol) at room temperature for 3 min
without prior fixation. Colonies were defined as clusters
containing ≥50 cells. The number of colonies was counted
manually.
Reverse transcription-quantitative PCR
(RT-qPCR)
HSC3 and FaDu cells were seeded into 6-well plates
at densities of 1×105 and 3×105 cells/well,
respectively. After 24 h, the cells were treated with 10 µM PD or
cisplatin, either as a single treatment or in combination, at 37°C
for 12 h in HSC3 cells and for 3 h in FaDu cells. Total RNA was
extracted using RiboEx™ (GeneAll Biotechnology Co., Ltd.),
according to the manufacturer's protocol. A NanoDrop
spectrophotometer (Thermo Fisher Scientific, Inc.) was used to
assess the RNA concentration and quality. Extracted RNA was reverse
transcribed into cDNA using a Tetro cDNA Synthesis Kit (Meridian
Bioscience, Inc.). Reverse transcription was performed at 45°C for
30 min, followed by enzyme inactivation at 85°C for 5 min,
according to the manufacturer's instructions. RT-qPCR was performed
using SYBR Green Premix Ex (Meridian Bioscience, Inc.) with the
following thermocycling conditions: Initial denaturation at 95°C
for 30 sec, followed by 40 cycles of denaturation at 95°C for 5 sec
and annealing at 60°C for 30 sec. The reaction was evaluated using
melting curve analysis. Each experiment was performed in triplicate
using independent biological replicates, and the data were analyzed
by relative quantitation using the 2−ΔΔCq method and
normalized to β-actin (30). The
following primer sequences were used: Heme oxygenase-1 (HO-1)
forward, 5′-CCAGGCAGAGAATGCTGAGTTC-3′ and reverse,
5′-AAGACTGGGCTCTCCTTGTTGC-3′; NAD(P)H quinone dehydrogenase 1
(NQO1) forward, 5′-CCTGCCATTCTGAAAGGCTGGT-3′ and reverse,
5′-GTGGTGATGGAAAGCACTGCCT-3′; superoxide dismutase 1 (SOD1)
forward, 5′-CTCACTCTCAGGAGACCATTGC-3′ and reverse,
5′-CCACAAGCCAAACGACTTCCAG-3′; sulfiredoxin 1 (SRXN1) forward,
5′-GCAGAGCCTCGTGGACACGAT-3′ and reverse,
5′-ATGGTCTCTCGCTGCAGTTGCT-3′; and β-actin forward,
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse,
5′-CTCCTTAATGTCACGCACGAT-3′. The means of three independent
biological replicates were calculated. The fold change was
calculated by dividing the mean expression of the treated groups by
that of the untreated control group. Statistical analysis of the
differences among cells treated with PD, cisplatin or their
combination was performed using one-way ANOVA followed by Tukey's
post hoc test.
ROS assay
HSC3 and FaDu cells were seeded into 6-well plates
at densities of 1×105 and 3×105 cells/well,
respectively. After 24 h of incubation, the cells were pretreated
with 100 nM rapamycin (37094; MilliporeSigma) at 37°C for 2 h as
indicated, followed by treatment with 10 µM PD and cisplatin,
either alone or in combination at 37°C for the designated time
points.
Based on preliminary time-course experiments
conducted to determine appropriate detection time points,
intracellular ROS levels were assessed at multiple time points (3,
6, 12, 24 and 48 h) following PD and cisplatin treatment. These
pilot experiments indicated that ROS accumulation peaked at ~12 h
in HSC3 cells and at 3 h in FaDu cells (data not shown).
After incubation, the culture medium was removed and
20 µM 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA; cat. no.
ab113851; Abcam) was added to each well to measure ROS levels. The
cells were then incubated at 37°C for 30 min, washed twice with
DPBS, detached using trypsin-EDTA and resuspended in DPBS, diluted
1:2 with DPBS, and adjusted to a final volume of 1 ml prior to
analysis. Flow cytometry analysis was performed using a FACSCalibur
flow cytometer (BD Biosciences) with CellQuest software (version
6.0; BD Biosciences).
Morphological analysis
HSC3 and FaDu cells were seeded at 5×104
and 1×105 cells, respectively. After 24 h of incubation,
cells were treated with PD (10 µM), cisplatin (10 µM), or the
combination at 37°C for 48 h. Cell morphology was then examined
under a light microscope to evaluate vesicular structure
accumulation.
Confocal microscopy
HSC3 and FaDu cells were seeded into 8-well plates
at a density of 5×103 cells/well. After 24 h of
incubation at 37°C, the cells were treated with 10 µM PD or 10 µM
cisplatin as a single treatment, or in combination, at 37°C. After
48 h of treatment, the cells were fixed with 4% paraformaldehyde at
room temperature for 15 min and washed three times with DPBS.
Permeabilization was then performed using 0.1% Triton X-100 diluted
in DPBS, followed by blocking with 5% BSA (cat. no. A0100-010;
GenDEPOT, LLC) at room temperature for 30 min. The cells were
incubated overnight at 4°C with primary antibodies against LC3B
(1:1,000; cat. no. 2775S; Cell Signaling Technology, Inc.) and
sequestosome 1 (SQSTM1)/p62 (1:1,000; cat. no. 5114S; Cell
Signaling Technology, Inc.). After incubation, the cells were
washed twice with DPBS. Secondary antibodies [Goat Anti-Rabbit IgG
H&L (Alexa Fluor® 488) cat. no. ab150077; Abcam]
were applied at a dilution of 1:500 in BSA at room temperature for
40 min, followed by three washes with PBS. The cells were then
stained with DAPI (Roche Diagnostics) at room temperature for 5
min. Cells were observed at a magnification of ×200 using a
confocal fluorescence microscope (Zeiss AG) in a dark room.
Fluorescence intensity analysis and puncta quantification were
performed using ZEN software (version 700; Zeiss AG), which is
integrated with the confocal imaging system.
Western blotting
Prior to western blotting, HNSCC cells were treated
with 10 µM PD and 10 µM cisplatin at 37°C for 48 h. When indicated,
Bafilomycin A1 (BafA1; 10 nM; cat. no. S1413; Selleck Chemicals), a
lysosomal inhibitor used as a positive control for late-stage
autophagy inhibition, was added 1 h prior to cell harvest. Lysates
from HNSCC cells were homogenized in RIPA buffer [1% Triton X-100,
1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl (pH
7.5) and 2 mM ethylenediaminetetraacetic acid (pH 8.0)] purchased
from Biosesang containing a protease inhibitor cocktail. The
protein concentration was then quantified using a Pierce Micro BCA
Protein Assay Kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Equal amounts of protein (10 µg per
lane) were mixed with loading dye (5X SDS-polyacrylamide gel
electrophoresis loading buffer; Intron Biotechnology, Inc.). After
boiling for 10 min, cell protein samples were added to each lane
and separated on a 10 or 15% SDS-polyacrylamide gel. Following
electrophoresis, proteins were transferred to polyvinylidene
difluoride membranes (MilliporeSigma) and blocked at room
temperature for 1 h in 5% skimmed milk and Tris-buffered saline
with 0.1% Tween-20. These membranes were then incubated with
appropriate primary antibodies overnight at 4°C. The following
antibodies were used: Anti-LC3A/B (1:1,000; cat. no. 12741S; Cell
Signaling Technology, Inc.), anti-SQSTM1/p62 (1:1,000; cat. no.
5114S; Cell Signaling Technology, Inc.), anti-poly(ADP-ribose)
polymerase (PARP; 1:1,000; cat. no. 9542S; Cell Signaling
Technology, Inc.), anti-caspase 3 (1:1,000; cat. no. 9662S; Cell
Signaling Technology, Inc.) and anti-β-actin (1:5,000; cat. no.
