Introduction
Super enhancers (SEs) are large cluster of
enhancers, characterized by abundant binding of transcription
factors, cofactors and other regulatory proteins (1). These regions are notably enriched with
acetylation at lysine 27 of histone H3 (H3K27ac), a histone
modification that is typically associated with open chromatin
regions and active transcription (1). SEs are key regulators that govern cell
states and identity, and lineage-specific gene expression programs
(2). Furthermore, SEs are
implicated in tumorigenesis by activating the expression of
pro-oncogenes that promote cell proliferation, metabolic
reprogramming, cell survival and adaptability, immune response
modulation and therapeutic resistance (3,4).
Notably, cancer cells are dependent on SE-driven genes and the
activity of these SEs require intact regulatory machinery.
Perturbation of the components that maintain SE landscapes and
activity can disrupt gene expression, highlighting SEs as a
potential target for novel cancer therapy (5).
Small molecule inhibitors have been developed to
target SE-associated regulatory proteins, including JQ1, which
inhibits bromodomain-containing 4 (BRD4) (6). Serving as an epigenetic reader, BRD4
serves an important role in SE organization and transcriptional
activation by binding acetylated lysine residues on histone
proteins and recruiting other transcription factors and co-factors
(7). Studies have shown that
inhibition of BRD4 leads to the downregulation of oncogenic drivers
and disrupts the SE landscape, which impairs cancer cell
proliferation and fitness (8,9). In
addition to BRD4, the histone acetyltransferase (HAT) p300 is also
enriched at SEs, where it regulates chromatin opening, thereby
facilitating the accessibility and recruitment of the
transcriptional activation machinery (10). Given the dependency of cancer cells
on SE-driven genes, p300 inhibition may perturb the transcriptional
activation of these genes, thus impairing cancer growth and
progression (11). p300 inhibitors,
such as A-485 and C646, not only hold potential as therapeutic
agents but also serve as tools for studying chromatin remodeling
and transcriptional regulation in cancer (11,12).
Originally identified as a bacterial adaptive immune
system, CRISPR/Cas9 technology has evolved, with notable
advancements extending its applications beyond traditional gene
editing. The development of dead Cas9 (dCas9) variants has enabled
the modulation of transcription processes and epigenetic
modifications, allowing their application in study of both normal
biological and pathophysiological processes (13). In particular, CRISPR/dCas9-mediated
locus-specific chromatin remodeling is achieved by fusing the dCas9
protein with histone-modifying enzymes such as HAT and histone
deacetylase (HDAC) (14). For
example, downregulation of the zinc finger protein 334
(ZNF334) is associated with poor prognosis in colorectal
cancer (CRC). Targeted acetylation of the ZNF334 promoter
using dCas9-p300 increases its expression, which impairs CRC cell
proliferation, likely through enhanced transcriptional activation
of this gene (15). By contrast,
dCas9-HDAC3-mediated deacetylation of liver metastasis-promoting
dipeptidyl peptidase 4 inhibits its expression and markedly
decreases CRC cell proliferation and metastatic potential (16). Similar CRISPR-mediated histone
deacetylation has been used to suppress mutant KRAS
expression in CRC, which suppresses CRC cell proliferation
(17). This highlights the value of
CRISPR-mediated epigenome editing for studying epigenetic
modification and gene expression regulation in cancer
pathogenesis.
Clear cell renal cell carcinoma (ccRCC) is the most
common subtype of renal cancer, characterized by biallelic von
Hippel-Lindau tumor suppressor inactivation and accumulation of
pro-oncogenic hypoxia-inducible factor 2α (18). The overall 5-year survival rate for
ccRCC patients is 70–80% for localized disease, but decreases in
advanced or metastatic cases to 10–20% despite available systemic
therapies, including receptor tyrosine kinase inhibitors, immune
checkpoint inhibitors and their combinations (19,20).
This poor outcome is due to widespread genetic heterogeneity, the
high aggressiveness of advanced-stage ccRCC and rapid development
of therapeutic resistance (21–23).
H3K27ac chromatin immunoprecipitation—sequencing (ChIP-Seq)
profiling has revealed that one of the strongest SEs in ccRCC,
characterized by large genomic locus size, high H3K27ac enrichment
and occupancies by transcriptional co-activators and Mediator
(1), is located near the
KLF6 locus. This SE region, which drives high expression of
KLF6 to support ccRCC pathogenesis, is insensitive to
perturbation. Many SEs can be substantially disrupted by chemical
or genetic perturbations, which can alter their landscape and
activity (24). In contrast, the
KLF6 SE in ccRCC remains largely unaffected by CRISPR
interference (CRISPRi)-mediated repression of its constituent
enhancers, indicating its particular stability (25). The role of BRD4 and p300 in
regulating this SE region and driving high expression of
KLF6 in ccRCC has not yet been fully elucidated. Therefore,
the present study investigated the effects of chemical inhibition
of BRD4 and p300 on ccRCC cell viability, KLF6 expression
and H3K27ac signals at several constituent enhancers within the
KLF6 SE locus. Additionally, the present study aimed to
determine whether CRISPR/dCas9-mediated deacetylation of
constituent enhancers could induce chromatin remodeling and
downregulation of KLF6 expression. To model ccRCC-specific
dependencies, three genetically diverse ccRCC cell lines (786-M1A,
OS-LM1B and UOK101) were used; these exhibit strong KLF6 SE
activity (25). These models were
used to examine the regulatory roles of BRD4 and p300 within a
disease-relevant epigenetic landscape. Understanding the regulation
of SEs in ccRCC is key, as these elements are pivotal in driving
oncogene expression and tumor progression, making them promising
targets for therapeutic intervention in this aggressive cancer
type.
