Introduction
Olfactory receptors (ORs) constitute the largest
family of G-protein-coupled receptors (GPCRs), accounting for ~400
and 1,200 intact genes in humans and mice, respectively (1–3). In
olfactory sensory neurons, activation of ORs triggers dissociation
of the olfactory Gα protein (Golf) from its Gβγ subunit, followed
by activation of type III adenylyl cyclase and an increase in
intracellular cAMP. Elevated cAMP levels induce the opening of
cAMP-gated Ca2+ channels, which in turn trigger membrane
depolarization and the generation of action potentials (4–7).
Most ORs are expressed in the nasal olfactory
epithelium, but accumulating evidence indicates that they are also
present in various non-nasal tissues (8,9). This
ectopic expression suggests potential roles for ORs beyond smell
perception, mediating diverse physiological processes (10–15).
For example, OR17-4 is expressed in human spermatozoa and plays an
essential role in efficient fertilization by mediating sperm
chemotaxis toward the oocyte (10–12).
The mouse olfactory receptor MOR23 (a homolog of human OR10J5) has
been shown to promote skeletal muscle regeneration by regulating
myoblast adhesion and migration, underscoring the role of ORs in
tissue remodeling and repair (13).
OR78 in renal juxtaglomerular cells responds to short-chain fatty
acids derived from gut microbiota, linking microbial signals to
renin secretion and systemic blood pressure regulation (14). In the liver, OR10J5 senses the
plant-derived compound α-cedrene and regulates hepatic lipid
metabolism and steatosis through the cAMP-PKA signaling pathway,
suggesting a metabolic role for ORs in energy homeostasis (15).
Notably, studies have reported that ORs are
abnormally expressed in various tumor cells, including those in
breast cancer (16), melanoma
(17), colon cancer (18), bladder cancer (19), neuroendocrine carcinomas (20), liver cancer (21), lung cancer (22) and brain cancer (23). In these cancers, ORs can regulate
cell invasion, migration, proliferation and apoptosis through their
own signaling cascades involving both canonical and non-canonical
signaling pathways (24). Olfactory
receptor 51E1 (OR51E1) is one of the most abundantly expressed
ectopic ORs in various tissues, including small intestine
neuroendocrine carcinoma (25,26),
lung carcinoid (27) and prostate
carcinoma (24,28–30).
Because OR51E1 is highly expressed in prostate tissues, it has also
been referred to as prostate-specific GPCR2 (28). Interestingly, OR51E1 gene expression
is significantly higher in prostate cancer (PC) tissues than in
normal prostate tissues (28–30),
although no correlation between OR51E1 expression and survival of
patients with PC has been identified. Furthermore, studies have
shown that activation of OR51E1 by short- and medium-chain fatty
acid ligands, such as butyrate, triggers intracellular signaling
cascades characterized by elevated cAMP levels and PKA activation.
This signaling promotes the upregulation of cell cycle inhibitors,
including p21 and p27, as well as the tumor suppressor p53, leading
to cell cycle arrest (29,30). However, the precise molecular
mechanisms underlying these effects remain to be fully
elucidated.
In the present study, the expression and prognostic
significance of OR51E1 and other GPCRs in PC were analyzed. Using
nonanoic acid (NA)-induced cAMP production as a functional readout,
we found that sphingosine-1-phosphate receptor 1 (S1PR1) can
regulate OR51E1-mediated signaling and tumor suppression. The
present findings provide mechanistic insights into the
tumor-suppressive effects of OR51E1 in PC and suggest the existence
of specific GPCR-mediated regulatory mechanisms for ectopic
olfactory receptors.
Materials and methods
Cell culture and reagents
LNCaP cells were obtained from the Korean Cell Line
Bank, and Du145 and PC3 cells were purchased from the American Type
Culture Collection. LNCaP, DU145 and PC3 cells were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS;
Hyclone; Cytiva), 100 U/ml penicillin, 100 µg/ml streptomycin, and
20 mM HEPES at 37°C in a humidified 5% CO2 incubator.
Hana3A cells, generously provided by Professor Hiroaki Matsunami
(Duke University School of Medicine), were cultured in Dulbecco's
modified Eagle's medium supplemented with 10% FBS, penicillin and
streptomycin. Butyric acid (BA) and NA were purchased from
MilliporeSigma. TC-G1006 (cat. no. 4747) was purchased from Tocris
Bioscience. Anti-S1PR1 (cat. no. 55133-1-AP) and anti-PARP1 (cat.
no. 51-6639GR) antibodies were purchased from Proteintech Group,
Inc. and BD Biosciences, respectively. Anti-ERK1/2 (cat. no. 9102),
anti-phospho-ERK1/2 (T202/Y204) (cat. no. 4370), anti-SRC (cat. no.
2109), anti-phospho-SRC (Tyr416; cat. no. 6943), anti-JNK (cat. no.
9252), anti-phospho-JNK (T183/Y185; cat. no. 4668), anti-p38 MAPK
(cat. no. 8690) and anti-phospho-p38 MAPK (T180/Y182; cat. no.
4511) antibodies were purchased from Cell Signaling Technology,
Inc. Anti-β-Actin-HRP (cat. no. sc-47778) and anti-rabbit IgG
HRP-conjugated (cat. no. AS014) antibodies were purchased from
Santa Cruz Biotechnology, Inc. and ABclonal Biotech Co., Ltd.,
respectively.
Plasmids
Human GPCR cDNAs were obtained from the Missouri
S&T cDNA Resource Center and cloned into pENTR/D-TOPO vectors
(Invitrogen; Thermo Fisher Scientific, Inc.) as previously
described (31). If necessary, a
stop codon was introduced via site-directed mutagenesis. N-terminal
lucy-flag-rho tag of OR51E1 was a kind gift from Jennifer L.
Pluznick (32). Flag, and Myc tags
were cloned at the N-terminus of each GPCR using one-step sequence-
and ligation-independent cloning (33). GPCRs were subcloned into pcDNA3.1 or
pLenti-CMV-Puro-DEST (Addgene, Inc.; cat. no. 17452; a gift from
Eric Campeau & Paul Kaufman) vectors using the Gateway cloning
system (Invitrogen; Thermo Fisher Scientific, Inc.), following the
manufacturer's instructions. pGL4.29[luc2P/CRE/Hygro] reporter
plasmid was purchased from Promega Corporation (cat. no. E8471).
GloSensor-22F plasmid was purchased from Promega Corporation (cat.
no. E2301) for measuring cAMP production. For CRISPR-Cas9 gene
editing, non-targeting control sgRNA and guide RNAs targeting
OR51E1 were designed using the OmniGuide RNA Design Tool
(GenScript; http://www.genscript.com/tools/gRNA-design-tool)
and were cloned into the lentiCRISPR-v2 vector (Addgene, Inc.; cat.
no. 52961; a gift from Feng Zhang).
Generation of stably transduced cells
using lentivirus
Lentiviral particles were produced using the
ViraPower Lentiviral Expression System (cat. no. K4960-00; Thermo
Fisher Scientific, Inc.). Lenti-X 293T cells were co-transfected
with the lentiviral expression vectors pLenti-CMV-Puro-S1PR1
(custom lentiviral expression construct encoding human S1PR1) or
pLentiCRISPR-v2 encoding OR51E1-targeting sgRNA or a non-targeting
control, together with the packaging plasmids (pLP1, pLP2 and
pVSVG), using Lipofectamine 2000 according to the manufacturer's
instructions. Viral supernatants were collected 48 h
post-transfection and filtered through a 0.45-µm membrane. Viral
titers were determined using HT1080 cells, and LNCaP cells were
transduced at a multiplicity of infection (MOI) of 5 in the
presence of polybrene (8 µg/ml; MilliporeSigma). Transduced cells
were selected with puromycin (1 µg/ml) for 7 days to establish
pooled stable cell populations. OR51E1-knockout and non-targeting
control LNCaP cells were generated by lentiviral transduction using
lentiCRISPR constructs encoding sgOR51E1 #1, sgOR51E1
#2, or a non-targeting control sgRNA (sgCTL). The sgRNA target
sequences were as follows: sgOR51E1 #1:
5′-TAGCAGACAAGCATCAAAC-3′; sgOR51E1 #2:
5′-GGCCATAGACGACATTGACC-3′; sgCTL: 5′-ACGGAGGCTAAGCGTCGCAA-3′
(34). Following puromycin
selection for 12 days, knockout efficiency was functionally
validated by assessing NA-induced CRE-luciferase reporter activity.
To assess the mutation of OR51E1 target, PCR was conducted using
the primers 5′-CCTCCCTGGTTTAGAAGAGGC-3′ and
5′-GGATGCGCTGTCGAATCTCC-3′. The resulting PCR amplicon was digested
with BsrI (New England BioLabs, Inc.) for OR51E1 target #1
and HpaII (Fermentas; Thermo Fisher Scientific, Inc.) for
OR51E1 target #2 to confirm the knockout of OR51E1.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted and purified from cultured
cells using the RNeasy Mini Kit (Qiagen, Inc.). First-strand cDNA
was synthesized with the ReverTra Ace® RT-qPCR Kit
(Toyobo Life Science) according to the manufacturer's instructions.
Quantitative PCR was performed using the Brilliant SYBR Green QPCR
Master Mix (Agilent Technologies, Inc.). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 3 min,
followed by 40 cycles of denaturation at 95°C for 15 sec, annealing
at 60°C for 30 sec, and extension at 72°C for 30 sec. Gene
expression levels were normalized to the housekeeping gene β-actin,
and relative quantification was performed using the
2−ΔΔCq method (35). The
primer sequences used for amplification were as follows: Actin
forward, 5′-GGAAATCGTGCGTGACATTAAG-3′ and reverse,
5′-AGCTCGTAGCTCTTCTCCA-3′; and OR51E1 forward,
5′-CCGCCATTGGCCTGGACTCA-3′ and reverse,
5′-CCAAGATGACGGGCAGCGGA-3′.
Cell proliferation assay
Cell proliferation was assessed using the Cell
Counting Kit-8 (CCK-8; Dojindo Laboratories, Inc.). Cells were
seeded in 96-well microplates at a density of 15,000 cells per
well. For pathway inhibitor experiments, cells were pre-treated for
1 h with the indicated pathway inhibitors (SQ-22536, 100 µM (cat.
no. 568500; MilliporeSigma; PubChem ID 5270); SU6656, 10 µM (cat.
no. 572635; MilliporeSigma; PubChem ID 5312137); dasatinib, 10 nM
(cat. no. CDS023389; MilliporeSigma; PubChem ID 3062316); SP600125,
10 µM (cat. no. HY-12041; MilliporeSigma; PubChem ID NSC75890);
SB203580, 10 µM (cat. no. S1076; Selleck Chemicals; PubChem ID
176155); U0126, 1 µM (cat. no. HY-12031A; MilliporeSigma; PubChem
ID 3006531); AG490, 10 µM (cat. no. HY-12000; MilliporeSigma;
PubChem ID 5328779), followed by incubation with or without NA for
48 h. Subsequently, 10 µl of CCK-8 reagent was added to each well
and incubated for 4 h at 37°C. Absorbance was measured at 450 nm
using an EnVision microplate reader (PerkinElmer, Inc.).