47778; Santa Cruz Biotechnology, Inc.). The membranes were then
washed with TBST containing 0.1% Tween-20 and incubated with
HRP-conjugated secondary antibodies for 1 h at room temperature.
Anti-rabbit IgG (cat. no. 7074S; Cell Signaling Technology, Inc.)
and anti-mouse IgG (cat. no. 7076S; Cell Signaling Technology,
Inc.) were used at dilutions of 1:5,000 and 1:10,000, respectively.
The protein-antibody complexes were detected using enhanced
chemiluminescence (cat. no. RPN2232; Cytiva) according to the
manufacturer's protocol. Band intensities were quantified by
densitometric analysis using ImageJ software (version 1.54;
National Institutes of Health) and normalized to β-actin.
Flow cytometry
For apoptosis analysis, HSC3 and FaDu cells were
treated with PD (10 µM), cisplatin (10 µM), or their combination
for 48 h at 37°C. Cells were washed with PBS, trypsinized and
centrifuged at 1,000 × g for 5 min at 4°C. The cell pellets were
washed with cold PBS and centrifuged again under the same
conditions. Cells were resuspended in 1× binding buffer and stained
with annexin V-FITC for 15 min at room temperature in the dark,
followed by the addition of propidium iodide (PI) immediately prior
to flow cytometric analysis under cold conditions. Annexin
V-FITC/PI staining was performed according to the manufacturer's
protocol using an apoptosis detection kit (Biobud; cat. no.
LS-02-100; http://www.biobud.com/). Apoptotic
cells were measured using a FACSCalibur cell analyzer (BD
Biosciences) and data were analyzed using CellQuest software
(version 6.0; BD Biosciences), and the apoptotic rate was
calculated as the sum of early apoptotic (annexin
V-positive/PI-negative), late apoptotic (annexin
V-positive/PI-positive), and necrotic (annexin
V-negative/PI-positive) cell populations.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD) from at least three independent experiments. The 50%
inhibitory concentration (IC50) was calculated from
dose-response curves using GraphPad Prism software (version 5.01;
Dotmatics). To evaluate the synergistic effect between PD and
cisplatin, CompuSyn software (ComboSyn, Inc.; version not specified
by the manufacturer) was used to calculate the combination index
(CI). A CI value <1 indicated synergy, CI=1 represented an
additive effect and CI>1 suggested antagonism. Statistical
analysis for multiple-group comparisons was performed using one-way
ANOVA followed by Tukey's post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Synergistic effect of PD and cisplatin
on HNSCC cell viability
To evaluate the cytotoxic and synergistic effects of
PD and cisplatin on HNSCC cells, viability was assessed in HSC3 and
FaDu cells following treatment with various concentrations of PD
(5, 10 and 15 µM) or cisplatin (5 and 10 µM), either alone or in
combination, for 48 h. The combined treatment with PD and cisplatin
induced cell death in a dose-dependent manner (Fig. 1A).
CI analysis revealed that all tested combinations
exhibited CI values <1, confirming a synergistic cytotoxic
effect of PD and cisplatin in both HSC3 and FaDu cells (Fig. 1B). These results suggested that,
despite their limited cytotoxic effects when used as single
treatments, the combination of PD and cisplatin markedly enhanced
cellular sensitivity to treatment, supporting a sensitizing role of
PD toward cisplatin.
In addition, to evaluate the potential cytotoxic
effects of PD on normal cells, HaCaT human keratinocytes were
treated with PD and cell viability was assessed. PD treatment did
not markedly reduce cell viability at concentrations ranging
between 0 and 10 µM, indicating minimal cytotoxic effects in normal
cells (Fig. S1).
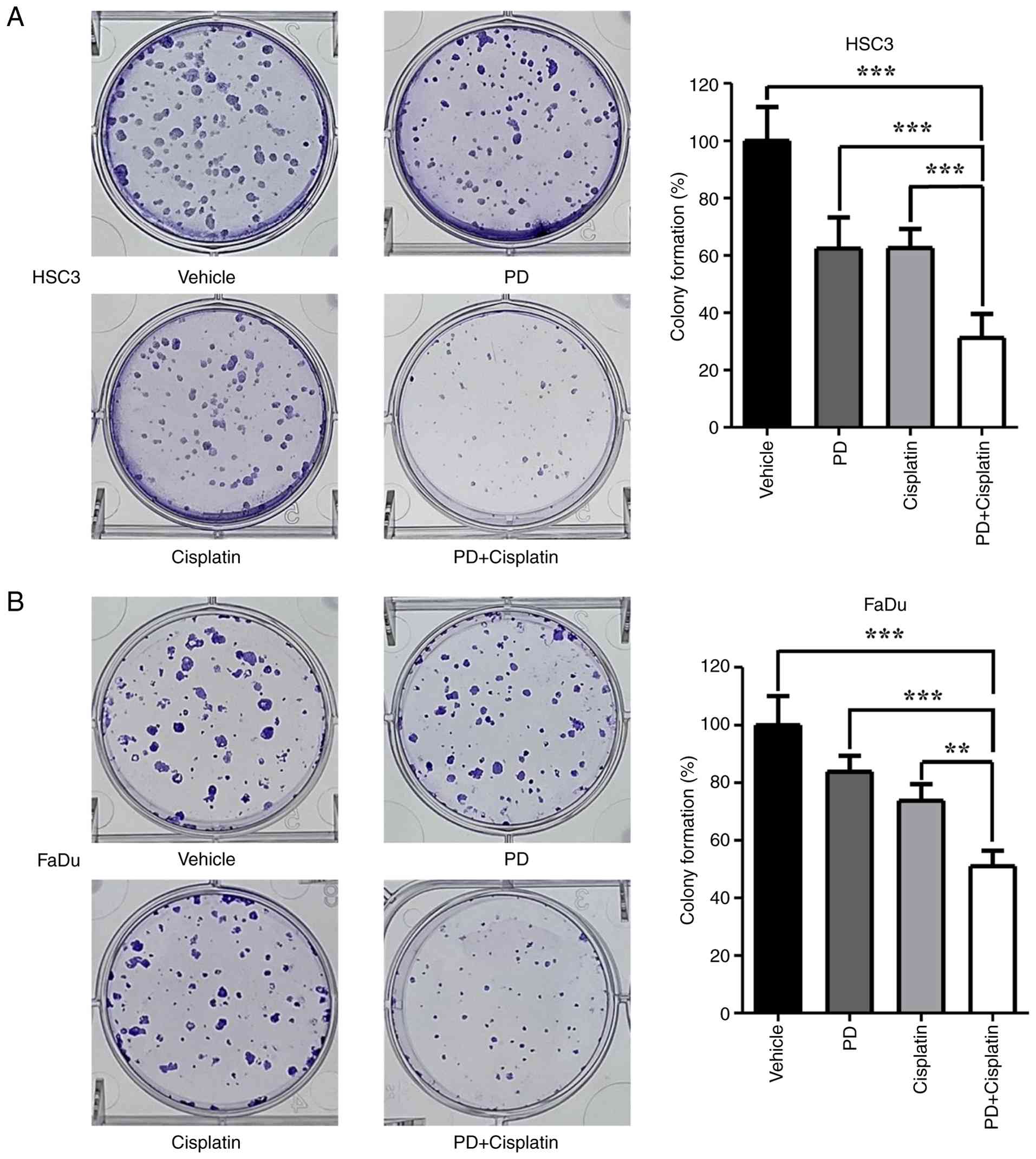
Inhibition of colony formation in
HNSCC cells by PD and cisplatin combination treatment
To assess long-term clonogenic survival beyond
short-term viability, colony formation assays were conducted in
HSC3 and FaDu cells treated with PD and cisplatin as a single
treatment or in combination. HSC3 and FaDu cells were treated with
PD and cisplatin (5 µM each) for 7–10 days. The results showed that
while PD and cisplatin alone reduced colony formation, the extent
of inhibition was substantially greater with combined treatment
using PD and cisplatin in both HSC3 and FaDu cells (Fig. 2A and B). This reduction in colony
formation was statistically significant (P<0.05), indicating a
strong synergistic effect of the combined treatment. Furthermore,
the combination treatment resulted in fewer and smaller colonies
compared with single-drug treatments and the control group. These
findings indicated that PD increased the sensitivity of HNSCC cells
to cisplatin and contributed to the suppression of colony
formation. Collectively, the results suggested that combined
treatment with PD and cisplatin markedly impairs the long-term
clonogenic survival of HNSCC cells, indicating sustained inhibition
of tumor cell proliferative potential beyond short-term cytotoxic
effects.