Materials and methods
Cell lines and reagents
The 786-M1A and OS-LM1B human ccRCC cell lines were
gifts from Professor Joan Massagué (Memorial Sloan Kettering Cancer
Center, NY, USA). These cell lines are metastatic derivatives of
786-O and OS-RC2 cells, respectively (26). The UOK101 human ccRCC cell line was
obtained from Dr Marston Linehan (National Cancer Institute, MD,
USA). The cell lines were cultured in RPMI-1640 (Nacalai Tesque,
Inc.) supplemented with 10% FBS (Tico Europe) and 1%
penicillin-streptomycin solution. For lentiviral production, 293T
cells (ATCC) were used and cultured in DMEM (Nacalai Tesque, Inc.)
supplemented with 10% FBS and 1% penicillin-streptomycin solution.
All cells were cultured at 37°C in a humidified atmosphere
containing 5% CO2. Blasticidin (InvivoGen) and
hygromycin (Nacalai Tesque, Inc.) were used for antibiotic
selection to isolate cells that were successfully transduced with
the dCas9-HDAC3 and sgRNA expression plasmid, respectively. JQ1 and
A-485 were purchased from Sigma-Aldrich (Merck KGaA). The
dCas9-HDAC3 (cat. no. 98591; Addgene, Inc.) (27) and the lentivirus packaging plasmids
psPAX2 (cat. no. 12260) and pMD2.G (cat. no. 12259) were obtained
from Addgene, Inc. All primers and single guide RNA (sgRNA)
constructs (Table I) were purchased
from Integrated DNA Technologies, Inc.
 | Table I.sgRNA and primer sequences. |
Table I.
sgRNA and primer sequences.
| Construct | Sequence
(5′-3′) |
|---|
| Tandem control
(NTC) |
ATCGAAGACAACACCGGAGTGTCGTCGTTGCTCCTAGTTTTAGAGCGCA |
|
|
GGTGTCGCCACCTGCGAAACACCGGGATACGGTGCGTCAATCTAGTTTT |
|
| ACAGTCTTCTCG |
| iSE_1_KLF6 |
ATCGAAGACAACACCGAGAATCGCTGAAGAAACGCGGTTTTAGAGCGC |
|
|
AGGTGTCGCCACCTGCGAAACACCGTACTGCACTGAAGACTCGGAGTT |
|
| TTACAGTCTTCTCG |
| iSE_2_KLF6 |
ATCGAAGACAACACCGACCAGCACAATTTGTCACCGGTTTTAGAGCGC |
|
|
AGGTGTCGCCACCTGCGAAACACCGTTGAAAAAAAACCTATCACAGTT |
|
| TTACAGTCTTCTCG |
| iSE_3_KLF6 |
ATCGAAGACAACACCGATGTGGCTCTGAATCACCATGTTTTAGAGCGCA |
|
|
GGTGTCGCCACCTGCGAAACACCGAACGGTGAGTTCCCGGTACAGTTT |
|
| TACAGTCTTCTCG |
|
dCas9-HDAC3_SDM_forward |
AGCGATGTGGAGATTAAATGTACAGGCAGTG |
|
dCas9-HDAC3_SDM_reverse |
CACTGCCTGTACATTTAATCTCCACATCGCT |
| ChIP qPCR iSE-1
forward |
GAAGTTGAGTCCCGGTGAAA |
| ChIP qPCR iSE-1
reverse |
ATACCCGTCCTGGGAAAATC |
| ChIP qPCR iSE-2
forward |
TCTGTAGCTGCTGAGGCTGA |
| ChIP qPCR iSE-2
reverse |
CACGGTGACAAATTGTGCTG |
| ChIP qPCR iSE-3
forward |
CAGGGAGTGGAAGCTGATGT |
| ChIP qPCR iSE-3
reverse |
CACGCTTGCTGATTTCAAAG |
Cell viability assay
The ccRCC cells, 786-M1A, OS-LM1B and UOK101, were
seeded at a density of 500–1,000 cells/well in a 96-well. On the
following day, the cell culture medium was replaced with fresh
medium containing the respective chemical inhibitors, JQ1 or A-485.