Apoptosis assay
LNCaP cells were plated in 12-well plates and
treated with the indicated compounds for 24 h. After treatment,
both the supernatant and adherent cells were collected using
Accutase and resuspended in 100 µl of DPBS containing 0.1% BSA
(cat. no. 11945; SERVA Electrophoresis GmbH) and annexin binding
buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2). Cells were stained with 10 µg/ml propidium
iodide and 40 µl of Annexin V Ready Flow conjugates (Invitrogen;
Thermo Fisher Scientific, Inc.) for 15 min at room temperature in
the dark. Stained cells were analyzed by FACS Canto II (BD
Biosciences), and data were processed using FlowJo software
(version 10; BD Biosciences).
cAMP assay
Intracellular cAMP levels were measured using the
pGloSensor-22F cAMP reporter plasmid (cat. no. E2301; Promega
Corporation). Hana3A cells were plated in 24-well plates 24 h prior
to transfection. One day before the assay, cells were transfected
with plasmids encoding OR51E1 (20 ng), RTP1s (5 ng), the indicated
GPCRs (5 ng), and the GloSensor-22F (50 ng) reporter using
Lipofectamine 2000 (1 µl per well; Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
The following day, cells were detached using DPBS containing EDTA
and resuspended in CO2-independent medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 0.1% BSA and 800 µg/ml
D-luciferin. Cells were transferred to white 96-well microplates
(cat. no. 3917; Corning, Inc.) and incubated at room temperature
for 2 h. Luminescence, reflecting intracellular cAMP levels, was
measured using a TriStar2 microplate reader (Berthold Technologies
GmbH) with a 540-nm filter.
CRE-luciferase reporter assay
LNCaP cells were seeded in 6-well cell culture
plates at 4×105 cells per well and transfected with the
pGL4.29[luc2P/CRE/Hygro] reporter plasmid (400 ng; cat. no. E8471;
Promega Corporation) using Lipofectamine 2000 (4 µl per well;
Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. After 48 h, cells were detached and
replated into 96-well white plates. The following day, cells were
starved with assay buffer (phenol red-free DMEM containing 0.1%
BSA) overnight. After starvation, cells were treated with NA for 12
h at 37°C. Luciferase activity was measured using the
Steady-Glo® Luciferase Assay System (10 µl per well;
Promega Corporation) for 5 min at room temperature in the dark
using a TriStar2 LB 942 multimode microplate reader (Berthold
Technologies GmbH).
Detection of cell surface expression
of GPCRs
For ELISA, Hana3A cells were transfected with
pcDNA-Lucy-FLAG-Rho-OR51E1 in 96 well plates. A total of 2 days
after transfection, cells were fixed with 4% paraformaldehyde for
15 min at room temperature under non-permeabilizing conditions.
Cells were incubated with anti-FLAG antibody (cat. no. F1804;
MilliporeSigma; 1:4,000) at 4°C overnight, followed by incubation
with an HRP-conjugated anti-mouse antibody (cat. no. A9044;
MilliporeSigma; 1:40,000) for 1 h in the dark. Cells were washed
three times with PBS containing 1% BSA, incubated with TMB
substrate (cat. no. 34028; Thermo Fisher Scientific, Inc.), and the
reaction was stopped with sulfuric acid. Absorbance was measured at
450 nm using an EnVision Multilabel Plate Reader (Perkin Elmer,
Inc.). For flow cytometry, LNCaP cells were dissociated using
Accutase (cat. no. A6964; MilliporeSigma). Cells were incubated
with an anti-S1PR1 rabbit polyclonal antibody (1:1,000) or control
rabbit IgG (1:1,000) for 1 h at 4°C, followed by incubation with a
FITC-conjugated anti-rabbit IgG secondary antibody (1:1,000) for 30
min at 4°C. Staining was performed in PBS containing 1% BSA. Cells
were analyzed using a BD FACSCanto II cytometer equipped with a 488
nm laser and a 525/50 nm bandpass filter for FITC detection. At
least 10,000 live events were collected per sample. Single cells
were identified by forward scatter area (FSC-A) versus forward
scatter height (FSC-H) gating, followed by exclusion of debris
using FSC-A vs. side scatter area. Fluorescence-positive
populations were gated based on unstained controls and
fluorescence-minus-one controls for FITC. A viability dye was not
used, and Fc-blocking was not applied because the cell lines used
do not express Fc receptors under our culture conditions. Data were
analyzed using FlowJo software.
Western blot analysis
Total protein was extracted using
radioimmunoprecipitation assay lysis buffer (50 mM Tris-HCl, pH
7.4; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS; 1
mM EDTA) supplemented with protease inhibitors (phenylmethyl
sulfonyl fluoride, 10 mM; pepstatin, 1 mM; leupeptin, 1 mM;
benzamide, 1 mM) and phosphatase inhibitors (sodium orthovanadate,
1 µM; β-glycerol phosphate, 10 mM; sodium pyrophosphate, 10 mM;
sodium fluoride, 10 mM). Protein concentrations were determined
using a Bio-Rad protein assay kit (Bio-Rad Laboratories Inc.).
Equal amounts of proteins (25 µg per sample) were separated by
SDS-PAGE using 10% acrylamide gels and transferred onto
nitrocellulose membranes. Membranes were blocked with 5% non-fat
milk in TBS containing 0.1% Tween-20 for 1 h and then incubated at
4°C overnight with the following primary antibodies: anti-FLAG
(1:4,000), phospho-Src (1:1,000), Src, phospho-SAPK/JNK (1:1,000),
SAPK/JNK, phospho-p38 MAPK (1:1,000), p38 MAPK, phospho-ERK
(1:1,000), ERK (1:1,000), PARP1 (1:2,000) and β-actin (1:20,000).
Immunoreactive bands were detected using a chemiluminescent imaging
system (AE-9150 Ez-Capture II; ATTO Corporation). Band intensities
were quantified using ImageJ software (version 1.54p; National
Institutes of Health).
Expression and survival analysis
Gene expression data for prostate adenocarcinoma
(PRAD) and normal prostate tissue were obtained from the UCSC Xena
Browser (https://xenabrowser.net/) using the
dataset ‘The Cancer Genome Atlas (TCGA) TARGET Genotype-Tissue
Expression (GTEx) transcript expression by RSEM using UCSC TOIL
RNA-seq recompute’. Transcript abundance was reported as RSEM
transcripts per million (TPM). All TCGA and GTEx samples were
processed through the UCSC TOIL RNA-seq recompute pipeline, which
applies a uniform workflow for read alignment, quantification,
normalization and quality control, thereby minimizing batch effects
and enabling direct comparison of expression values between tumor
and normal tissues. OR51E1 expression levels were compared between
GTEx prostate samples and TCGA PRAD samples, including
stratification by pathological stage where indicated. Expression
levels were visualized using violin plots, and progression-free
interval was assessed using Kaplan-Meier survival analysis. All
analyses were performed using R software (version 4.1.2; R
Foundation for Statistical Computing).
Statistical analysis
All experimental data are presented as the mean ±
standard error of the mean (SEM). Statistical significance was
determined using unpaired two-tailed Student's t-test, or two-way
ANOVA followed by Tukey's multiple comparison test, as appropriate.
P<0.05 was considered to indicate a statistically significant
difference. Analyses were performed using GraphPad Prism 9 software
(GraphPad Software; Dotmatics).
Results
OR51E1 suppresses PC cell
proliferation, but its expression is not associated with patient
survival
Among patients with cancer, OR51E1 expression is
markedly higher in PRAD than in other cancer types (Fig. S1). To assess the effect of OR51E1
on PC cell survival, LNCaP cells were treated for 48 h with its
saturated fatty acid ligands, NA and BA, and cell viability was
measured. Consistent with previous studies (29,30),
both NA and BA reduced LNCaP cell survival in a dose-dependent
manner (Fig. 1A). To directly
assess the role of OR51E1 in mediating NA-induced effects, OR51E1
knockout LNCaP cells were generated using CRISPR/Cas9-mediated gene
editing. Successful knockout of OR51E1 was validated using
restriction enzymes that target the PAM sites for sgOR51E1
#1 and #2 (Fig. S2A and B). In
addition, CRE-luciferase reporter assay showed a significant
decrease in NA-induced reporter activity in OR51E1 knockout cells
generated with two independent guide RNA (sgOR51E1 #1 and
#2), compared with control cells expressing a non-targeting sgRNA
(sgCTL) (Fig. S2C). This confirms
the effective ablation of NA-induced OR51E1 signaling. Notably,
NA-induced reduction in cell viability observed in control LNCaP
cells was completely abolished in OR51E1-knockout cells (Fig. 1B), demonstrating that NA-mediated
growth inhibition is strictly dependent on OR51E1 expression.
Consistent with this finding, NA-induced cytotoxicity was observed
exclusively in LNCaP cells, which express OR51E1, but not in DU145
or PC3 cells, which lack OR51E1 expression (Fig. 1C and D). These results demonstrated
that activation of OR51E1 by its agonist suppresses PC cell
proliferation and that this anti-survival effect in PC cells is
dependent on the presence of OR51E1.
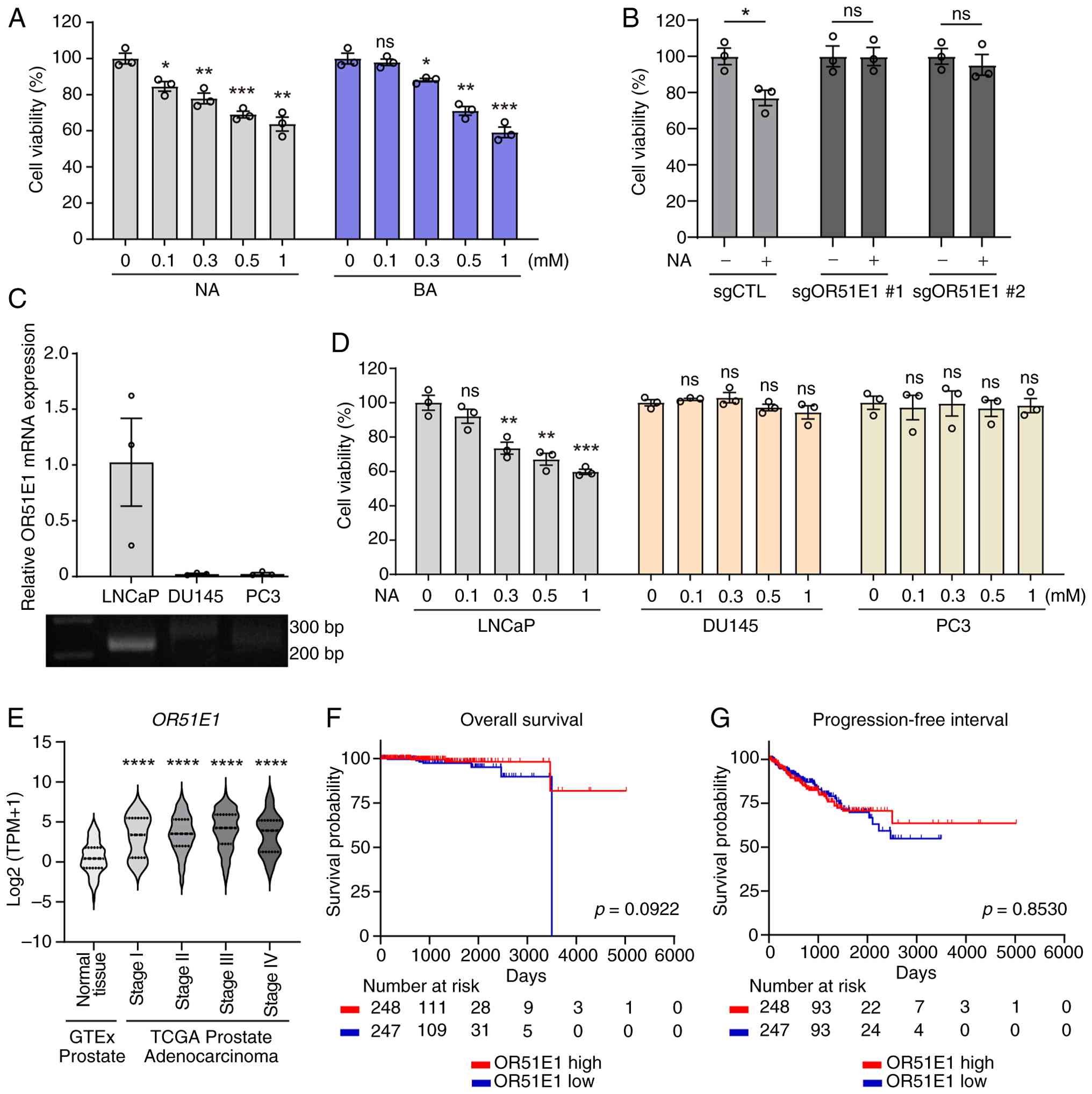 | Figure 1.OR51E1 agonists reduce LNCaP cell
viability, but expression alone does not predict patient outcome.