ROS-associated cytotoxic effect of PD
and cisplatin combination treatment
Several studies have reported that cisplatin induces
DNA damage in cancer cells, leading to an increase in intracellular
ROS levels (31–33). Additionally, numerous studies have
demonstrated that PD also promotes ROS accumulation (34,35).
Considering these studies, the present study aimed to determine
whether the suppression of cell proliferation and induction of cell
death observed following PD and cisplatin combination treatment in
HNSCC cells were associated with increased ROS levels.
HO-1, NQO1, SOD1 and SRXN1 are well-known
antioxidant enzymes involved in maintaining cellular redox
homeostasis (36,37). To evaluate the effect of PD and
cisplatin on the expression of these antioxidant genes, the present
study conducted RT-qPCR in HSC3 and FaDu cells following treatment
with either drug alone or in combination. The expression levels of
HO-1, NQO1, SOD1 and SRXN1 showed variable changes following single
treatment, with some genes exhibiting increased or decreased
expression compared with the vehicle control, and no consistent
pattern was observed. By contrast, combination treatment with PD
and cisplatin resulted in a significant decrease in the expression
of all four antioxidant genes compared with either single treatment
alone in both HSC3 and FaDu cells (Fig.
3A and B), indicating a pronounced impairment of the cellular
antioxidant defense system.
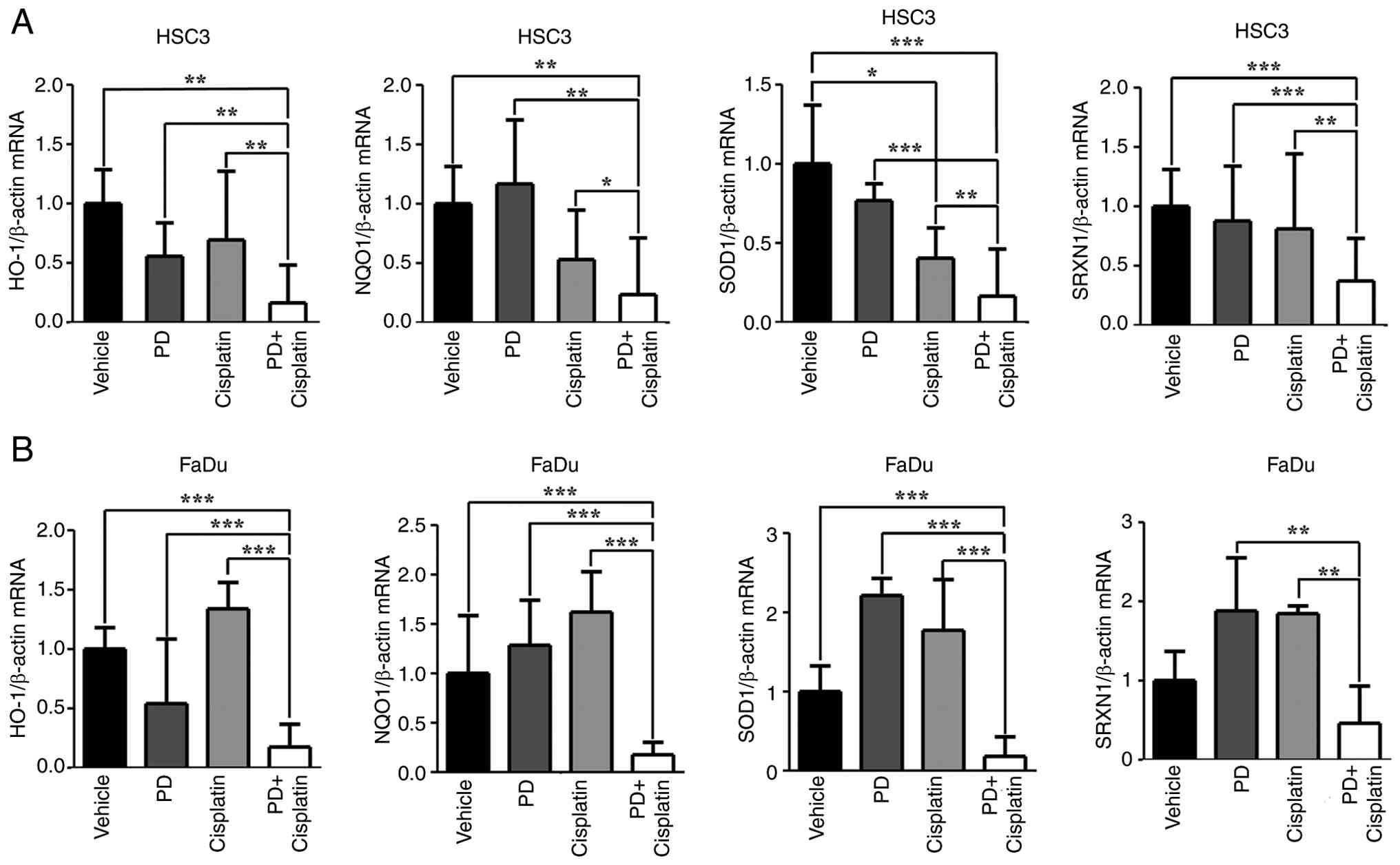 | Figure 3.Combination of PD and cisplatin
downregulates antioxidant gene expression in head and neck squamous
cell carcinoma cells. (A) HSC3 and (B) FaDu cells were treated with
PD (10 µM), cisplatin (10 µM) or combination treatment for 12 h
(HSC3 cells) or 3 h (FaDu cells). mRNA expression levels of
antioxidant genes (HO-1, NQO1, SOD1 and SRXN1) were measured by
reverse transcription-quantitative PCR and normalized to β-actin
expression. Combination treatment markedly reduced the expression
of all four genes compared with single treatments. Statistical
significance was determined by one-way ANOVA followed by Tukey's
post hoc test. *P<0.05, **P<0.01, ***P<0.001. PD,
Platycodin D; HO-1, heme oxygenase-1; NQO1, NAD(P)H quinone
dehydrogenase 1; SOD1, superoxide dismutase 1; SRXN1, sulfiredoxin
1. |
To further assess the regulation of ROS levels by PD
and cisplatin, DCF-DA staining followed by flow cytometry was
performed in HSC3 and FaDu cells treated with each drug (10 µM), as
a single treatment or in combination. No significant differences in
ROS levels were observed between the vehicle control and single
treatment groups. By contrast, ROS levels were significantly
increased in the combination treatment group compared with those in
the individual drug treatment groups (Fig. 4A and B). Collectively, these results
indicated that PD and cisplatin synergistically enhanced ROS
accumulation in HNSCC cells, potentially contributing to their
synergistic cytotoxic effects. The significant downregulation of
antioxidant genes along with increased ROS levels demonstrated a
potential mechanism by which combination treatment exacerbated
oxidative stress, ultimately leading to enhanced cell death and
impaired proliferation.
 | Figure 4.Combination of PD and cisplatin
synergistically increases ROS levels in head and neck squamous cell
carcinoma cells. (A) HSC3 and (B) FaDu cells were treated with PD
(10 µM), cisplatin (10 µM) or combination treatment, and
intracellular ROS levels were assessed by DCF-DA staining.