For JQ1 treatment, concentrations of 1, 10, and 20 µM were tested,
whereas A-485 was used at concentrations of 1 and 10 µM. Fresh
medium containing an equivalent volume of DMSO was added to control
wells, where DMSO served as the vehicle for chemical inhibitor
dissolution. The treated cells were incubated at 37°C for 72 h. The
medium was replaced with 120 µl fresh medium containing AlamarBlue™
Reagent (Thermo Fisher Scientific, Inc.) at a 1:10 dilution. The
cells were incubated for 2 h at 37°C in a humidified atmosphere
with 5% CO2, protected from light. A total of 100 µl
medium from each well was transferred to a black 96-well plate and
fluorescence was measured using a Varioskan Flash microplate reader
(Thermo Fisher Scientific, Inc.) at an excitation wavelength of 560
nm and an emission wavelength of 590 nm. A total of three wells
containing medium without cells were included as blank controls.
Each treatment group consisted of five replicate wells. To
determine the number of plated cells, baseline fluorescence
measurements were taken on day 0 (24 h after initial seeding). The
mean blank control readings was subtracted from those of each
treatment well to eliminate background fluorescence. The
fluorescence readings after 72 h incubation at 37°C, were
normalized to the day 0 values. Finally, the relative fluorescence
of inhibitor-treated cells was calculated against that of the
DMSO-treated control group. Images were acquired using an inverted
light microscope (Nikon TS100).
Colony formation assay
The ccRCC cells, 786-M1A, OS-LM1B and UOK101, were
seeded at a density of 100–500 cells/well in a 6-well plate. The
following day, the medium was replaced with medium supplemented
with either JQ1 or A-485 (1 and 10 µM). Control cells were
maintained in medium containing DMSO. Following 7 days of treatment
at 37°C, the culture medium was aspirated and cells were rinsed
twice with 1 ml 1X PBS. The PBS solution was then removed and the
cells were fixed with 500 µl 1:1 mixture of 10% methanol and 10%
acetic acid at room temperature for 15 min. The 6-well plates were
placed on a horizontal shaker for 20 min at room temperature.
Colonies, defined as visible cell clusters formed from a single
cell, were stained with 500 µl 0.5% crystal violet staining
solution (Sigma-Aldrich; Merck KGaA) for 30 min at room
temperature. Excess stain was discarded and the wells were washed
under running tap water. The plates were then air-dried in an oven
until dry. Colony images were captured using the ChemiDoc MP
imaging system (Bio-Rad Laboratories, Inc.).
For quantitative analysis of crystal violet
staining, 600 µl of 10% acetic acid were added to each well,
followed by incubation at room temperature on a shaker for 10 min
to elute the bound dye. A 200-µl aliquot of the eluted solution was
transferred to a clear 96-well plate and the absorbance was
measured at 590 nm using the Varioskan Flash microplate reader
(Thermo Fisher Scientific, Inc.). Absorbance values were normalized
to the control wells and expressed as a percentage relative to the
control group.
RNA extraction, cDNA synthesis and
reverse transcription--quantitative PCR (RT-qPCR)
Total RNA was extracted from ccRCC cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The yield and purity of
total RNA were quantified using the NanoDrop™ 2000c
Spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was
synthesized from total RNA using a LunaScript RT SuperMix kit (New
England BioLabs, Inc.) as follows: 25°C for 2 min, 55°C for 10 min
and 95°C for 1 min. RT-qPCR was performed using TaqMan chemistry
with pre-designed TaqMan probes (Thermo Fisher Scientific, Inc.;
cat. nos. KLF6 Hs00810569_m1, β-actin Hs01060665_g1) and Luna
Universal Probe qPCR Master Mix (New England BioLabs, Inc.). qPCR
reactions were run on the 7500 Fast Real-Time System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with the following
cycling conditions: 95°C for 1 min for initial denaturation,
followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. β-actin
served as the reference gene for normalization and gene expression
fold-changes were calculated using the 2−ΔΔCq method
(28).
Site-directed mutagenesis
Site-directed mutagenesis of the original
dCas9-HDAC3 plasmid was performed using a PCR-based whole-plasmid
amplification approach to substitute the stop codon TAA with AAA.
Complementary forward and reverse primers were designed to anneal
to the target region of the dCas9-hHDAC3 plasmid. This primer pair
carries the nucleotide to be substituted, located in the middle of
the primers (Table I).
PCR amplification was performed using Accuprime™ Pfx
Supermix (Thermo Fisher Scientific, Inc.) on the Thermal Cycler
T100 (Bio-Rad Laboratories, Inc.). The thermocycling conditions
were as follows: Initial denaturation at 95°C for 2 min, followed
by 40 cycles of denaturation at 95°C for 15 sec, annealing at
50–65°C for 30 sec and extension at 68°C for 15 min. During
amplification, the forward and reverse primers anneal to opposite
strands of the plasmid in their respective 5′-3′ orientations and
extend in opposite directions, resulting in amplification of the
entire plasmid while incorporating the intended nucleotide
substitution.