(A) LNCaP cells were treated with increasing concentrations (0.1–1
mM) of NA or BA for 48 h, and cell viability was measured using the
Cell Counting Kit-8 assay. Values were normalized to untreated
controls. (B) The effect of NA on cell viability was assessed in
control and OR51E1 knockout LNCaP cells after 48 h of NA (0.5 mM)
treatment. (C) Relative OR51E1 mRNA expression in LNCaP, DU145, and
PC3 prostate cancer cell lines was assessed by reverse
transcription-quantitative PCR. Expression levels were normalized
to β-actin. Representative PCR products were visualized by agarose
gel electrophoresis. (D) LNCaP, DU145 and PC3 cells were cultured
with various concentrations of NA for 48 h, and cell viability was
measured. Data represent the mean ± SEM of three independent
experiments. Statistical significance was determined using an
unpaired Student's t-test. (E) OR51E1 expression across
pathological stages of prostate cancer. OR51E1 mRNA expression
levels were compared between normal prostate tissues from the GTEx
dataset and prostate adenocarcinoma samples from TCGA stratified by
pathological stage: Stage I (T2b), Stage II (T2b and T2c), Stage
III (T3a and 3b), and Stage IV (T4). Transcript expression values
(RSEM TPM) were obtained from the UCSC Xena Browser using the
TCGA-TARGET-GTEx TOIL RNA-seq recompute dataset. Statistical
significance was determined using an unpaired Student's t-test. (F
and G) Kaplan-Meier survival curves for (F) overall survival and
(G) progression-free interval stratified by OR51E1 expression
levels. Red and blue lines indicate high- and low-expression
groups, respectively. Survival probabilities were compared using
the log-rank test, and P-values are shown in each panel.
*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. OR51E1,
olfactory receptor 51E1; BA, butyric acid; NA, nonanoic acid; GTEx,
Genotype-Tissue Expression; TCGA, The Cancer Genome Atlas; ns, not
significant. |
Previous studies have reported that OR51E1 is
significantly overexpressed in PC tissues compared with normal
prostate tissues (28–30). To validate this observation, RNA-seq
data from TCGA and the Genotype-Tissue Expression (GTEx) project
were analyzed. The analysis confirmed a significant increase in
OR51E1 mRNA expression in all stages of PC tissues relative to
normal prostate tissues (Fig. 1E).
However, OR51E1 expression levels revealed no significant
association with overall survival (Fig.
1F), progression-free interval (Fig. 1G), disease-specific survival
(Fig. S3A), or disease-free
interval (Fig. S3B). These
findings led us to hypothesize that although OR51E1 is an
overexpressed anti-survival GPCR in PC, it may require interactions
with other regulatory GPCRs to exert its tumor-suppressive effects.
To test this possibility, a screening was performed to identify
GPCRs that modulate OR51E1-mediated signaling.
S1PR1 enhances OR51E1-mediated cAMP
signaling through increased surface expression
Previous studies have shown that class A GPCRs can
interact with ectopically expressed ORs, suggesting a potential
regulatory role in OR expression and signaling (23,36–39).
To identify GPCRs that could potentially regulate OR51E1 signaling
in PC, class A GPCRs were ranked based on their expression levels
in the TCGA PRAD dataset. Receptors in the top quartile were then
selected for functional screening, with the aim of prioritizing
GPCRs that are robustly expressed in tumor tissues and more likely
to act as physiologically relevant modulators of OR51E1 signaling
(Fig. 2A). To evaluate their
effects on OR51E1 signaling, Hana3A cells were co-transfected with
OR51E1 and each selected GPCR, and then measured cAMP production
upon stimulation with NA. Hana3A cells are a 293T-derived line that
stably express RTP1, RTP2, REEP1 and Gαolf-accessory proteins that
facilitate OR trafficking and function, and are widely used for
heterologous OR expression studies (36–39).
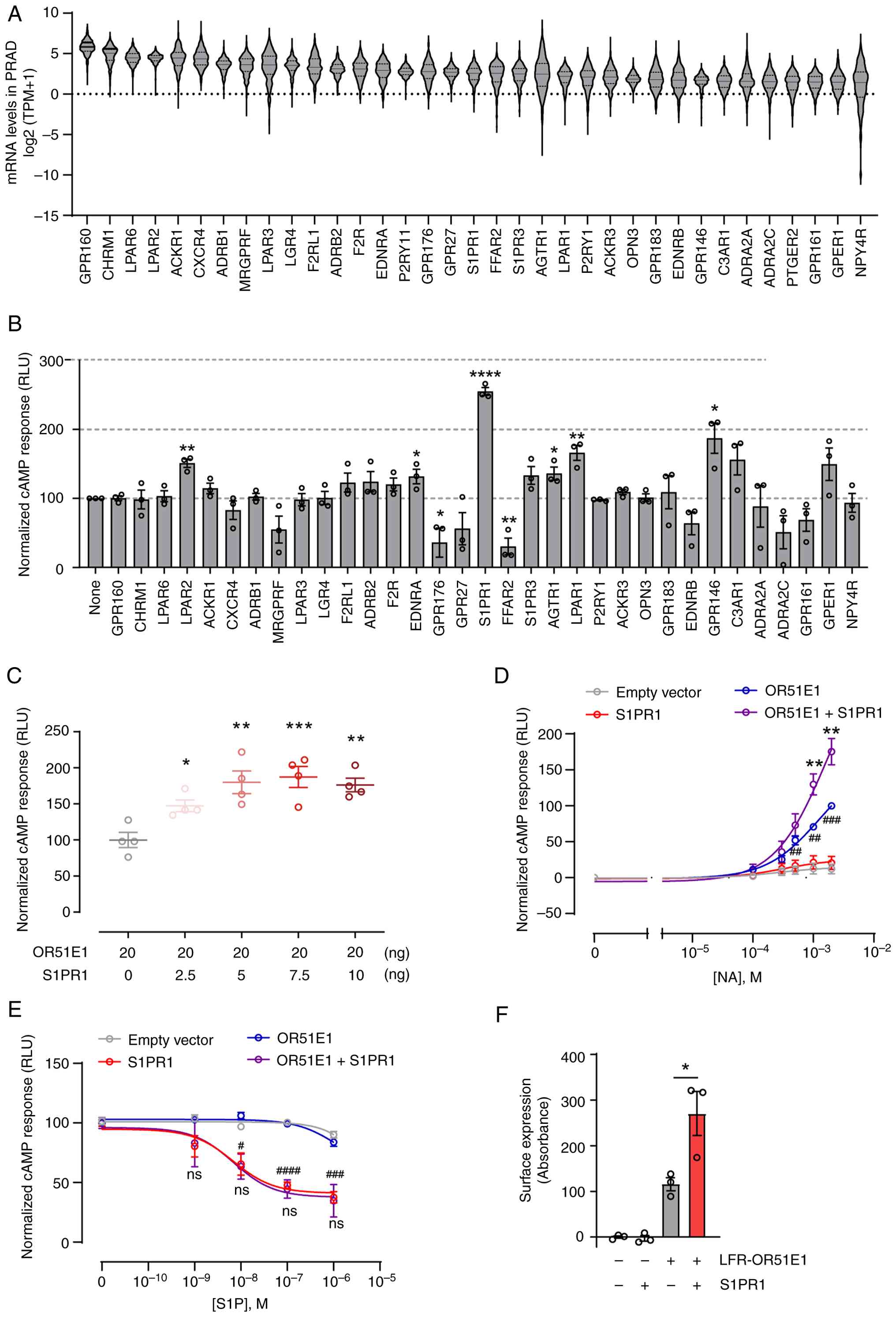 | Figure 2.Identification of S1PR1 as a GPCR
enhancer of OR51E1 signaling. (A) Class A GPCRs with the highest
quartile of mRNA expression in The Cancer Genome Atlas PRAD. Violin
plots show median TPM values for each GPCR. (B) Hana 3A cells were
transfected with Rho-Flag-OR51E1 alone or co-transfected with
individual candidate GPCRs from (A) NA (1 mM)-induced cAMP
production was measured using the Glosensor-22F cAMP reporter assay
and normalized to OR51E1-only controls. Data were analyzed by an
unpaired Student's t-test. (C) Dose-dependent effect of S1PR1 on
OR51E1 signaling. Hana3A cells were transfected with a constant
amount of OR51E1 plasmid (20 ng) and increasing amounts of S1PR1
plasmid (0–10 ng). cAMP production was analyzed using an unpaired
Student's t-test. (D) Hana3A cells were transfected with empty
vector, OR51E1, S1PR1, or OR51E1 together with S1PR1 along with the
GloSensor-22F cAMP reporter. Cells were stimulated with increasing
concentrations of NA (0.1–2 mM), and cAMP production was measured.
Statistical significance was determined using an unpaired Student's
t-test. Asterisks (*) indicate significance differences between
OR51E1 and OR51E1 + S1PR1 groups, and hash symbols (#) indicate
significance differences between empty vector and OR51E1 groups.
(E) Hana3A cells were transfected as described in (D) and treated
with increasing concentrations of S1P (1–1,000 nM) in the presence
of forskolin (1 µM). Statistical significance was determined using
an unpaired Student's t-test. Asterisks (*) indicate significance
differences between S1PR1 and OR51E1 + S1PR1 groups, and hash
symbols (#) indicate significance differences between empty vector
and S1PR1 groups. (F) Surface expression of Lucy-Rho-FLAG
(LFR)-tagged OR51E1 was measured by ELISA under non-permeabilized
conditions in the presence or absence of co-transfected S1PR1.