Combination treatment markedly increased ROS levels compared with
single treatments. Statistical significance was determined by
one-way ANOVA followed by Tukey's post hoc test. In the graphs,
vehicle, PD, cisplatin and combination treatment groups are
represented by black, green, blue and red lines, respectively. FL
indicates fluorescence intensity measured by flow cytometry and
reflects intracellular ROS levels. *P<0.05, **P<0.01,
***P<0.001. PD, Platycodin D; ROS, reactive oxygen species;
DCF-DA, 2′,7′-dichlorodihydrofluorescein diacetate. |
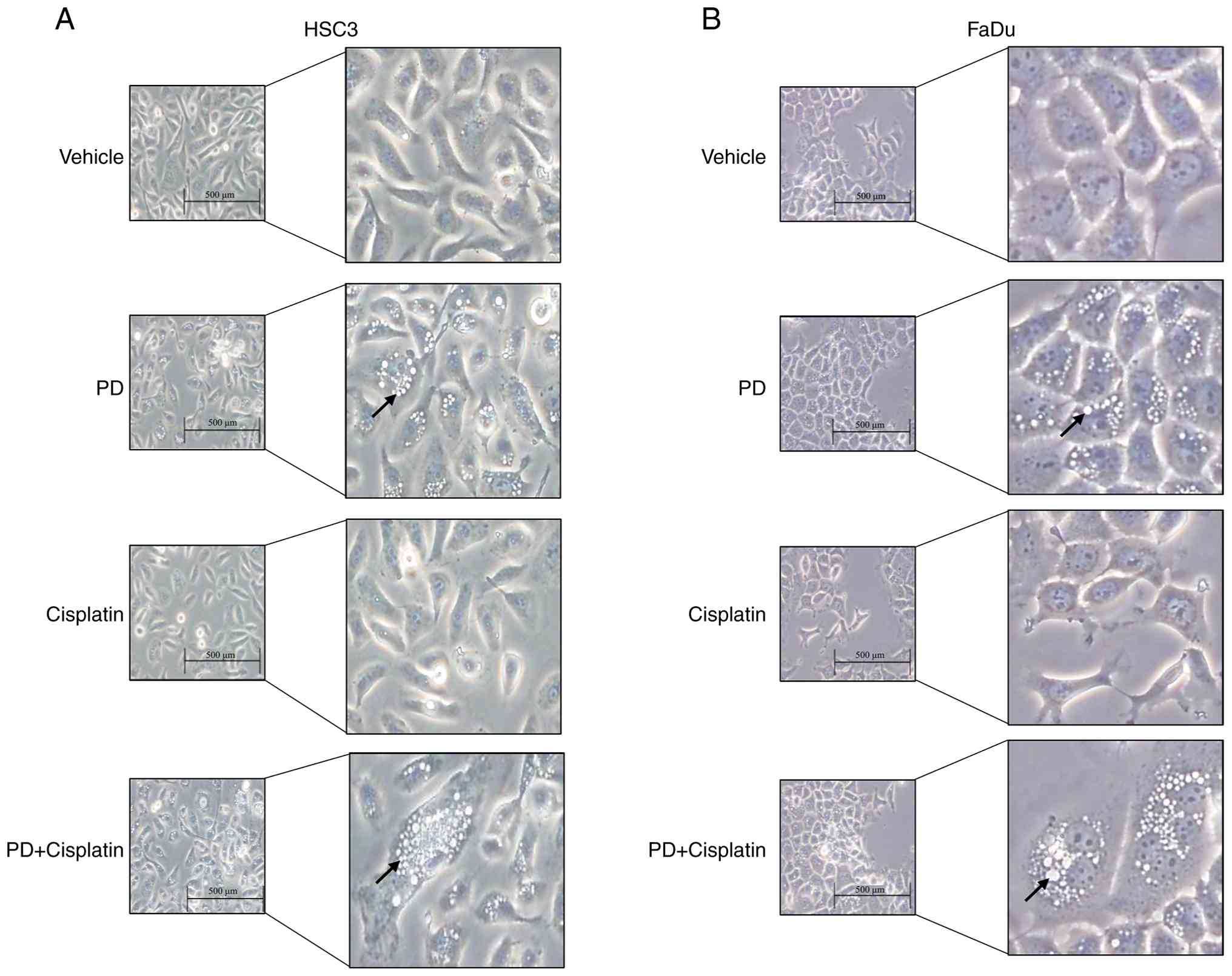
Induction of autophagy arrest
following combination treatment with PD and cisplatin in HNSCC
cells
Previous experiments have shown that ROS serve a
crucial role in cellular stress responses and the regulation of
autophagy (38). To analyze the
effect of PD and cisplatin combination treatment on cellular
phenotype modifications, HSC3 and FaDu cells were treated for 48 h,
followed by microscopy to observe cell morphology modifications. An
excessive increase in vesicular structures was observed in cells
treated with PD alone or in combination with cisplatin (Fig. 5). To identify the type of these
vesicles, additional experiments were conducted (Fig. 5).
Since PD has already been reported to be associated
with autophagy (39), the present
study considered the possibility that the increased ROS induced by
PD and cisplatin combination treatment could influence the
autophagy process. ROS can activate the early signaling pathways of
autophagy; however, excessive ROS accumulation can impair
autophagic flux, resulting in autophagy arrest (38). To determine whether ROS accumulation
induced by combined PD and cisplatin treatment disrupts autophagic
flux progression, leading to flux arrest, the present study
analyzed changes in key autophagy markers.
During the initiation of autophagy, the cytosolic
form of LC3 (LC3-I) is lipidated through conjugation with
phosphatidylethanolamine to generate LC3-II, which associates with
the autophagosomal membrane and is essential for autophagosome
formation.. In the later stages, p62 functions as an adaptor
protein that mediates the sequestration of ubiquitinated proteins
into autophagosomes and is subsequently degraded upon fusion of
autophagosomes with lysosomes (40). Therefore, LC3 and p62 serve as major
markers for evaluating autophagy. HSC3 and FaDu cells were treated
with 10 µM PD and cisplatin for 48 h, followed by staining with
LC3B and p62 antibodies and visualization using confocal
microscopy. The results revealed that PD treatment significantly
increased LC3B levels in both cell lines, with a further and more
pronounced increase observed following combined treatment with
cisplatin (Fig. 6A and C).
Furthermore, p62 expression was significantly increased by PD
treatment and was markedly further elevated in the combination
treatment group compared with the single treatment groups (Fig. 6B and D). The increase in p62
expression suggested excessive accumulation of ubiquitinated
proteins, indicating that combination treatment may disrupt the
degradation process of autophagosomes.
To confirm these findings, western blot analysis was
performed, which showed results consistent with the results of
confocal microscopy. PD treatment induced the accumulation of LC3B,
whereas combination treatment with cisplatin resulted in LC3B
accumulation accompanied by increased p62 levels compared with the
single treatment groups (Fig. 7A and
B). The accumulation of LC3B can result from either enhanced
autophagy initiation or impaired degradation in the later autophagy
stages, while p62 accumulation occurs when the fusion of
autophagosomes with lysosomes is blocked, preventing degradation
(41,42). Given the marked increase in p62
expression observed in the present study, cells were treated with
the lysosomal inhibitor Bafilomycin A1, a well-established positive
control for late-stage autophagy inhibition (43), to further validate this
interpretation. Bafilomycin A1 treatment resulted in marked
accumulation of both LC3B and p62, mimicking the expression
patterns observed in PD and cisplatin co-treated cells (Fig. 7A and B). Collectively, these
findings suggested that combination treatment with PD and cisplatin
induced autophagy arrest by inhibiting autophagosome degradation at
the late stage of autophagy, thereby contributing to the
suppression of HNSCC cell proliferation.