Following PCR amplification, the original and
amplified plasmids were treated with DpnI restriction enzyme
(New England BioLabs, Inc.), which selectively digests the
methylated original plasmid template, for 1 h at 37°C. The
undigested plasmids were transformed into Escherichia coli
(New England BioLabs, Inc.) and selected on LB agar plates
containing ampicillin. The plasmids were extracted from the
transformed bacteria using the Monarch Plasmid Miniprep kit (New
England BioLabs, Inc.) and subjected to Sanger sequencing (Apical
Scientific, Inc.) to confirm the successful site-directed
mutagenesis.
CRISPR sgRNA design
SE constituent regions at the KLF6 locus were
identified based on our previously published H3K27ac ChIP-seq
dataset (accession number GSE115749) (25). Regions with the highest H3K27ac
signal intensity and closest proximity to the KLF6 locus
were annotated as SE_1-3. The sgRNAs targeting these regions,
iSE_1, iSE_2 and iSE_3, were described in our previous study using
the webtool GPP sgRNA Designer
(portals.broadinstitute.org/gppx/crispick/public) (25). The sgRNA sequences are listed in
Table I. The dCas9-HDAC3 plasmid
used for CRISPR-mediated deacetylation was obtained from Addgene,
Inc (cat. no. 98591).
Bacteria transformation and plasmid
extraction
Plasmids were introduced into chemically competent
E. coli cells by adding the plasmid DNA directly to the
competent cells, followed by incubation on ice for 30 min. Heat
shock was then performed at 42°C for 1 min, after which the tubes
were immediately returned to ice for 5 min. Subsequently, 300 µl
fresh Luria Bertani (LB; Sigma-Aldrich; Merck KGaA) medium was
added to each tube and the cultures were incubated at 37°C with
shaking at 200 rpm for 1 h. The transformed cells were spread onto
LB agar plates supplemented with ampicillin (1:1,000) for selection
of successfully transformed cells and incubated overnight at 37°C.
Single colonies from LB + ampicillin plates were inoculated into 3
ml LB medium containing ampicillin (1:1,000) and cultured overnight
at 37°C in a shaking incubator at 200 rpm. Plasmids were extracted
using the Monarch Plasmid Miniprep kit (New England BioLabs, Inc.)
according to the manufacturer's protocol. The concentration and
purity of the extracted plasmids were assessed using a NanoDrop
2000c Spectrophotometer (Thermo Fisher Scientific, Inc.).
Lentivirus production and
transduction
Lentivirus was produced by co-transfecting 293T
cells (ATCC) with second generation lentivirus packaging plasmids
psPAX2 and envelope plasmid pMD2.G together with the plasmid of
interest using the Attractene transfection reagent (Qiagen GmbH).
The lentivirus plasmids were obtained from Addgene, Inc. Briefly,
1.3 µg psPAX2, 0.5 µg pMD2.G and 1.5 µg plasmid of interest were
mixed, followed by the addition of 10 µl Attractene transfection
reagent. The mixture was incubated at room temperature for 30 min
and added to the 293T cells. Transfected cells were incubated 37°C
overnight, after which the media was replaced with fresh complete
media. Lentivirus-containing media was collected 72 h
post-transfection and filtered through a 0.45 µm Minisart NML
syringe filter (Sartorius AG).
Cells at 60–70% confluency were transduced with
filtered lentivirus-containing media in the presence of 8 µg/ml
Polybrene (MilliporeSigma) and incubated at 37°C overnight. Cells
transduced with dCas9-HDAC3 plasmid were selected with 30 µg/ml
blasticidin for approximately 5 days, while cells transduced with
the sgRNA expression vector were selected with 1,000 µg/ml
hygromycin for approximately 7 days. For validation of dCas9-HDAC3
expression by western blotting, non-transduced parental cells were
maintained in parallel and used as a negative control. In addition,
untransduced cells were included during antibiotic selection to
monitor selection efficiency. Selection was considered complete
once all untransduced cells had died, after which the
antibiotic-containing medium was replaced with standard culture
medium.
Protein extraction and western
blotting
The transduced ccRCC cells were lysed on ice in 1X
RIPA buffer supplemented with 1:100 protease inhibitor cocktail
(Nacalai Tesque, Inc.). The protein concentration was quantified
using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Equal amounts
of protein (50 µg per lane) were denatured in 1X Laemmli SDS sample
buffer (GeneTex, Inc.) containing 8% β-mercaptoethanol and resolved
via 8% SDS-PAGE. Proteins were transferred onto nitrocellulose
membranes (GE Healthcare), which were blocked with 5% non-fat milk
in 1× TBS containing 0.1% Tween-20 for 1 h at room temperature. The
membrane was incubated with primary antibodies overnight at 4°C.