Surface expression was calculated by subtracting optical density
values from vector-only controls. Data represent the mean ± SEM of
three independent experiments and were analyzed using an unpaired
Student's t-test. *P<0.05, **P<0.01, ***P<0.001 and
****P<0.0001; #P<0.05, ##P<0.01,
###P<0.001 and ####P<0.0001. S1PR1,
sphingosine-1-phosphate receptor 1; GPCR, G-protein-coupled
receptor; PRAD, prostate adenocarcinoma; TPM, transcripts per
million; ns, not significant. |
Among the GPCRs tested, LPAR2, EDNRA, S1PR1, ATGR1,
LPAR1 and GPR146 significantly enhanced NA-induced cAMP production,
whereas GPR176 and FFAR2 suppressed it (Fig. 2B). Notably, S1PR1 produced the
strongest enhancement despite being a Gαi-coupled receptor, which
typically does not directly increase cAMP levels, highlighting it
as a key candidate for further study. Co-expression of OR51E1 with
increasing amounts of S1PR1 plasmid DNA led to a dose-dependent
enhancement of NA-induced cAMP signaling (Fig. 2C). By contrast, co-expression of
OR51E1 with dopamine receptor D2 (DRD2), another Gαi-coupled
receptor, failed to augment NA-induced cAMP production (Fig. S4), confirming that the effect is
specific to S1PR1 rather than a general property of Gαi-coupled
GPCRs.
NA did not induce cAMP responses in cells
transfected with an empty vector, whereas robust cAMP production
was observed in cells overexpressing OR51E1 (Fig. 2D), indicating that NA-induced cAMP
signaling is specifically mediated by OR51E1. OR51E1-mediated cAMP
responses increased in a dose-dependent manner at higher NA
concentrations (1–2 mM) and were further enhanced by S1PR1
co-expression (Fig. 2D). To assess
potential reciprocal regulation, S1PR1 signaling was examined in
the presence of OR51E1. In S1PR1-expressing cells,
forskolin-induced cAMP production was suppressed by S1P, and
co-expression of OR51E1 did not alter this suppression (Fig. 2E), indicating that OR51E1 does not
modulate S1PR1 signaling. Finally, to investigate the mechanism by
which S1PR1 enhances OR51E1 signaling, OR51E1 surface expression
under non-permeabilized conditions was measured using an ELISA.
OR51E1 was tagged with a Lucy-Flag-Rho sequence, which promotes OR
surface trafficking (32,40,41).
Co-expression of S1PR1 significantly increased the surface
localization of OR51E1 (Fig. 2F).
By contrast, OR51E1 mRNA levels were unaffected by S1PR1
co-expression (Fig. S5),
suggesting that S1PR1 enhances OR51E1 function primarily by
facilitating post-transcriptional trafficking to the plasma
membrane rather than by transcriptional upregulation in Hana3A
cells.
S1PR1 expression enhances
OR51E1-mediated inhibition of PC cell survival
To examine the role of S1PR1 in OR51E1-mediated
inhibition of PC cell survival, LNCaP cells, which lack detectable
S1PR1 expression (Fig. S6A), were
transfected with an S1PR1 expression construct. NA-induced
inhibition of cell survival was significantly enhanced in
S1PR1-transfected LNCaP cells, but not in cells transfected with
DRD2 (Fig. 3A). Similarly, BA
enhanced OR51E1-mediated inhibition of LNCaP survival only in the
presence of S1PR1 (Fig. S7). To
further clarify the mechanism, a stable LNCaP cell line
overexpressing S1PR1 (LNCaP-S1PR1) was generated via lentiviral
transduction. Flow cytometric analysis confirmed robust surface
expression of S1PR1 in these cells (Fig. S6B). NA treatment led to a
dose-dependent decrease in cell survival, with a more pronounced
effect in LNCaP-S1PR1 cells compared with parental cells (Fig. 3B). These findings suggested that
S1PR1 expression enhances the anti-survival effect of OR51E1
activation in PC cells. To determine whether reduced cell survival
was attributable to apoptosis, Annexin-V/PI staining was performed.
NA treatment induced both early and late apoptosis in a
dose-dependent manner, and this effect was significantly amplified
in LNCaP-S1PR1 cells (Figs. 3C and
S8). Consistently, NA treatment
increased caspase-3 activation and PARP1 cleavage, with more
pronounced effects observed in the presence of S1PR1 (Fig. 3D and E).
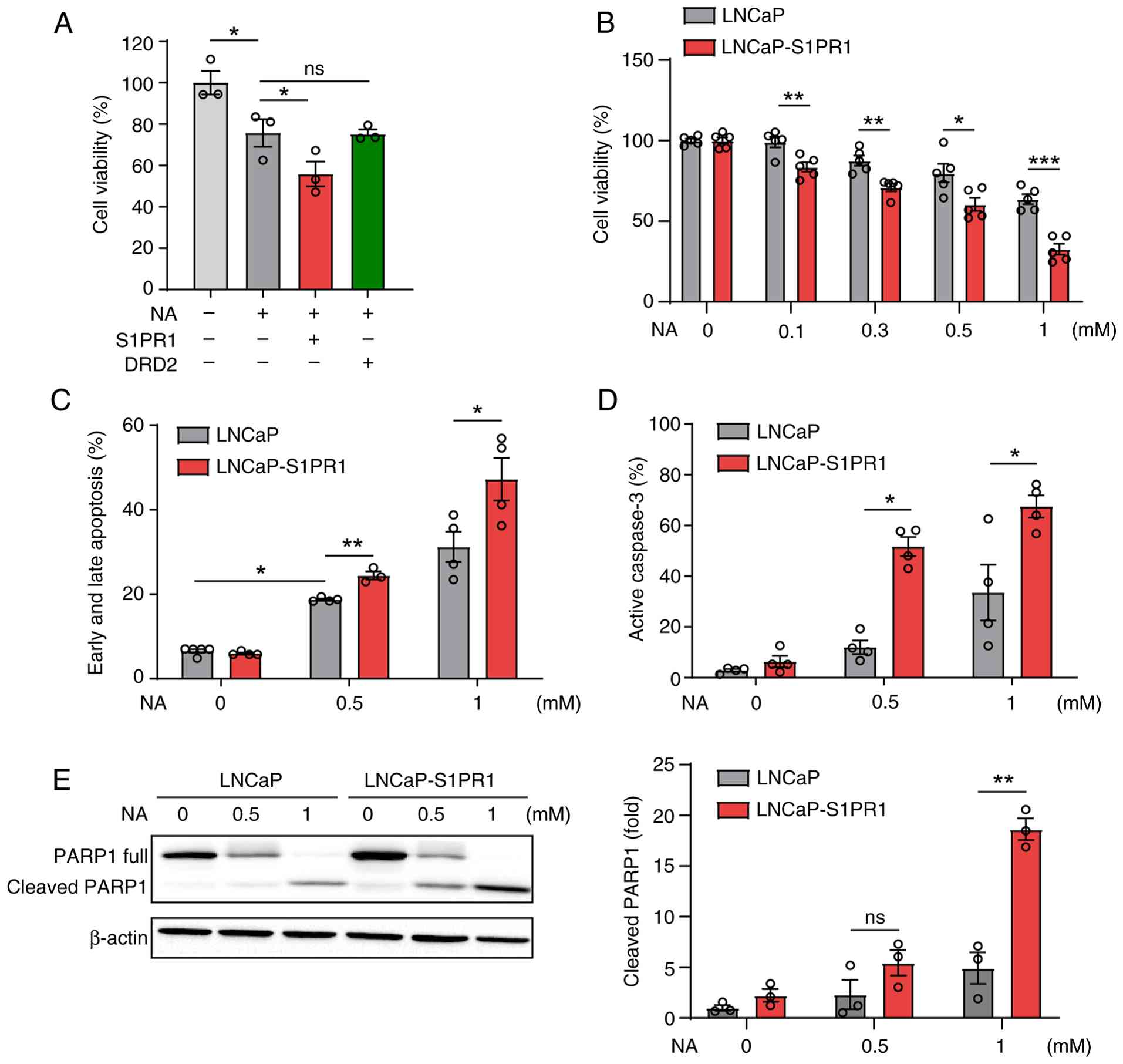 | Figure 3.S1PR1 enhances OR51E1-mediated
cytotoxicity and apoptosis in LNCaP cells. (A) LNCaP cells were
transfected with an empty vector, S1PR1, or DRD2. After 48 h, cells
were treated with NA (0.5 mM) for an additional 48 h, and cell
viability was measured using the Cell Counting Kit-8 assay. (B)
LNCaP cells were transduced with lentivirus encoding S1PR1 and
selected with puromycin (1 µg/ml). Cell viability of parental LNCaP
and LNCaP-S1PR1 cells was measured 48 h after treatment with
increasing concentrations of NA (0.1–1 mM). (C) Apoptosis was
assessed in LNCaP cells treated with NA for 48 h using annexin V/PI
staining. Apoptotic cells were quantified as the percentage of
annexin V-positive cells (early apoptosis) and annexin V/PI
double-positive cells (late apoptosis) relative to the total cell
population. (D) Caspase-3 activation was measured by flow cytometry
following NA treatment. (E) PARP1 cleavage was analyzed by western
blotting in NA-treated cells. Band intensities were quantified
using ImageJ software. Data represent the mean ± SEM of at least
three independent experiments. Statistical significance was
determined using an unpaired Student's t-test. *P<0.05,
**P<0.01 and ***P<0.001. S1PR1, sphingosine-1-phosphate
receptor 1; OR51E1, olfactory receptor 51E1; DRD2, dopamine
receptor D2; NA, nonanoic acid; PI, propidium iodide; ns, not
significant. |
To assess whether S1PR1 activation further modulates
OR51E1 function, the effect of NA on apoptosis was examined in the
presence or absence of TC-G1006, a selective S1PR1 agonist.
TC-G1006 significantly potentiated NA-induced apoptosis only in
S1PR1-overexpressing cells, indicating that ligand-induced S1PR1
activation enhances OR51E1-mediated apoptosis (Figs. 4A and S9A-C). Notably, TC-G1006 alone did not
induce apoptosis in either parental or S1PR1-expressing cells,
suggesting that S1PR1 activation alone is insufficient to trigger
PC cell apoptosis but can potentiate the effect of NA. In
LNCaP-S1PR1 cells, TC-G1006 also potentiated NA-induced growth
inhibition, and this effect was abolished by co-treatment with
either pertussis toxin or NIBR-0213, a selective antagonist of
S1PR1 (Fig. 4B), confirming that
the enhancement requires ligand-induced S1PR1 activation. However,
neither pertussis toxin nor NIBR-0213 reversed NA-induced growth
inhibition in LNCap-S1PR1 cells, indicating that S1PR1 augments
OR51E1 function not only through agonist-induced activation but
also via its basal activity, independent of Gi signaling. Taken
together, these findings demonstrate that both S1PR1 expression and
activation enhance OR51E1-mediated proliferation inhibition and
apoptosis in PC cells.