The present study examined whether autophagy
modulation serves a causal role in ROS accumulation induced by PD
and cisplatin. HSC3 and FaDu cells were pretreated with the
autophagy activator rapamycin prior to PD and cisplatin treatment,
followed by DCF-DA staining and flow cytometry. Consistent with
previous observations, treatment with PD or cisplatin alone
resulted in a slight increase in ROS levels, whereas combination
treatment markedly enhanced ROS accumulation. Notably, rapamycin
pretreatment markedly reduced ROS levels induced by PD and
cisplatin co-treatment (Fig. S2A).
This reduction in ROS upon autophagy activation suggested that
autophagic flux inhibition contributed to excessive oxidative
stress under combination treatment conditions. These findings
indicated that impaired autophagy served a functional role in ROS
accumulation induced by PD and cisplatin, supporting a mechanistic
association between autophagic flux arrest and enhanced oxidative
stress in HNSCC cells.
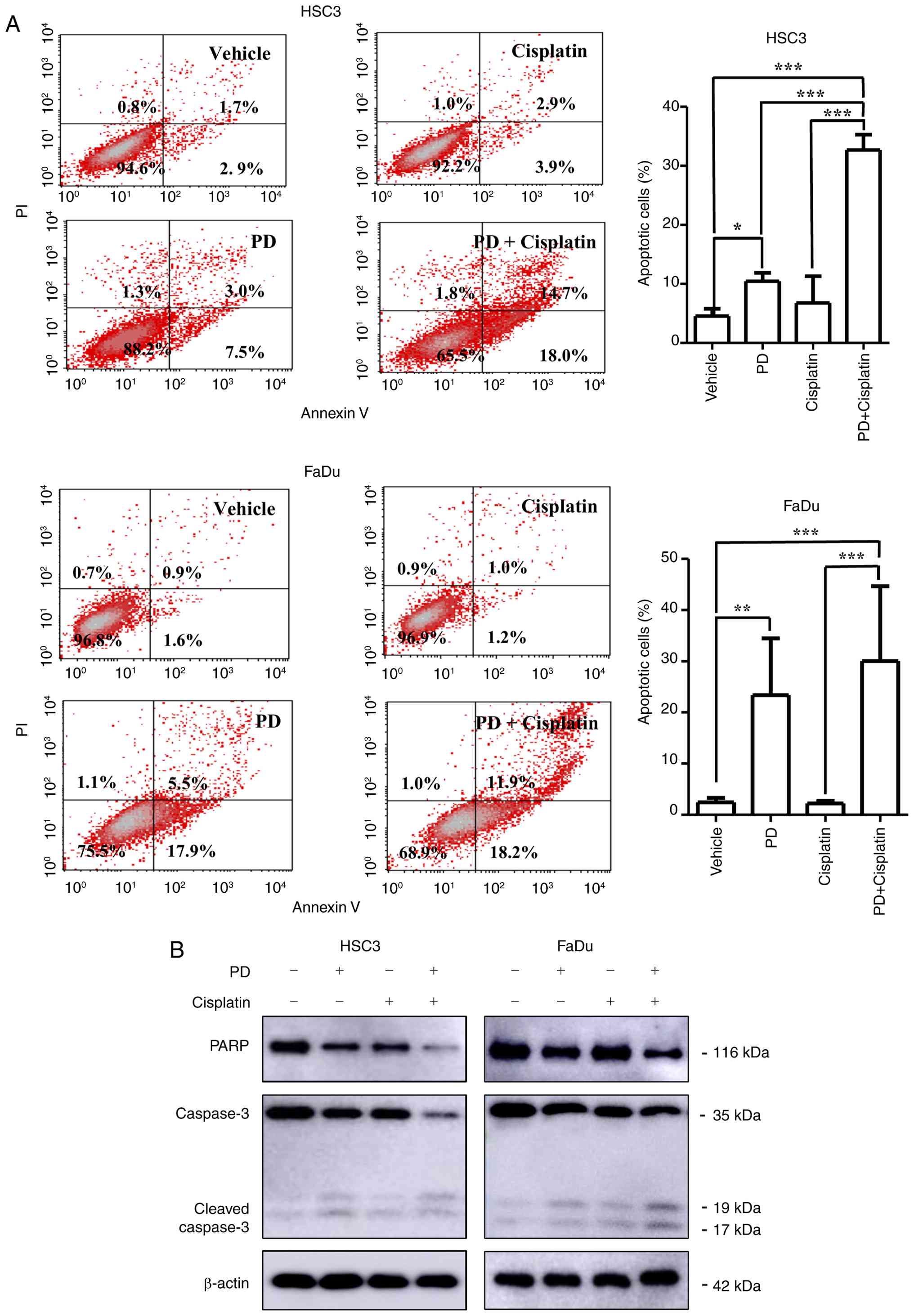
Autophagy arrest-mediated apoptotic
cell death induced by PD and cisplatin co-treatment
Given that ROS accumulation and autophagic flux
inhibition are known to be closely associated with cellular stress
(44–46), the present study investigated
whether the reduced cell viability observed following PD and
cisplatin co-treatment was mediated by the induction of apoptosis.
HSC3 and FaDu cells were treated with 10 µM PD, cisplatin or their
combination for 48 h and apoptosis was assessed by annexin V/PI
double staining. In HSC3 cells, the proportion of apoptotic cells
increased from 10.46±1.42% with PD treatment and 6.79±4.51% with
cisplatin treatment to 32.69±2.60% following combination treatment,
which was statistically significant. Similarly, in FaDu cells, the
proportion of apoptotic cells was elevated from 23.41±11.08% with
PD treatment and 2.23±0.51% with cisplatin treatment to
30.04±14.64% following combined treatment, which was also
statistically significant (Fig.
8A). These results were consistent with the cell viability
assay findings.
Apoptosis is regulated by the activation of caspase
3, which subsequently cleaves downstream substrates such as PARP.
Therefore, the present study analyzed the levels of caspase 3 and
PARP as representative apoptosis markers (47). Combined treatment with PD and
cisplatin resulted in a decrease in the levels of total caspase 3
and PARP, accompanied by an increase in cleaved-caspase 3 levels,
indicating enhanced apoptotic cell death (Fig. 8B).
Collectively, these results demonstrated that
co-treatment with PD and cisplatin induced apoptotic cell death
through ROS accumulation and autophagic flux inhibition in HNSCC
cells.
Discussion
The present study aimed to assess whether the
combination of PD and cisplatin enhanced antitumor efficacy and to
evaluate the potential of PD as a sensitizer to cisplatin. The
findings of the present study were as follows: i) Combination
treatment with PD and cisplatin reduced cell viability and a
synergistic effect between the two agents was confirmed; ii) the
combination treatment also markedly suppressed colony formation of
HNSCC cells; iii) combination treatment led to a significant
increase in intracellular ROS levels; iv) furthermore, it impaired
autophagic flux, as evidenced by the accumulation of LC3B and p62
proteins, indicating defective autophagosome degradation; and v)
combination treatment with PD and cisplatin induced autophagy
arrest-associated cell death in HNSCC cells. Collectively, the
results suggested that the combination of PD and cisplatin induced
cell death in HNSCC cells by increasing oxidative stress and
inhibiting autophagic flux.
HNSCC is primarily treated with definitive
radiotherapy or surgery followed by adjuvant chemotherapy to
prevent recurrence (4). Cisplatin,
a platinum-based chemotherapeutic agent, is widely utilized as a
key component of concurrent chemoradiotherapy in patients with
HNSCC (8). However, cisplatin
treatment is associated with adverse side effects, including hair
loss, nephrotoxicity and immunosuppression, which can compromise
long-term therapeutic efficacy and markedly reduce the quality of
life of patients (48).