Following incubation with rabbit anti-mouse IgG-HRP (1:1,000; cat.
no. P0260; Dako; Agilent Technologies, Inc.) for 1 h at room
temperature, signals were detected using Pierce™ ECL substrate
(Thermo Fisher Scientific, Inc.) and imaged using the ChemiDoc Go
Imaging System (Bio-Rad Laboratories, Inc.). Primary antibodies
against Cas9 (1:500; cat. no. sc-517386) and β-actin (1:5,000; cat.
no. sc-69879) (both Santa Cruz Biotechnology, Inc.) were used. The
Cas9 antibody was used to detect the dCas9-HDAC3 fusion protein
expressed in transduced cells. dCas9 is a catalytically inactive
variant of Cas9 generated by introducing two point mutations in the
nuclease domains (29). As dCas9
retains the Cas9 protein sequence, it is recognized by Cas9
antibodies and detected by western blotting.
ChIP
Cells were cross-linked with 10 ml 1% formaldehyde
for 10 min at room temperature with gentle rotation to preserve
protein-DNA interactions. The cross-linking reaction was quenched
by adding 1 ml 0.125 M glycine and incubating cells for 5 min at
room temperature with rotation. Cells were pelleted by
centrifugation at 1,200 × g for 5 min at room temperature and
washed twice with 15 ml cold 1X PBS. After the final wash, cell
pellets were either snap-frozen and stored at −80°C or used
immediately for chromatin extraction. Frozen cells were thawed on
ice and lysed in 2 ml lysis buffer composed of 20 mM Tris-HCl (pH
8.0), 150 mM NaCl, 2mM EDTA (pH 8.0), 0.1% SDS and 1% Triton X-100.
The cell suspension was pipetted 10 times and incubated on ice for
5 min. Homogenization was performed on ice using a Dounce
homogenizer. The homogenate was transferred to a 15-ml centrifuge
tube for sonication. Chromatin was fragmented by sonication using a
Branson Ultrasonics Sonifier at a frequency of 20 kHz (30 sec
on/off for 15 cycles) while keeping samples on ice to minimize heat
damage. The lysate was centrifuged at 18,000 × g for 20 min at 4°C
and the supernatant containing sheared chromatin was collected. A
10 µl aliquot of lysate was set aside as input control for
downstream qPCR.
For IP, 100 µl magnetic protein A/G beads (Thermo
Fisher Scientific, Inc.) were equilibrated three times with 1 ml
0.5% BSA (Sigma-Aldrich; Merck KGaA) in 1X PBS. The beads were
resuspended in 500 µl 0.5% BSA and conjugated with 5 µg
anti-H3K27ac (cat. no. ab4729) or control IgG antibody (cat. no.
ab37415) (both Abcam) for ≥4 h at 4°C on a rotating platform. The
antibody-conjugated beads were washed three times with 0.5% BSA and
incubated overnight at 4°C with 600 µl chromatin lysate on a
rotating platform. Beads were washed three times with low-salt wash
buffer (50 mM HEPES, pH 7.5, 140 mM NaCl, 1% Triton) and once with
high-salt wash buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 1%
Triton). All washing steps were performed on a magnetic rack
(Thermo Fisher Scientific, Inc.). To reverse cross-links and elute
bound DNA, 100 µl elution buffer was added and samples were
incubated at 65°C with shaking at 1,000 rpm for 3 h. DNA was
purified using the Monarch PCR & DNA Cleanup kit (New England
BioLabs, Inc.) according to the manufacturer's protocol.
qPCR was performed by adding the purified,
de-crosslinked DNA to a reaction mix containing 2X PowerUp SYBR
Green Master Mix (Thermo Fisher Scientific, Inc.) and 10 µM forward
and reverse ChIP-qPCR iSE_1, ChIP qPCR iSE_2 and ChIP qPCR iSE_3
primers (Table I). The reaction was
performed using the 7500 Fast Real-Time System (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with the following conditions: 50°C
for 2 min, 95°C for 2 min for initial denaturation, followed by 40
cycles of 95°C for 3 sec and 60°C for 30 sec. Enrichment of ChIP
DNA was quantified using the percent input (% input) method
(30).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software (version 10.6.0; Dotmatics). Data are presented as
the mean ± SD from three independent experiments unless otherwise
specified. Statistical significance was tested using one-way ANOVA
followed by Tukey's or Dunnett's post hoc test or an unpaired
t-test with Welch's correction. P<0.05 was considered to
indicate a statistically significant difference.