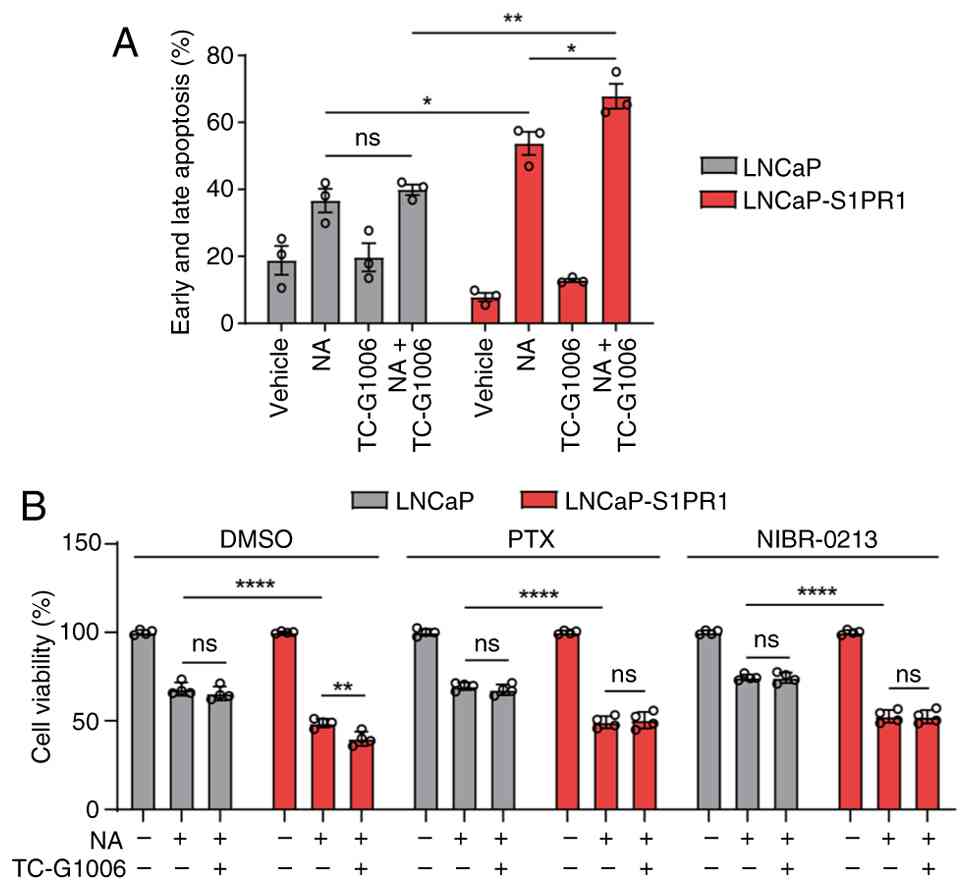 | Figure 4.Activation of S1PR1 enhances
OR51E1-mediated apoptosis in LNCaP cells. (A) LNCaP and LNCaP-S1PR1
cells were treated with NA, TC-G1006 (a selective S1PR1 agonist),
or both for 48 h, and apoptosis was assessed using annexin V/PI
staining. (B) LNCaP and LNCaP-S1PR1 cells were preincubated with
DMSO, PTX, or NIBR-0213 (a selective S1PR1 antagonist) for 1 h and
then treated with NA for 48 h. Cell viability was measured using
the CCK-8 assay. Data represent the mean ± SEM of at least three
independent experiments. Statistical analysis was performed using
two-way ANOVA with Tukey's multiple comparison test. *P<0.05,
**P<0.01 and ****P<0.0001. S1PR1, sphingosine-1-phosphate
receptor 1; OR51E1, olfactory receptor 51E1; NA, nonanoic acid; PI,
propidium iodide; DMSO, dimethyl sulfoxide; PTX, pertussis toxin;
ns, not significant. |
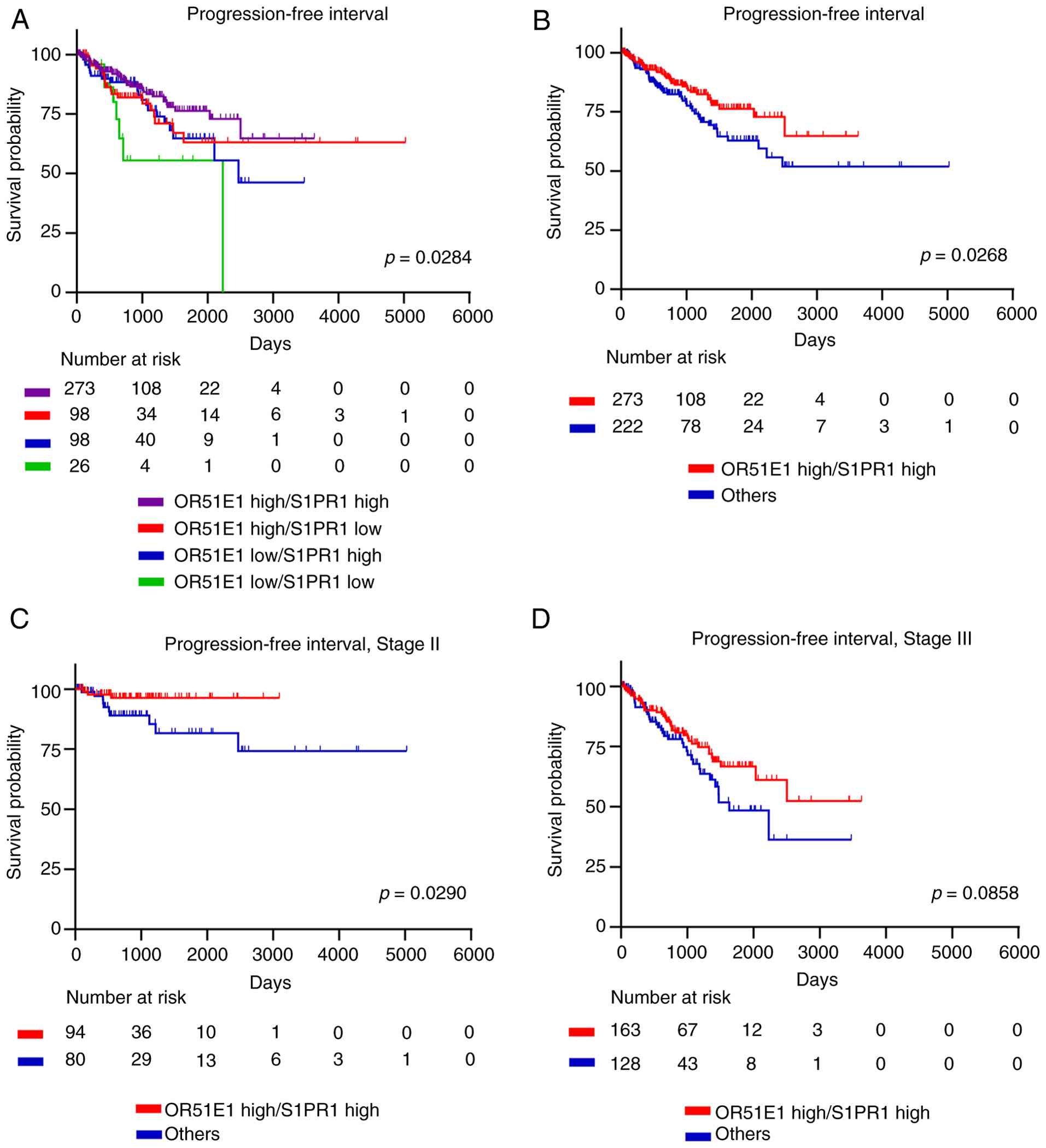
Co-expression of OR51E1 and S1PR1
correlates with improved prognosis in patients with PC
To assess the clinical relevance of S1PR1 and OR51E1
in PC, RNA-seq data from the TCGA PRAD cohort were analyzed.
Patients were stratified into four groups based on OR51E1 and S1PR1
mRNA expression levels: High expression of both (OHSH), high OR51E1
and low S1PR1 (OHSL), low OR51E1 and high S1PR1 (OLSH), and low
expression of both (OLSL). Progression-free interval differed
significantly among the four groups, with the OHSH group showing
the highest survival probability and the OLSL group the lowest
(Fig. 5A). When patients were
divided into the ‘both high (OHSH)’ group versus all other groups
combined, the OHSH group again exhibited significantly improved
progression-free interval (Fig.
5B). Stage-stratified analyses revealed that high OR51E1-S1PR1
co-expression was significantly associated with improved
progression-free interval in stage II patients (Fig. 5C), while a similar trend was
observed in stage III patients but did not reach statistical
significance (Fig. 5D). Analyses of
stage I and IV patients were limited by small sample sizes,
precluding definitive conclusions (Fig. S10). Collectively, these results
suggested that co-expression of OR51E1 and S1PR1 is associated with
improved prognosis in PC regardless of tumor stages.
These clinical observations are consistent with the
experimental findings that OR51E1 suppresses PC cell survival and
that S1PR1 enhances OR51E1-mediated apoptosis. Collectively, these
results suggested that S1PR1 co-expression potentiates the
tumor-suppressive function of OR51E1 and contributes to improved
clinical outcomes in PC. Targeting this axis may represent a novel
therapeutic strategy to limit disease progression.
S1PR1 enhances OR51E1-mediated Src-JNK
signaling to inhibit PC cell survival
To investigate the downstream signaling pathways
involved in NA-induced cell death in PC, LNCaP cells were
pretreated with pharmacological inhibitors targeting key nodes of
the canonical Gs/olf-cAMP pathway and alternative signaling
pathways. Inhibition of adenylate cyclase with SQ22536, which
blocks canonical Gs/olf-mediated cAMP production, failed to rescue
NA-induced inhibition of proliferation (Fig. 6A). Similarly, inhibition of PKA with
H-89 and inhibition of the exchange protein directly activated by
cAMP (EPAC) with ESI-09 did not attenuate NA-induced suppression of
proliferation. By contrast, inhibition of Src kinase with SU6656
significantly but only partially restored cell viability in
NA-treated cells. These results indicated that canonical
Gs/olf-cAMP signaling is dispensable for OR51E1-mediated
anti-survival effects and instead support the involvement of an
alternative Src-dependent signaling pathway in NA-induced PC cell
death.