Furthermore, the high level of drug resistance reduces therapeutic
efficacy and ultimately leads to tumor recurrence (49). As a result of these therapeutic
limitations, the 5-year survival rate for patients with HNSCC
remains <50% worldwide, based on global epidemiological data
reported in recent decades (50).
Therefore, there is a critical need to develop effective
therapeutic strategies that can enhance antitumor effects while
reducing cytotoxicity toward normal cells. In this context, the
present study also evaluated the cytotoxic effects of PD in normal
human keratinocytes. Notably, PD treatment did not markedly reduce
cell viability in normal cells at the concentrations used, as
assessed using an MTT assay. This finding suggested that PD exerted
limited cytotoxicity toward normal cells while enhancing antitumor
effects in HNSCC cells, highlighting its potential as a selective
cisplatin sensitizer.
Cisplatin is a widely used chemotherapeutic agent
that induces cell death primarily by causing DNA damage and
disrupting redox homeostasis, leading to increased intracellular
ROS levels (31,51,52).
Cancer cells respond to cytotoxic stress by activating protective
mechanisms such as autophagy. Autophagy is a catabolic process that
degrades and recycles damaged organelles and proteins to maintain
cellular homeostasis (51–53). Therefore, autophagy may serve as a
crucial target for regulating the therapeutic efficacy of
anticancer treatments.
PD, a triterpenoid saponin derived from the root of
PG, has been reported to demonstrate pharmacological effects across
various types of cancer, including lung and colorectal cancers
(18,54), particularly through
autophagy-related anticancer mechanisms (55). However, the effects of PD have not
been fully examined in HNSCC, particularly in combination with
cisplatin. Based on this, we hypothesized that PD could sensitize
HNSCC cells to cisplatin-induced cytotoxicity by inhibiting
autophagy, and a series of in vitro experiments were
conducted to verify this hypothesis. Combination treatment with PD
and cisplatin markedly reduced cell viability compared with single
treatments, demonstrating a synergistic effect. Microscopic
observation showed a substantial increase in intracellular vesicle
structures in cells treated with the combination of PD and
cisplatin compared with the vehicle control and single treatments,
which were assumed to be associated with autophagic activity.
Accordingly, the expression of the autophagy-related markers LC3B
and p62 was analyzed. The levels of both proteins were upregulated
following combination treatment, with a particularly notable
accumulation of p62.
Autophagy is initiated in response to cellular
stress, leading to the formation of double-membraned autophagosomes
that enclose damaged organelles and proteins. These autophagosomes
subsequently fuse with lysosomes to form autolysosomes, where the
enclosed contents are degraded and recycled (56). LC3B is a key component of the
autophagosome membrane and serves a crucial role in its formation,
while p62 functions as a selective cargo adaptor that is typically
degraded during autophagic flux (56,57).
Therefore, accumulation of p62 indicates disrupted autophagic
degradation. The present study observed increased levels of both
LC3B and p62 following combination treatment with PD and cisplatin,
suggesting that PD inhibited autophagosome-lysosome fusion and
disrupted autophagic flux, thereby impairing the clearance of
damaged cellular components. This interpretation was further
supported by the observation that the effects of PD on LC3B and p62
accumulation were comparable to those induced by Bafilomycin A1, a
late-stage autophagy inhibitor that blocks autophagosome-lysosome
fusion.
Cisplatin induces the generation of ROS, thereby
promoting cytotoxicity through oxidative stress (31), while autophagy can serve as a
protective mechanism by eliminating ROS and supporting cell
survival (58). However, in the
present study, PD-mediated inhibition of autophagy may have
compromised this protective mechanism, leading to the accumulation
of cisplatin-induced ROS within the cells. To verify this
hypothesis, the present study analyzed the mRNA expression levels
of antioxidant genes such as HO-1, NQO1, SOD1 and SRXN1 (59–61).
The expression of these representative antioxidant markers was
markedly decreased following combination treatment, suggesting that
cancer cells failed to effectively activate their antioxidant
defense system in response to severe oxidative stress. This
impaired response may have increased the susceptibility of the
cells to ROS-mediated damage. Furthermore, measurement of
intracellular ROS levels using DCF-DA staining followed by FACS
analysis demonstrated a significant increase in ROS levels in cells
treated with the combination of PD and cisplatin. These results
indicated that PD-mediated inhibition of autophagic flux disrupted
the clearance of cisplatin-induced ROS, thereby exacerbating
oxidative stress and ultimately promoting cell death. To further
support this hypothesis, the present study observed that activation
of autophagy by rapamycin markedly reduced ROS accumulation induced
by PD and cisplatin co-treatment. Although these findings are
promising, they require further validation in vivo. Further
studies should assess the pharmacokinetics, toxicity and
therapeutic effects of PD in animal models of HNSCC, in addition to
further clarifying the molecular mechanisms associated with
autophagy inhibition. Nevertheless, the present study provided a
compelling rationale for the development of PD-based combination
therapies as a novel strategy to enhance the efficacy of current
chemotherapeutic regimens in HNSCC.
In conclusion, the present study proposed a
mechanism in which PD inhibited autophagic flux, thereby enhancing
the cytotoxic effects of cisplatin-induced ROS stress. This led to
ROS accumulation, downregulation of antioxidant gene expression,
increased oxidative stress and ultimately accelerated cell death.
These findings suggested that the combination of PD (10 µM) and
cisplatin (10 µM), selected based on cell viability assays to allow
evaluation of synergistic effects, may exhibit a potent synergistic
effect in the treatment of HNSCC and provide a potential strategy
for the development of innovative combination therapies.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank Dr. Jin-Woo Lee (Kyung Hee Medical
Center, Seoul, South Korea) for kindly providing the HaCaT cell
line.
Funding
The present study was supported by the National Research
Foundation of Korea, funded by the Korean government, under grant
nos. RS-2020-NR049559, RS-2024-00461726 and RS-2025-02243112.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
MB and YGE conceived the study. MGJ and SGK
developed the methodology. MB conducted the experiments. MKL and
SIK performed the data analysis. MB drafted the original manuscript
and YGE reviewed and edited the manuscript. YL and HJ contributed
to data visualization and participated in data interpretation. YCL
and JWL contributed to data validation and interpretation and
critically reviewed the manuscript for important intellectual
content. YGE supervised the project. MB and YGE confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kim SI, Woo SR, Noh JK, Lee MK, Lee YC,
Lee JW, Ko SG and Eun YG: Clinical significance of FAT1 gene
mutation and mRNA expression in patients with head and neck
squamous cell carcinoma. Mol Oncol. 16:1661–1679. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seol S, Choi JR, Choi B, Kim S, Jeon JY,
Park KN, Park JH, Park MW, Eun YG, Park JJ, et al: Effect of statin
use on head and neck cancer prognosis in a multicenter study using
a Common Data Model. Sci Rep. 13:197702023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barsouk A, Aluru JS, Rawla P, Saginala K
and Barsouk A: Epidemiology, risk factors, and prevention of head
and neck squamous cell carcinoma. Med Sci (Basel).
11:422023.PubMed/NCBI
|
|
4
|
Bourdier S, Fisch AS, Alp KM, Das R,
Mertins P and Tinhofer I: High Ano1 expression as key driver of
resistance to radiation and cisplatin in HPV-negative head and neck
squamous cell carcinoma. Sci Rep. 15:15552025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee MK, Noh JK, Woo SR, Kong M, Lee YC,
Lee JW, Ko SG and Eun YG: Prognostic significance of the
BIRC2-BIRC3 gene signature in head and neck squamous cell
carcinoma. Cancer Genomics Proteomics. 19:591–605. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rauf S, Ullah S, Abid MA, Ullah A, Khan G,
Khan AU, Ahmad G, Ijaz M, Ahmad S and Faisal S: A computational
study of gene expression patterns in head and neck squamous cell
carcinoma using TCGA data. Future Sci OA. 10:23805902024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Noh JK, Woo SR, Yun M, Lee MK, Kong M, Min
S, Kim SI, Lee YC, Eun YG and Ko SG: SOD2- and NRF2-associated gene
signature to predict radioresistance in head and neck cancer.