Results
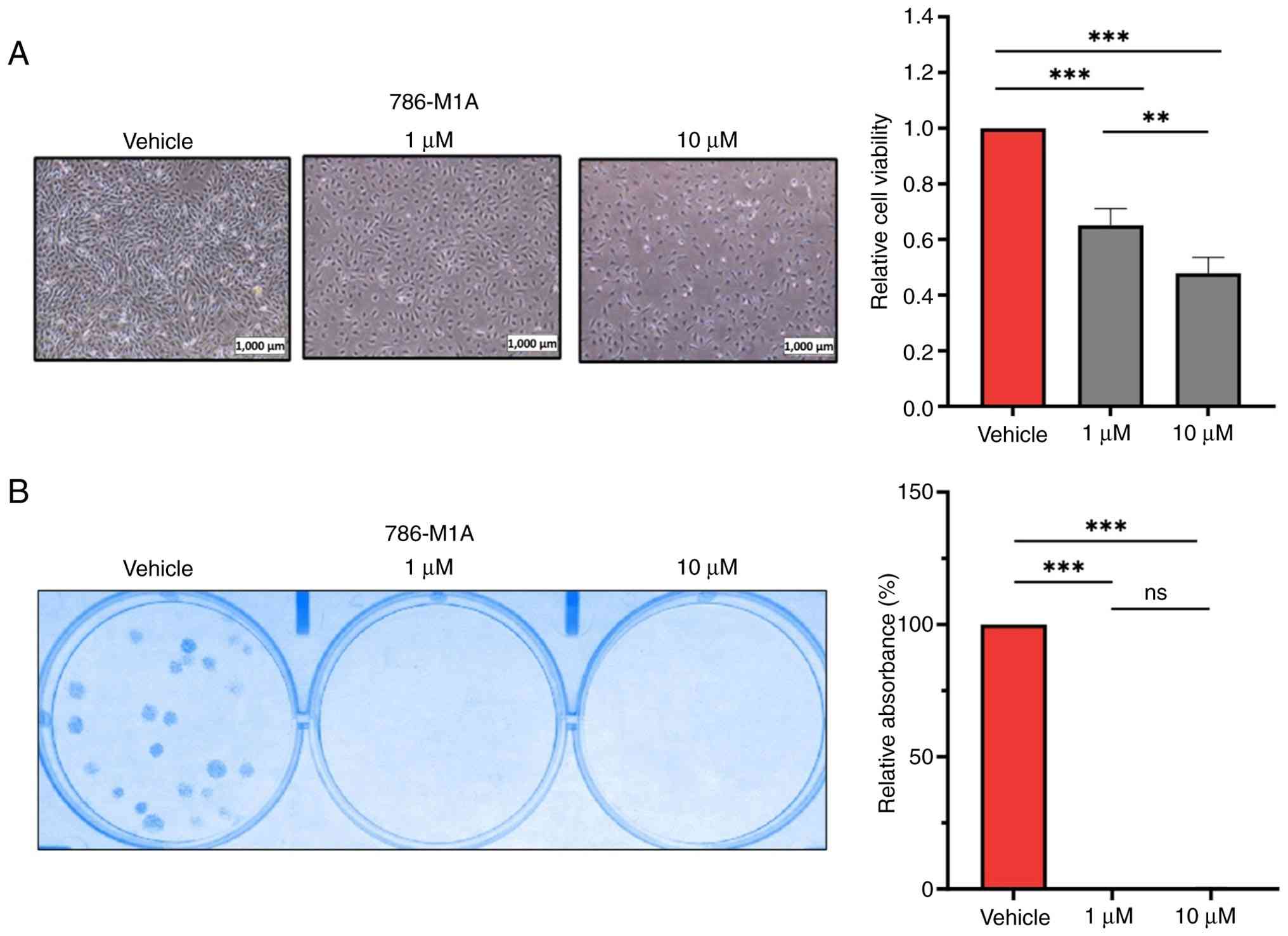
BRD4 inhibition suppresses ccRCC cell
viability
As BRD4 serves pivotal roles in regulating the SE
landscape and transcription of downstream genes (8), the present study evaluated whether
ccRCC cells are sensitive to BRD4 inhibition by JQ1. 786-M1A,
OS-LM1B and UOK101 human ccRCC cells were treated with increasing
concentrations of JQ1 before cell viability assays at 72 h
post-treatment. BRD4 inhibition significantly decreased ccRCC cell
viability in a dose—dependent manner compared with that of the
vehicle-treated cells (Figs. 1A and
S1A). The present study also
investigated the effect of BRD4 inhibition on the colony formation
of ccRCC cells. JQ1 treatment, even at a concentration of 1 µM,
significantly suppressed the clonogenic potential of ccRCC cells
(Figs. 1B and S1B). Overall, ccRCC cells were sensitive
to JQ1-mediated BRD4 inhibition, highlighting the key role of BRD4
in supporting ccRCC cell viability.