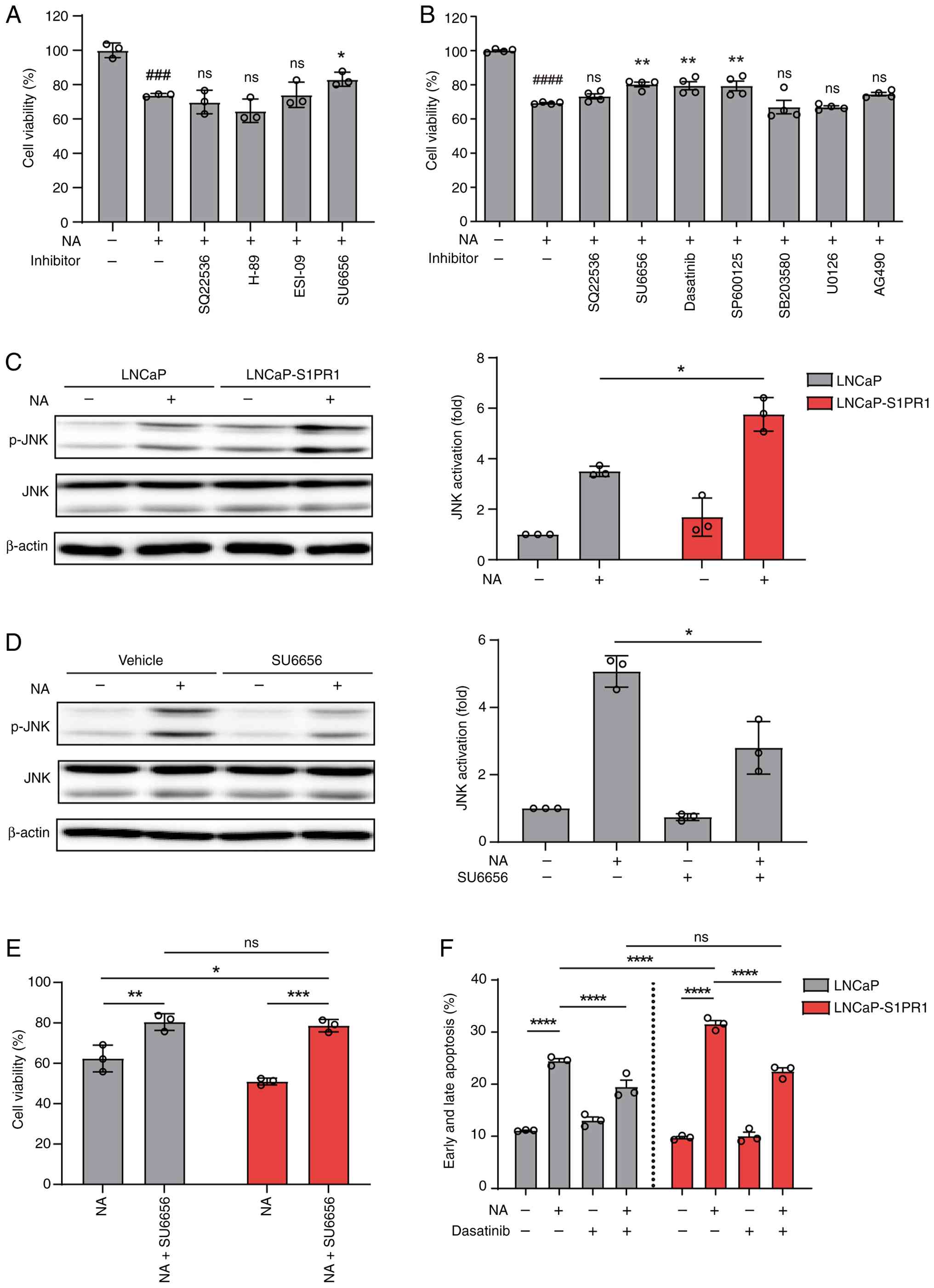 | Figure 6.Co-expression of S1PR1 amplifies
OR51E1-Src-JNK signaling and suppression of proliferation. (A and
B) Inhibitor profiling reveals signaling pathways involved in
NA-induced reduction of LNCaP cell viability. LNCaP cells were
preincubated for 1 h with (A) canonical OR pathway inhibitors
(SQ-22536, 100 µM; H-89, 3 µM; ESI-09, 3 µM; SU6656, 10 µM) or with
(B) other, less well-characterized pathway inhibitors (SU6656, 10
µM; Dasatinib, 10 nM; SP600125, 3 µM; SB203580, 3 µM; U0126, 10 µM;
AG-490, 10 µM), followed by treatment with NA for 48 h. SQ-22536
and SU6656 were included in both panels for comparison. Cell
viability was measured using the CCK-8 assay. Data represent the
mean ± SEM of at least three independent experiments. Statistical
analysis was performed using an unpaired Student's t-test. Hash
symbols (#) indicate statistical significance between vehicle- and
NA-treated cells, whereas asterisks (*) indicate statistical
significance between cells treated with NA alone and those
co-treated with NA and the indicated inhibitors. (C) Western blot
analysis of JNK activation in LNCaP and LNCaP-S1PR1 cells treated
with NA for 1 h. Protein levels of p-JNK and total JNK were
analyzed. (D) Effect of Src inhibition on NA-induced JNK
activation. LNCaP cells were pretreated with the Src inhibitor
SU6656 (10 µM) for 1 h and then treated with NA for 1 h. Protein
levels of p-JNK and total JNK were assessed. Band intensities were
quantified using ImageJ software. Data represent the mean ± SEM of
three independent experiments. Statistical significance was
determined using an unpaired Student's t-test. (E) Effect of Src
inhibition on NA-mediated suppression of proliferation. LNCaP and
LNCaP-S1PR1 cells were pretreated with SU6656 for 1 h, and cell
viability was measured using the CCK-8 assay after NA treatment.
Data represent the mean ± SEM of at least three independent
experiments and were analyzed using two-way ANOVA with Tukey's
multiple comparison test. (F) Effect of Src inhibition on
NA-induced apoptosis. LNCaP and LNCaP-S1PR1 cells were pretreated
with dasatinib (100 nM) for 1 h and then treated with NA. Apoptosis
was assessed using annexin V/PI staining. Data represent the mean ±
SEM of three independent experiments and were analyzed using
two-way ANOVA with Tukey's multiple comparison test.
###P<0.001 and ####P<0.0001;
*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. S1PR1,
sphingosine-1-phosphate receptor 1; OR51E1, olfactory receptor
51E1; NA, nonanoic acid; CCK-8, Cell Counting Kit-8; p-,
phosphorylated; PI, propidium iodide; ns, not significant. |
To further delineate the underlying mechanisms,
activation of key signaling pathways including Src, JNK, p38 MAPK
and ERK, was examined following NA treatment. Immunoblotting
revealed a rapid increase in Src phosphorylation, peaking at 15 min
(Fig. S11A and B), and a gradual
increase in JNK phosphorylation up to 1 h (Fig. S11C). Phosphorylation of p38 MAPK
was also detected as early as 5 min after NA treatment (Fig. S11D), whereas ERK phosphorylation
remained unchanged (Fig.
S11E).
To determine which pathway contributes to NA-induced
inhibition of proliferation, cells were treated with NA in the
presence of specific inhibitors: SU6656 and Dasatinib (Src
inhibitors), SP600125 (JNK inhibitor), U0126 (MEK/ERK inhibitor),
SB203580 (p38 inhibitor) and AG490 (JAK/STAT3 inhibitor).
NA-induced cell death was attenuated by SU6656, dasatinib and
SP600125, but not by SB203580, U0126, or AG490 (Fig. 6B), indicating that Src and JNK, but
not p38, ERK, or STAT3, play a role in NA-mediated inhibition of
proliferation. Although NA robustly activated p38, its inhibition
did not affect cell viability, suggesting that p38 is dispensable
in this context.
Consistent with the enhanced apoptosis observed in
S1PR1-overexpressing LNCaP cells, NA-induced JNK phosphorylation
was more pronounced in LNCaP-S1PR1 cells than in parental cells
(Fig. 6C). Moreover, SU6656
treatment reduced NA-induced JNK phosphorylation (Fig. 6D), indicating that JNK activation
occurs downstream of Src signaling. Both SU6656 and dasatinib
partially restored cell viability and suppressed NA-induced
apoptosis in LNCaP cells regardless of S1PR1 expression (Fig. 6E and F; Fig. S12A-C), confirming the critical role
of the Src-JNK axis in mediating NA-induced apoptosis. Taken
together, these findings demonstrated that NA-induced inhibition of
proliferation in PC cells is mediated primarily through Src
activation followed by JNK phosphorylation, rather than through
classical G protein-cAMP signaling.
Discussion
Previous studies have reported that certain ORs
overexpressed in cancer can exert either pro-tumorigenic or
anti-tumorigenic effects (42–45).
However, a clear relationship between OR overexpression and patient
survival has rarely been established. OR51E1 is highly expressed in
PC cells and has been identified to inhibit cancer cell
proliferation (29,30). Consistent with these findings, it
was confirmed that NA- or BA-induced reduction in cell survival
occurred specifically in LNCaP cells, which endogenously express
OR51E1 (Fig. 1A). Importantly, this
anti-survival effect was abrogated in OR51E1-knockout LNCaP cells,
demonstrating that NA-mediated suppression of proliferation is
dependent on the presence of OR51E1 (Fig. 1B). By contrast, NA-induced
cytotoxicity was not observed in DU145 or PC3 PC cell lines, which
lack OR51E1 expression (Fig. 1C and
D). Although ectopic overexpression of OR51E1 in these cell
lines would provide a direct test of whether OR51E1-mediated
anti-survival effects are broadly conserved across PC models, such
experiments were beyond the scope of the present study.
Nevertheless, the current data provide strong evidence that OR51E1
is required for NA-induced suppression of proliferation in PC cells
that express this receptor.
In the present analysis of the TCGA dataset, OR51E1
was significantly overexpressed across all stages of prostate
carcinoma compared with normal prostate tissue; however, its
expression level was not significantly associated with patient
survival (Fig. 1F and G). Based on
these findings, it was hypothesized that the tumor-suppressive
function of OR51E1-potentially shared by other ORs-may be regulated
by additional GPCRs. To identify GPCRs that modulate OR51E1
function in PC cells, the expression profiles of class A GPCRs were
first examined in PC tissues (Fig.
2A) and the effects of highly expressed GPCRs on NA-induced
OR51E1 activation were subsequently evaluated in Hana3A cells
(Fig. 2B). Through this screening
approach, S1PR1 was identified as a candidate GPCR that enhances
OR51E1 function by amplifying OR51E1-mediated cAMP signaling
(Fig. 2B-D). Expression of S1PR1 in
LNCaP cells, which endogenously express OR51E1, significantly
increased NA-induced inhibition of proliferation and apoptosis
(Fig. 3). Furthermore, activation
of S1PR1 with its selective agonist TC-G1006 further potentiated
OR51E1-mediated inhibition of proliferation (Fig. 4). Importantly, analysis of TCGA
patient dataset revealed that high expression of both OR51E1 and
S1PR1 was associated with significantly improved progression-free
interval, whereas OR51E1 expression alone had no prognostic value
(Fig. 1E and F; Fig. 5). Collectively, these findings
suggested that S1PR1 enhances the tumor-suppressive function of
OR51E1 and may contribute to improved clinical outcomes in PC.
Mechanistically, it was found that NA-induced,
OR51E1-mediated inhibition of proliferation in LNCaP cells was
dependent on the Src-JNK axis rather than the canonical Gs/olf-cAMP
pathway (Fig. 6). Inhibitors of Src
and JNK abrogated both NA-induced apoptosis and the enhancing
effects of S1PR1, suggesting that S1PR1 promotes OR51E1 function
through a shared signaling pathway. Importantly, inhibition of Src
or JNK only partially attenuated NA-induced inhibition of
proliferation and apoptosis, indicating that although the Src-JNK
axis represents a major downstream pathway of OR51E1 activation,
additional signaling mechanisms are likely to contribute to the
full anti-survival response. While it remains unclear whether
Src-JNK signaling directly induces apoptosis or indirectly
facilitates OR51E1 function by increasing trafficking or membrane
localization, the current data support both possibilities. In
Hana3A cells, co-expression of S1PR1 significantly increased the
surface expression of OR51E1 without affecting OR51E1 mRNA levels
(Figs. 2G and S3), indicating a post-transcriptional
regulatory mechanism. Unfortunately, due to the lack of suitable
antibodies, it was not possible to detect endogenous OR51E1 surface
expression in LNCaP cells.