Cancer Genomics Proteomics. 18:675–684. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu T, Yang Y, Chen Z, Wang J, Wang X,
Zheng Y, Wang C, Wang Y, Zhu Z, Ding X, et al: TNFAIP2 confers
cisplatin resistance in head and neck squamous cell carcinoma via
KEAP1/NRF2 signaling. J Exp Clin Cancer Res. 42:1902023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tong T, Qin X, Jiang Y, Guo H, Wang X, Li
Y, Xie F, Lu H, Zhai P, Ma H and Zhang J: A novel CREB5/TOP1MT axis
confers cisplatin resistance through inhibiting mitochondrial
apoptosis in head and neck squamous cell carcinoma. BMC Med.
20:2312022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu S, Ren B, Gao H, Liao S, Zhai YX, Li
S, Su XJ, Jin P, Stroncek D, Xu Z, et al: Over-expression of BAG-1
in head and neck squamous cell carcinomas (HNSCC) is associated
with cisplatin-resistance. J Transl Med. 15:1892017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khayer N, Shabani S, Jalessi M, Joghataei
MT and Mahjoubi F: A dynamic co-expression approach reveals Gins2
as a potential upstream modulator of HNSCC metastasis. Sci Rep.
15:33222025. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hering K, Kuhnt T, Kossack N, Richter LM,
Schultze M, Osowski U, Henkel L, Gaupel AC, Solbes MN, Zolyniak B,
et al: Real-world treatment patterns and survival outcomes in
patients with locally advanced squamous cell carcinoma of the head
and neck in Germany using claims data. Front Oncol. 15:15473112025.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leonard BC, Lee ED, Bhola NE, Li H,
Sogaard KK, Bakkenist CJ, Grandis JR and Johnson DE: ATR inhibition
sensitizes HPV- and HPV+ head and neck squamous cell carcinoma to
cisplatin. Oral Oncol. 95:35–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han SH, Lee JH, Woo JS, Jung GH, Jung SH,
Han EJ, Park YS, Kim BS, Kim SK, Park BK and Jung JY: Platycodin D
induces apoptosis via regulating MAPK pathway and promotes
autophagy in colon cancer cell. Biomed Pharmacother.
172:1162162024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Androutsopoulos V, Arroo RR, Hall JF,
Surichan S and Potter GA: Antiproliferative and cytostatic effects
of the natural product eupatorin on MDA-MB-468 human breast cancer
cells due to CYP1-mediated metabolism. Breast Cancer Res.
10:R392008. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim TY, Jeon S, Jang Y, Gotina L, Won J,
Ju YH, Kim S, Jang MW, Won W, Park MG, et al: Platycodin D, a
natural component of Platycodon grandiflorum, prevents both
lysosome- and TMPRSS2-driven SARS-CoV-2 infection by hindering
membrane fusion. Exp Mol Med. 53:956–972. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang CH, Baskaran R, Ng SS, Wang TF, Li
CC, Ho TJ, Hsieh DJ, Kuo CH, Chen MC and Huang CY: Platycodin D
confers oxaliplatin Resistance in Colorectal Cancer by activating
the LATS2/YAP1 axis of the hippo signaling pathway. J Cancer.
14:393–402. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Tian S, Yi B, Feng Z, Chu T, Liu J,
Zhang C, Zhang S and Wang Y: Platycodin D sensitizes KRAS-mutant
colorectal cancer cells to cetuximab by inhibiting the PI3K/Akt
signaling pathway. Front Oncol. 12:10461432022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi YJ, Lee SJ, Kim HI, Lee HJ, Kang SJ,
Kim TY, Cheon C and Ko SG: Platycodin D enhances LDLR expression
and LDL uptake via down-regulation of IDOL mRNA in hepatic cells.
Sci Rep. 10:198342020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee SJ, Choi YJ, Kim HI, Moon HE, Paek SH,
Kim TY and Ko SG: Platycodin D inhibits autophagy and increases
glioblastoma cell death via LDLR upregulation. Mol Oncol.
16:250–268. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi B, Chen J, Guo H, Shi X, Tai Q, Chen
G, Yao H, Mi X, Zhong R, Lu Y, et al: ACOX1 activates autophagy via
the ROS/mTOR pathway to suppress proliferation and migration of
colorectal cancer. Sci Rep. 15:29922025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kong L, Deng J, Zhou X, Cai B, Zhang B,
Chen X, Chen Z and Wang W: Sitagliptin activates the p62-Keap1-Nrf2
signalling pathway to alleviate oxidative stress and excessive
autophagy in severe acute pancreatitis-related acute lung injury.
Cell Death Dis. 12:9282021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang T, Yang Q, Lai Q, Zhao J, Nie L, Liu
S, Yang J and Chu C: AP39 inhibits ferroptosis by inhibiting
mitochondrial autophagy through the PINK1/parkin pathway to improve
myocardial fibrosis with myocardial infarction. Biomed
Pharmacother. 165:1151952023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo Y, Liu R, Zhang H, Wang H, Yin H, Tian
G, Wang B, Yan Y, Ding Z, Dai J, et al: Amantadine against glioma
via ROS-mediated apoptosis and autophagy arrest. Cell Death Dis.
15:8342024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang S, Huang Y, Pi S, Chen H, Ye F, Wu
C, Li L, Ye Q, Lin Y and Su Z: Autophagy-amplifying nanoparticles
evoke immunogenic cell death combined with anti-PD-1/PD-L1 for
residual tumors immunotherapy after RFA. J Nanobiotechnology.
21:3602023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan J, Ren D, Wang J, Liu X, Zhang H, Wu M
and Yang G: Bruceine D induces lung cancer cell apoptosis and
autophagy via the ROS/MAPK signaling pathway in vitro and in vivo.
Cell Death Dis. 11:1262020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Folkerts H, Hilgendorf S, Vellenga E,
Bremer E and Wiersma VR: The multifaceted role of autophagy in
cancer and the microenvironment. Med Res Rev. 39:517–560. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin L, Chen Y, Cheng D, He Z, Shi X, Du B,
Xi X, Gao Y and Guo Y: YAP inhibits autophagy and promotes
progression of colorectal cancer via upregulating Bcl-2 expression.
Cell Death Dis. 12:4572021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao Q, Liu Y, Zhong J, Bi Y, Liu Y, Ren
Z, Li X, Jia J, Yu M and Yu X: Pristimerin induces apoptosis and
autophagy via activation of ROS/ASK1/JNK pathway in human breast
cancer in vitro and in vivo. Cell Death Discov. 5:1252019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang X, Liao X, Wang M, Liu J, Han J, An
D, Zheng T, Wang X, Cheng H and Liu P: Inhibition of
palmitoyltransferase ZDHHC12 sensitizes ovarian cancer cells to
cisplatin through ROS-mediated mechanisms. Cancer Sci.
115:1170–1183. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie X, Wu F, Tian J, Liu Z, He H, Bao D,
Li G, Li H, Chen J, Lai Y, et al: Pyrocatechol Alleviates
Cisplatin-Induced acute kidney injury by inhibiting ROS production.