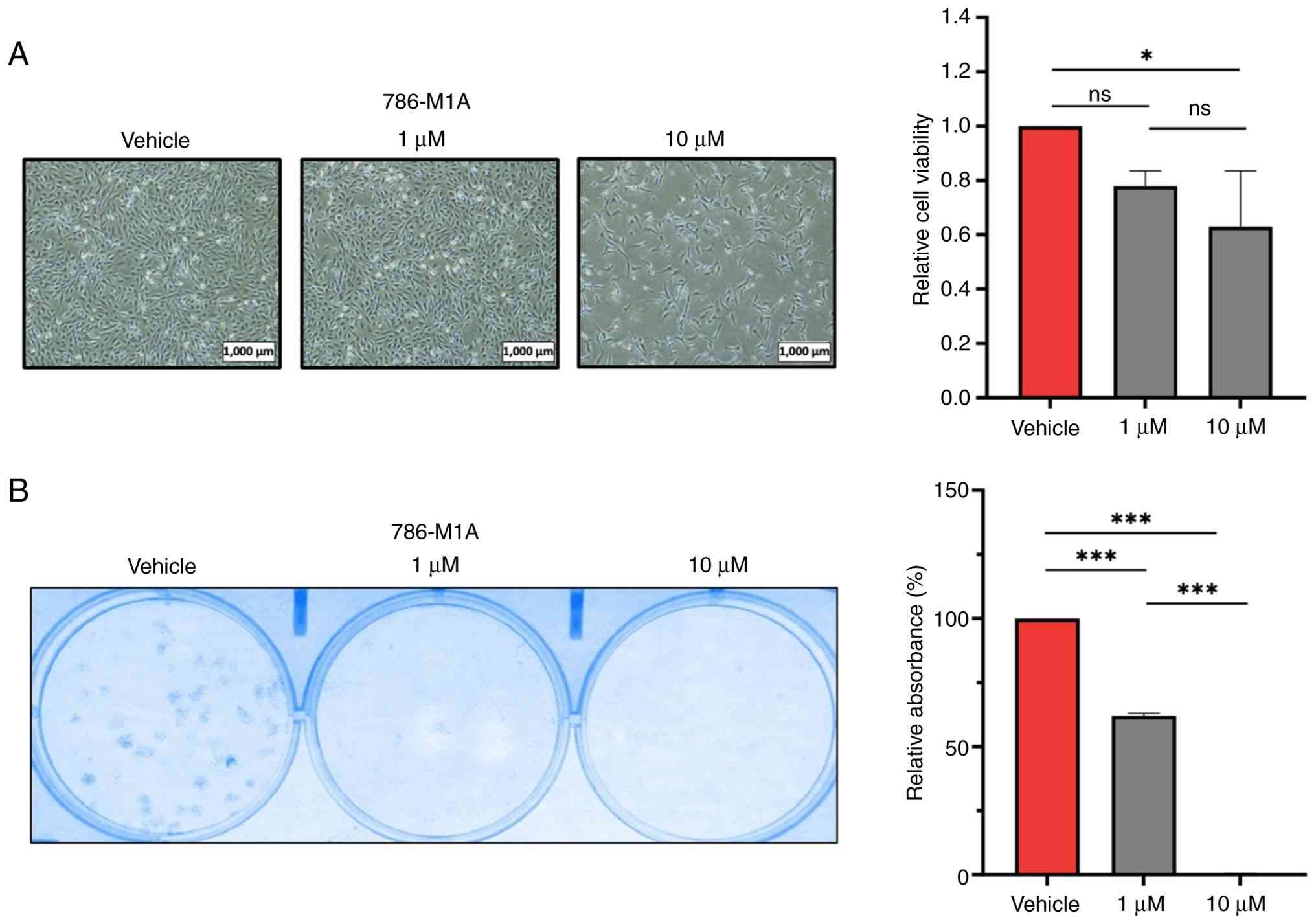
Targeting p300 HAT decreases ccRCC
cell fitness
The p300 HAT is involved in regulating chromatin
accessibility and transcription activation (12). Furthermore, p300 binding is also
enriched at the SE regions, driving the expression of pro-oncogenes
that promote cancer progression (12). The present study aimed to assess the
effect of targeting p300 activity on ccRCC cell viability by
treating 786-M1A and OS-LM1B cells with 1 and 10 µM A-485 for 72 h.
In both ccRCC cell lines, inhibition of p300 with 10 µM A-485
significantly decreased the viability compared with that of
vehicle-treated controls, whereas treatment with a lower
concentration of A-485 did not result in a significant decrease in
cell viability (Figs. 2A and
S2A). By contrast, colony
formation assays revealed that p300 inhibition with 1 or 10 µM
A-485 significantly impaired the clonogenic capacity of ccRCC
cells, which was consistent with the overall trend observed in the
cell viability assays (Figs. 2B and
S2B). Overall, ccRCC cells
exhibited sensitivity to A-485-mediated p300 inhibition, which
resulted in a marked reduction in cell viability and colony
formation. This suggested that p300 serves a role in controlling
the expression of genes that promote ccRCC cell viability.