Although NA-induced cytotoxicity is mediated by
Src-JNK signaling, OR51E1-driven cAMP signaling remains a robust
and reproducible response in both Hana3A cells and LNCaP cells
endogenously expressing OR51E1. However, pharmacological inhibition
of adenylyl cyclase, PKA, or EPAC did not attenuate NA-induced
inhibition of proliferation or apoptosis, indicating that canonical
Gs/olf-cAMP signaling is dispensable for the anti-survival effects
of OR51E1 activation. These findings suggested that OR51E1 engages
parallel signaling pathways in PC cells, with non-canonical Src-JNK
signaling mediating cytotoxicity, while the functional role of cAMP
signaling may be context-dependent and warrants further
investigation.
In Hana3A cells, expression of S1PR1 enhanced
NA-induced cAMP responses when co-expressed with OR51E1 (Fig. 2C and D), despite S1PR1 being a
Gαi-coupled receptor that does not independently stimulate cAMP
production. In LNCaP cells, overexpression of S1PR1 alone did not
produce a statistically significant increase in apoptosis (Fig. 3C-E). These findings suggested that
S1PR1 by itself is insufficient to induce apoptosis but can amplify
NA-induced OR51E1-dependent anti-survival effects. Because the
apoptosis-inducing effect of NA is independent of the canonical
Gs/olf-cAMP pathway, and Src and JNK activation was not directly
examined in Hana3A cells co-expressing OR51E1 and S1PR1, it remains
unclear whether S1PR1 expression alone, in the absence of OR51E1,
can activate the Src/JNK pathway in this heterologous system.
Several studies have demonstrated that class A GPCRs
can form heteromers with ORs both in vitro and in
vivo (23,36–39).
For example, the β2-adrenergic receptor, purinergic
receptors (P2Y1 and P2Y2), and the adenosine 2A receptor promote
both ERK activation and surface expression of OR-M71 through
heteromerization (36,37). Similarly, the muscarinic
acetylcholine receptor 3 forms heteromers with OR-S6, enhancing OR
signaling via a β-arrestin-dependent mechanism without altering
surface localization (38,39). While S1PR1 has been reported to
interact with other GPCRs (46,47),
the observed enhancement of OR51E1 signaling could arise from
multiple mechanisms, including receptor heteromerization or
indirect effects on receptor trafficking. At present, there is no
direct evidence for a physical interaction or signaling crosstalk
between OR51E1 and S1PR1, and both possibilities therefore remain
speculative.
An alternative hypothesis for the enhancement of
OR51E1′s tumor-suppressive function by S1PR1 is that S1PR1
influences OR51E1 localization and activity through signaling
crosstalk. Previous studies have revealed that S1PR1 activation
regulates various vesicular trafficking systems, including internal
vesicles in hippocampal neurons (46), exosomal multivesicular bodies
(47), and transport carriers in
the trans-Golgi network (48).
These findings suggest that S1PR1 may regulate the intracellular
trafficking and membrane localization of OR51E1 in PC cells.
The JNK pathway is known to promote apoptosis and
inhibit proliferation in PC (49–52).
For example, mitogen-activated kinase phosphatase 1, which is
upregulated by oncogenes, suppresses apoptosis by inhibiting JNK
activity in PC (53). In the
present study, inhibition of JNK attenuated NA-induced inhibition
of proliferation, supporting its role in mediating OR51E1-induced
apoptosis. This finding is consistent with previous findings
showing that JNK phosphorylation is absent in well-, moderately-,
and poorly differentiated tumors in the transgenic adenocarcinoma
mouse prostate model, despite activation of ERK and p38 in
well-differentiated tumors (54).
Moreover, studies on OR51E1 and OR51E2 in PC cells
have demonstrated their tumor-suppressive activity through Src
kinase-dependent, rather than G-protein-mediated, signaling
(29,30,55).
The findings of the present study align with this model: Adenylyl
cyclase inhibition did not affect OR51E1 function, whereas NA
stimulation induced rapid Src phosphorylation within 5 min.
Inhibition of Src not only blocked JNK activation but also restored
cell viability and suppressed NA-induced apoptosis. These results
indicated that OR51E1 mediates tumor suppression in PC via a
Src-JNK-dependent pathway.
In addition to these mechanistic insights, emerging
evidence suggests that the metabolite-sensing functions of ORs may
link tumor metabolism to signaling outcomes. ORs are increasingly
recognized as metabolite-sensing GPCRs that can link cellular
metabolism to signaling outcomes. In PC, metabolic reprogramming,
particularly involving lipid and short-chain fatty acid metabolism,
plays a critical role in disease progression and therapy
resistance. Notably, acetate utilization has been shown to promote
hormone therapy resistance through metabolic reprogramming and
neuroendocrine differentiation, highlighting the pathological
relevance of short-chain fatty acids in PC progression (56). Furthermore, metabolic inputs, such
as pyruvate flux through the mitochondrial pyruvate carrier, can
influence cell fate decisions and antitumor function in immune
cells, underscoring the broader principle that metabolic signals
can regulate cellular behavior (57). Collectively, these findings suggest
that OR51E1, as a fatty acid-sensing receptor, may function as a
metabolic sensor that links changes in tumor metabolite
availability to non-canonical signaling pathways, including Src-JNK
activation, thereby influencing PC cell survival and
progression.
The present study demonstrates the involvement of
the OR51E1 signaling axis in PC using LNCaP cells as an in
vitro model. Importantly, transcriptomic analyses of TCGA and
GTEx datasets revealed that OR51E1 is not only highly expressed in
PRAD but also shows significantly elevated expression in other
cancer types such as kidney renal clear cell carcinoma (KIRC) and
glioblastoma multiforme (GBM) compared with corresponding normal
tissues. These observations suggest that OR51E1′s ectopic
expression and potential functional roles may extend beyond PC.
Supporting this notion, previous studies have reported OR51E1
expression in neuroendocrine tumors of the small intestine and lung
carcinoid tumors, where it may serve as a biomarker and contribute
to tumor biology (25,27). Furthermore, recent single-cell
transcriptomic analyses in GBM have indicated cell type-specific
expression of OR51E1 within the tumor microenvironment, implicating
it in processes such as vascular remodeling and tumor progression
(58). Overall, these observations
raise the possibility that the OR51E1 signaling axis characterized
in LNCaP cells may also play a role in other OR51E1-expressing
cancers such as KIRC and GBM. However, given that the current
functional analyses were limited to a single cell line, further
investigations using additional in vitro and in vivo
models are necessary to confirm the broader relevance and
mechanistic involvement of OR51E1 in diverse tumor types.
A limitation of the present study is the absence of
rescue experiments in which an sgRNA-resistant OR51E1 is
re-expressed in OR51E1-knockout cells. Such experiments would
further strengthen the causal link between OR51E1 and NA-induced
Src/JNK activation and apoptosis and will be addressed in future
studies. It is also acknowledged that the survival analyses were
stratified only by pathological stage and did not account for other
clinical covariates, such as Gleason score, PSA at diagnosis, or
patient age. Future studies using larger and more comprehensively
annotated clinical cohorts will be required to determine whether
OR51E1-S1PR1 co-expression is independently associated with
prognosis after multivariate adjustment for these factors.
In conclusion, the current findings reveal that
although OR51E1 is overexpressed in PC and exerts anti-tumorigenic
effects, its impact on patient prognosis is not apparent when
considered in isolation. However, the identification of S1PR1 as a
co-regulator that enhances OR51E1-mediated signaling and apoptosis
provides new insight into the contextual regulation of ORs in
cancer. The observed synergy between OR51E1 and S1PR1, both in
vitro and in patient datasets, suggests that the
tumor-suppressive activity of OR51E1 may depend on co-expression
with specific GPCR partners. These results highlight the importance
of receptor interactions and signaling context in evaluating the
functional and clinical relevance of ectopically expressed ORs in
cancer. Targeting such receptor networks may offer novel
therapeutic strategies for PC and other malignancies in which ORs
are dysregulated.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Jae-Yeon Jeong
(GPCR Therapeutics Inc.) for helpful comments and discussions.
Funding
The present study was supported by the National Research
Foundation of Korea (grant nos. RS-2020-NR049538 and
2021R1A2C1013718) and Samsung Science and Technology Foundation
(grant no. SSTF-BA1901-09).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
KK, JL, CC, JB, HJC and WKH conceived and designed
experiments. KK performed experiments. KK and WKH analyzed data and
wrote the manuscript. All authors reviewed and commented on the
manuscript. All authors read and approved the final version of the
manuscript. KK and WKH confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
WKH is a shareholder in GPCR Therapeutics Inc. The
other authors declare that they have no competing interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript or to generate images, and subsequently,
the authors revised and edited the content produced by the
artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
Glossary
Abbreviations
Abbreviations:
|
BA
|
butyric acid
|
|
CCK-8
|
Cell Counting Kit-8
|
|
DRD2
|
dopamine receptor D2
|
|
FBS
|
fetal bovine serum
|
|
Golf
|
olfactory Gα protein
|
|
GPCR
|
G-protein-coupled receptor
|
|
NA
|
nonanoic acid
|
|
OR
|
olfactory receptor
|
|
OR51E1
|
olfactory receptor 51E1
|
|
PRAD
|
prostate adenocarcinoma
|
|
S1PR1
|
sphingosine-1-phosphate receptor 1
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TPM
|
transcripts per million
|
References
|
1
|
Bjarnadóttir TK, Gloriam DE, Hellstrand
SH, Kristiansson H, Fredriksson R and Schiöth HB: Comprehensive
repertoire and phylogenetic analysis of the G protein-coupled
receptors in human and mouse. Genomics. 88:263–273. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Godfrey PA, Malnic B and Buck LB: The
mouse olfactory receptor gene family. Proc Natl Acad Sci USA.
101:2156–2161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malnic B, Godfrey PA and Buck LB: The
human olfactory receptor gene family. Proc Natl Acad Sci USA.
101:2584–2589. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boekhoff I, Tareilus E, Strotmann J and
Breer H: Rapid activation of alternative second messenger pathways
in olfactory cilia from rats by different odorants. EMBO J.
9:2453–2458. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reed RR: Signaling pathways in odorant
detection. Neuron. 8:205–209. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fleischer J, Breer H and Strotmann J:
Mammalian olfactory receptors. Front Cell Neurosci. 3:92009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klasen K, Corey EA, Kuck F, Wetzel CH,
Hatt H and Ache BW: Odorant-stimulated phosphoinositide signaling
in mammalian olfactory receptor neurons. Cell Signal. 22:150–157.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Buck L and Axel R: A novel multigene
family may encode odorant receptors: A molecular basis for odor
recognition. Cell. 65:175–187. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parmentier M, Libert F, Schurmans S,
Schiffmann S, Lefort A, Eggerickx D, Ledent C, Mollereau C, Gérard
C, Perret J, et al: Expression of members of the putative olfactory
receptor gene family in mammalian germ cells. Nature. 355:453–455.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spehr M, Gisselmann G, Poplawski A,
Riffell JA, Wetzel CH, Zimmer RK and Hatt H: Identification of a
testicular odorant receptor mediating human sperm chemotaxis.