Oxid Med Cell Longev. 2022:21586442022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li P, Li D, Lu Y, Pan S, Cheng F, Li S,
Zhang X, Huo J, Liu D and Liu Z: GSTT1/GSTM1 deficiency aggravated
cisplatin-induced acute kidney injury via ROS-triggered
ferroptosis. Front Immunol. 15:14572302024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fu Y, Xin Z, Liu B, Wang J, Wang J, Zhang
X, Wang Y and Li F: Platycodin D inhibits inflammatory response in
LPS-Stimulated primary rat microglia cells through activating
LXRα-ABCA1 signaling pathway. Front Immunol. 8:19292017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeng CC, Zhang C, Yao JH, Lai SH, Han BJ,
Li W, Tang B, Wan D and Liu YJ: Platycodin D induced apoptosis and
autophagy in PC-12 cells through mitochondrial dysfunction pathway.
Spectrochim Acta A Mol Biomol Spectrosc. 168:199–205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ngo V and Duennwald ML: Nrf2 and oxidative
stress: A general overview of mechanisms and implications in human
disease. Antioxidants (Basel). 11:23452022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kanzaki H, Shinohara F, Kanako I,
Yamaguchi Y, Fukaya S, Miyamoto Y, Wada S and Nakamura Y: Molecular
regulatory mechanisms of osteoclastogenesis through cytoprotective
enzymes. Redox Biol. 8:186–191. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chang TM, Chi MC, Chiang YC, Lin CM, Fang
ML, Lee CW, Liu JF and Kou YR: Promotion of ROS-mediated apoptosis,
G2/M arrest, and autophagy by naringenin in non-small cell lung
cancer. Int J Biol Sci. 20:1093–1109. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Khan M, Maryam A, Zhang H, Mehmood T and
Ma T: Killing cancer with platycodin D through multiple mechanisms.
J Cell Mol Med. 20:389–402. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Galindo-Moreno M, Giraldez S, Saez C,
Japon MA, Tortolero M and Romero F: Both p62/SQSTM1-HDAC6-dependent
autophagy and the aggresome pathway mediate CDK1 degradation in
human breast cancer. Sci Rep. 7:100782017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bjorkoy G, Lamark T, Pankiv S, Overvatn A,
Brech A and Johansen T: Monitoring autophagic degradation of
p62/SQSTM1. Methods Enzymol. 452:181–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liebl MP, Meister SC, Frey L, Hendrich K,
Klemmer A, Wohlfart B, Untucht C, Nuber J, Pohl C and Lakics V:
Robust LC3B lipidation analysis by precisely adjusting autophagic
flux. Sci Rep. 12:792022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Redmann M, Benavides GA, Berryhill TF,
Wani WY, Ouyang X, Johnson MS, Ravi S, Barnes S, Darley-Usmar VM
and Zhang J: Inhibition of autophagy with bafilomycin and
chloroquine decreases mitochondrial quality and bioenergetic
function in primary neurons. Redox Biol. 11:73–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu J, Lu S, Zheng L, Guo Q, Cao L, Xiao
Y, Chen D, Zou Y, Liu X, Deng C, et al: ATM-CHK2-TRIM32 axis
regulates ATG7 ubiquitination to initiate autophagy under oxidative
stress. Cell Rep. 42:1134022023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang YT, Liu TY, Shen CH, Lin SY, Hung CC,
Hsu LC and Chen GC: K48/K63-linked polyubiquitination of ATG9A by
TRAF6 E3 ligase regulates oxidative stress-induced autophagy. Cell
Rep. 38:1103542022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guo W, Zhou H, Wang J, Lu J, Dong Y, Kang
Z, Qiu X, Ouyang X, Chen Q, Li J, et al: Aloperine suppresses
cancer progression by interacting with VPS4A to inhibit
autophagosome-lysosome fusion in NSCLC. Adv Sci (Weinh).
11:e23083072024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Boulares AH, Yakovlev AG, Ivanova V,
Stoica BA, Wang G, Iyer S and Smulson M: Role of poly(ADP-ribose)
polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP
mutant increases rates of apoptosis in transfected cells. J Biol
Chem. 274:22932–22940. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wongpan A, Panvongsa W, Krobthong S, Nutho
B, Kanjanasirirat P, Jearawuttanakul K, Khumpanied T, Phlaetita S,
Chabang N, Munyoo B, et al: Cleistanthin A derivative disrupts
autophagy and suppresses head and neck squamous cell carcinoma
progression via targeted vacuolar ATPase. Sci Rep. 14:225822024.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Karapurkar JK, Colaco JC, Suresh B, Tyagi
A, Woo SH, Jo WJ, Ko N, Singh V, Hong SH, Oh SJ, et al: USP28
promotes tumorigenesis and cisplatin resistance by deubiquitinating
MAST1 protein in cancer cells. Cell Mol Life Sci. 81:1452024.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sheykhbahaei N, Tameemi AHA and Koopaie M:
Effect of short-term fasting on the cisplatin activity in human
oral squamous cell carcinoma cell line HN5 and chemotherapy side
effects. BMC Cancer. 24:9892024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu W, Xu L, Wang X, Zhang D, Sun G, Wang
M, Wang M, Han Y, Chai R and Wang H: PRDX1 activates autophagy via
the PTEN-AKT signaling pathway to protect against cisplatin-induced
spiral ganglion neuron damage. Autophagy. 17:4159–4181. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yu S, Yue Z and Liu Q: Pectinose induces
cell cycle arrest in luminal A and triple-negative breast cancer
cells by promoting autophagy through activation of the p38 MAPK
signaling pathway. BMC Cancer. 24:6392024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li T, Chen X, Chen X, Ma DL, Leung CH and
Lu JJ: Platycodin D potentiates proliferation inhibition and
apoptosis induction upon AKT inhibition via feedback blockade in
non-small cell lung cancer cells. Sci Rep. 6:379972016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li T, Xu XH, Tang ZH, Wang YF, Leung CH,
Ma DL, Chen XP, Wang YT, Chen Y and Lu JJ: Platycodin D induces
apoptosis and triggers ERK- and JNK-mediated autophagy in human
hepatocellular carcinoma BEL-7402 cells. Acta Pharmacol Sin.
36:1503–1513. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jiao L, Zhang HL, Li DD, Yang KL, Tang J,
Li X, Ji J, Yu Y, Wu RY, Ravichandran S, et al: Regulation of
glycolytic metabolism by autophagy in liver cancer involves
selective autophagic degradation of HK2 (hexokinase 2). Autophagy.
14:671–684. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee M, Nam HY, Kang HB, Lee WH, Lee GH,
Sung GJ, Han MW, Cho KJ, Chang EJ, Choi KC, et al: Epigenetic
regulation of p62/SQSTM1 overcomes the radioresistance of head and
neck cancer cells via autophagy-dependent senescence induction.
Cell Death Dis. 12:2502021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xing Y, Wei X, Liu Y, Wang MM, Sui Z, Wang
X, Zhu W, Wu M, Lu C, Fei YH, et al: Autophagy inhibition mediated
by MCOLN1/TRPML1 suppresses cancer metastasis via regulating a
ROS-driven TP53/p53 pathway. Autophagy. 18:1932–1954. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ma J, Zhang J, Xi L, Qu J, Ma S, Yao S,
Liu J and Ren W: Tertiary butylhydroquinone regulates oxidative
stress in spleen injury induced by gas explosion via the Nrf2/HO-1
signaling pathway. Sci Rep. 15:119872025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Guo T, Wang X, Zhang G, Xia T, Zhu R and
Tou J: Dihydromyricetin functions as a tumor suppressor in
hepatoblastoma by regulating SOD1/ROS pathway. Front Oncol.
13:11605482023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ishaq M, Evans MD and Ostrikov KK:
Atmospheric pressure gas plasma-induced colorectal cancer cell
death is mediated by Nox2-ASK1 apoptosis pathways and oxidative
stress is mitigated by Srx-Nrf2 anti-oxidant system. Biochim
Biophys Acta. 1843:2827–2837. 2014. View Article : Google Scholar : PubMed/NCBI
|