Science. 299:2054–2058. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Milardi D, Colussi C, Grande G, Vincenzoni
F, Pierconti F, Mancini F, Baroni S, Castagnola M, Marana R and
Pontecorvi A: Olfactory receptors in semen and in the male tract:
From proteome to proteins. Front Endocrinol (Lausanne). 8:3792018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eisenbach M and Giojalas LC: Sperm
guidance in mammals-an unpaved road to the egg. Nat Rev Mol Cell
Biol. 7:276–285. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Griffin CA, Kafadar KA and Pavlath GK:
MOR23 promotes muscle regeneration and regulates cell adhesion and
migration. Dev Cell. 17:649–661. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pluznick JL, Protzko RJ, Gevorgyan H,
Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, et al:
Olfactory receptor responding to gut microbiota-derived signals
plays a role in renin secretion and blood pressure regulation. Proc
Natl Acad Sci USA. 110:4410–4415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tong T, Ryu SE, Min Y, de March CA,
Bushdid C, Golebiowski J, Moon C and Park T: Olfactory receptor
10J5 responding to alpha-cedrene regulates hepatic steatosis via
the cAMP-PKA pathway. Sci Rep. 7:94712017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weber L, Maßberg D, Becker C, Altmüller J,
Ubrig B, Bonatz G, Wölk G, Philippou S, Tannapfel A, Hatt H and
Gisselmann G: Olfactory receptors as biomarkers in human breast
carcinoma tissues. Front Oncol. 8:332018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gelis L, Jovancevic N, Veitinger S, Mandal
B, Arndt HD, Neuhaus EM and Hatt H: Functional characterization of
the odorant receptor 51E2 in human melanocytes. J Biol Chem.
291:17772–17786. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weber L, Al-Refae K, Ebbert J, Jägers P,
Altmüller J, Becker C, Hahn S, Gisselmann G and Hatt H: Activation
of odorant receptor in colorectal cancer cells leads to inhibition
of cell proliferation and apoptosis. PLoS One. 12:e01724912017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weber L, Schulz WA, Philippou S, Eckardt
J, Ubrig B, Hoffmann MJ, Tannapfel A, Kalbe B, Gisselmann G and
Hatt H: Characterization of the olfactory receptor OR10H1 in human
urinary bladder cancer. Front Physiol. 9:4562018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leja J, Essaghir A, Essand M, Wester K,
Oberg K, Tötterman TH, Lloyd R, Vasmatzis G, Demoulin JB and
Giandomenico V: Novel markers for enterochromaffin cells and
gastrointestinal neuroendocrine carcinomas. Mod Pathol. 22:261–272.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu C, Jia Y, Lee JH, Kim Y, Sekharan S,
Batista VS and Lee SJ: Activation of OR1A1 suppresses PPAR-γ
expression by inducing HES-1 in cultured hepatocytes. Int J Biochem
Cell Biol. 64:75–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kalbe B, Schulz VM, Schlimm M, Philippou
S, Jovancevic N, Jansen F, Scholz P, Lübbert H, Jarocki M, Faissner
A, et al: Helional-induced activation of human olfactory receptor
2J3 promotes apoptosis and inhibits proliferation in a
non-small-cell lung cancer cell line. Eur J Cell Biol. 96:34–46.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vadevoo SMP, Gunassekaran GR, Lee C, Lee
N, Lee J, Chae S, Park JY, Koo J and Lee B: The macrophage odorant
receptor Olfr78 mediates the lactate-induced M2 phenotype of
tumor-associated macrophages. Proc Natl Acad Sci USA.
118:e21024341182021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chung C, Cho HJ, Lee C and Koo J: Odorant
receptors in cancer. BMB Rep. 55:72–80. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cui T, Tsolakis AV, Li SC, Cunningham JL,
Lind T, Öberg K and Giandomenico V: Olfactory receptor 51E1 protein
as a potential novel tissue biomarker for small intestine
neuroendocrine carcinomas. Eur J Endocrinol. 168:253–261. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Almobarak B, Amlani V, Inge L, Hofving T,
Muth A, Nilsson O, Johansson M, Arvidsson Y and Elias E: Exposure
to nonanoic acid alters small intestinal neuroendocrine tumor
phenotype. BMC Cancer. 23:2672023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Giandomenico V, Cui T, Grimelius L, Öberg
K, Pelosi G and Tsolakis AV: Olfactory receptor 51E1 as a novel
target for diagnosis in somatostatin receptor-negative lung
carcinoids. J Mol Endocrinol. 51:277–286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Weng J, Cai Y, Penland R, Liu M
and Ittmann M: The prostate-specific G-protein coupled receptors
PSGR and PSGR2 are prostate cancer biomarkers that are
complementary to alpha-methylacyl-CoA racemase. Prostate.
66:847–857. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maßberg D, Jovancevic N, Offermann A,
Simon A, Baniahmad A, Perner S, Pungsrinont T, Luko K, Philippou S,
Ubrig B, et al: The activation of OR51E1 causes growth suppression
of human prostate cancer cells. Oncotarget. 7:48231–48249. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pronin A and Slepak V: Ectopically
expressed olfactory receptors OR51E1 and OR51E2 suppress
proliferation and promote cell death in a prostate cancer cell
line. J Biol Chem. 296:1004752021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choi EW, Seen DS, Song YB, Son HS, Jung
NC, Huh WK, Hahn JS, Kim K, Jeong JY and Lee TG: AdHTS: A
high-throughput system for generating recombinant adenoviruses. J
Biotechnol. 162:246–252. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shepard BD, Natarajan N, Protzko RJ, Acres
OW and Pluznick JL: A cleavable N-terminal signal peptide promotes
widespread olfactory receptor surface expression in HEK293T cells.
PLoS One. 8:e687582013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jeong JY, Yim HS, Ryu JY, Lee HS, Lee JH,
Seen DS and Kang SG: One-step sequence- and ligation-independent
cloning as a rapid and versatile cloning method for functional
genomics studies. Appl Environ Microbiol. 78:5440–5443. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hong JM, Lee JW, Seen DS, Jeong JY and Huh
WK: LPA1-mediated inhibition of CXCR4 attenuates CXCL12-induced
signaling and cell migration. Cell Commun Signal. 21:2572023.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−delta delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hague C, Uberti MA, Chen Z, Bush CF, Jones
SV, Ressler KJ, Hall RA and Minneman KP: Olfactory receptor surface
expression is driven by association with the beta2-adrenergic
receptor. Proc Natl Acad Sci USA. 101:13672–13676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bush CF, Jones SV, Lyle AN, Minneman KP,
Ressler KJ and Hall RA: Specificity of olfactory receptor
interactions with other G protein-coupled receptors. J Biol Chem.
282:19042–19051. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li YR and Matsunami H: Activation state of
the M3 muscarinic acetylcholine receptor modulates mammalian
odorant receptor signaling. Sci Signal. 4:ra12011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang Y, Li YR, Tian H, Ma M and Matsunami
H: Muscarinic acetylcholine receptor M3 modulates odorant receptor
activity via inhibition of β arrestin-2 recruitment. Nat Commun.
6:64482015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Krautwurst D, Yau KW and Reed RR:
Identification of ligands for olfactory receptors by functional
expression of a receptor library. Cell. 95:917–926. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhuang H and Matsunami H: Synergism of
accessory factors in functional expression of mammalian odorant
receptors. J Biol Chem. 282:15284–15293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li M, Schweiger MW, Ryan DJ, Nakano I,
Carvalho LA and Tannous BA: Olfactory receptor 5B21 drives breast
cancer metastasis. iScience. 24:1035192021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thomsen MT, Busk M, Zhang D, Chiu CL, Zhao
H, Garcia-Marques FJ, Bermudez A, Pitteri S, Borre M, Brooks JD and
Nyengaard JR: The olfactory receptor OR51E2 regulates prostate
cancer aggressiveness and modulates STAT3 in prostate cancer cells
and in xenograft tumors. BMC Cancer. 25:5352025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang F, Yang J, Zhu C, Ding T, Zhang X and
Zhang H: Olfactory receptor OR51B5 suppressed esophageal cancer
progression through activates Calcium / N-Ras signaling. Cell Death
Dis. 16:4502025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Masjedi S, Zwiebel LJ and Giorgio TD:
Olfactory receptor gene abundance in invasive breast carcinoma. Sci
Rep. 9:137362019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kajimoto T, Okada T, Yu H, Goparaju SK,
Jahangeer S and Nakamura S: Involvement of sphingosine-1-phosphate
in glutamate secretion in hippocampal neurons. Mol Cell Biol.
27:3429–3440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kajimoto T, Okada T, Miya S, Zhang L and
Nakamura S: Ongoing activation of sphingosine 1-phosphate receptors
mediates maturation of exosomal multivesicular endosomes. Nat
Commun. 4:27122013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Okada T, Nishida S, Zhang L, Ibrahim
Mohamed NN, Wang T, Ijuin T, Kajimoto T and Nakamura SI:
Constitutive activation of S1P receptors at the trans-golgi
network is required for surface transport carrier formation.
iScience. 24:1033512021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rodriguez-Berriguete G, Fraile B,
Martinez-Onsurbe P, Olmedilla G, Paniagua R and Royuela M: MAP
kinases and prostate cancer. J Signal Transduct. 2012:1691702012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lorenzo PI and Saatcioglu F: Inhibition of
apoptosis in prostate cancer cells by androgens is mediated through
downregulation of c-Jun N-terminal kinase activation. Neoplasia.
10:418–428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xu R and Hu J: The role of JNK in prostate
cancer progression and therapeutic strategies. Biomed Pharmacother.
121:1096792020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
El-Haibi CP, Singh R, Sharma PK, Singh S
and Lillard JW Jr: CXCL13 mediates prostate cancer cell
proliferation through JNK signalling and invasion through ERK
activation. Cell Prolif. 44:311–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Magi-Galluzzi C, Mishra R, Fiorentino M,
Montironi R, Yao H, Capodieci P, Wishnow K, Kaplan I, Stork PJ and
Loda M: Mitogen-activated protein kinase phosphatase 1 is
overexpressed in prostate cancers and is inversely related to
apoptosis. Lab Invest. 76:37–51. 1997.PubMed/NCBI
|
|
54
|
Uzgare AR, Kaplan PJ and Greenberg NM:
Differential expression and/or activation of P38MAPK, erk1/2, and
jnk during the initiation and progression of prostate cancer.
Prostate. 55:128–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Spehr J, Gelis L, Osterloh M, Oberland S,
Hatt H, Spehr M and Neuhaus EM: G protein-coupled receptor
signaling via Src kinase induces endogenous human transient
receptor potential vanilloid type 6 (TRPV6) channel activation. J
Biol Chem. 286:13184–13192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gao D, Shen Y, Xu L, Sun Y, Hu H, Xu B,
Wang Z and Xu H: Acetate utilization promotes hormone therapy
resistance in prostate cancer through neuroendocrine
differentiation. Drug Resist Updat. 77:1011582024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wenes M, Jaccard A, Wyss T,
Maldonado-Pérez N, Teoh ST, Lepez A, Renaud F, Franco F, Waridel P,
Yacoub Maroun C, et al: The mitochondrial pyruvate carrier
regulates memory T cell differentiation and antitumor function.
Cell Metab. 34:731–746.e9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cho HJ, Yeo DJ, Yang H and Koo J:
Comprehensive transcriptomic analysis reveals cell-type-specific
roles of human odorant receptors in glioblastoma and the tumor
microenvironment. Int J Mol Sci. 25:133822024. View Article : Google Scholar : PubMed/NCBI
|