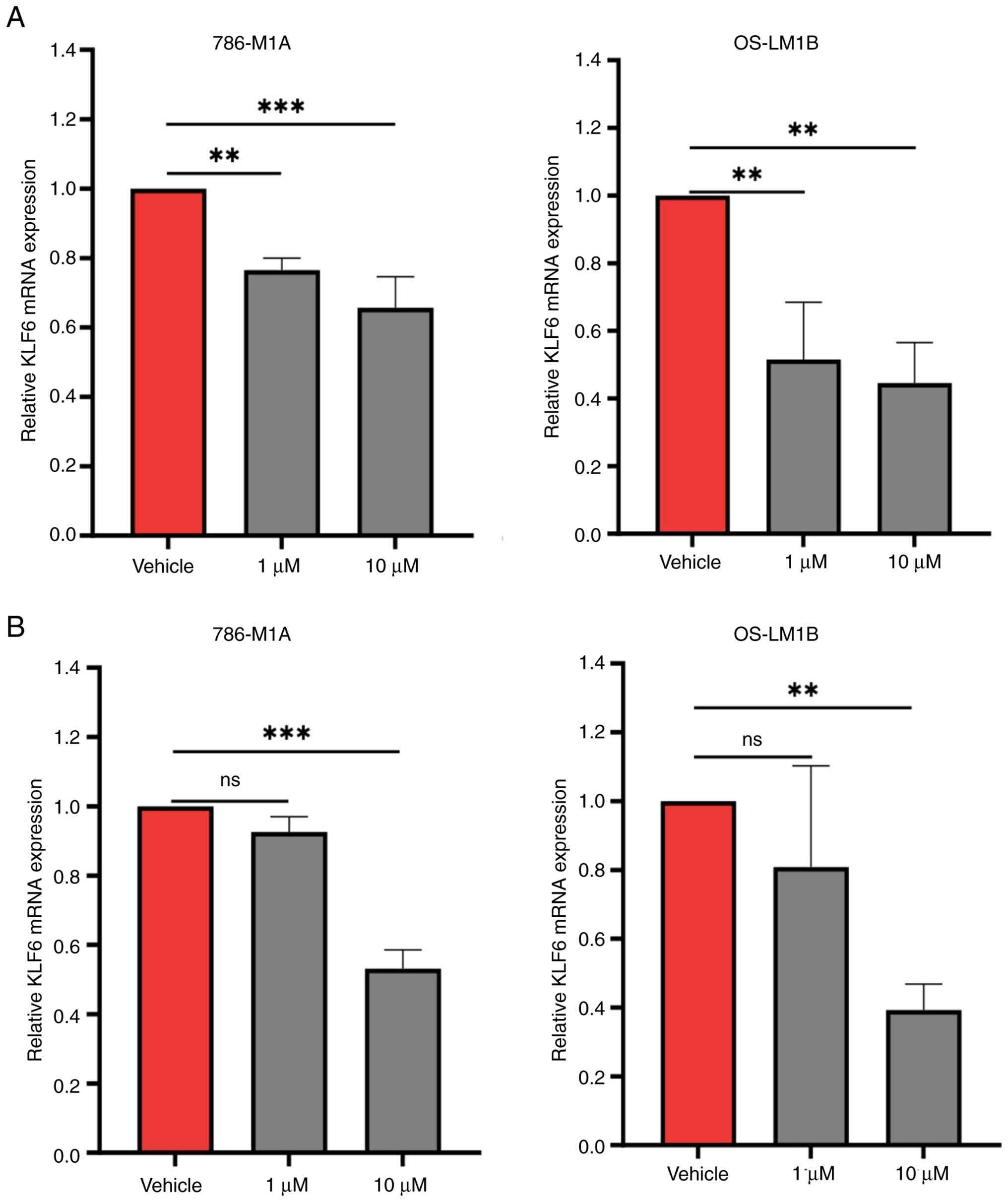
Expression of SE-driven KLF6 is
dependent on BRD4 and p300 activity
KLF6 supports ccRCC cell proliferation by regulating
cell networks through the transcriptional activation of
platelet-derived growth factor β (PDGFβ), linking mTOR
complex 1 (mTORC1) signaling pathway activity to lipid metabolism
(25). While high expression of
KLF6 is driven by one of the largest SEs in ccRCC (25), the roles of BRD4 and p300, key
regulators of SEs and transcription activity, remain underexplored.
Therefore, the present study investigated whether targeting of BRD4
and p300 with their respective inhibitors impairs KLF6
transcriptional activation in ccRCC cells. For BRD4 inhibition,
786-M1A and OS-LM1B cells were treated with 1 or 10 µM JQ1 for 6 h,
which was a time point shown to not affect cell viability (data not
shown), followed by assessment of KLF6 expression via qPCR
analysis. Treatment with either 1 or 10 µM JQ1 significantly
decreased KLF6 expression in both ccRCC cell lines compared
with vehicle-treated controls. Although KLF6 downregulation
was dose-dependent, the difference in the extent of reduction
between 1 and 10 µM JQ1 was not significant (Fig. 3A).
To examine the role of p300, 786-M1A and OS-LM1B
cells were treated with 1 or 10 µM A-485 for 16 h. qPCR analysis
revealed that treatment with 1 µM A-485 did not significantly
downregulate KLF6 expression in either ccRCC cell line.
However, treatment with the higher concentration of A-485 decreased
KLF6 expression by 50–60% in both 786-M1A and OS-LM1B cells
(Fig. 3B). These findings underline
the involvement of BRD4 and p300 in the regulation of KLF6
expression in ccRCC.
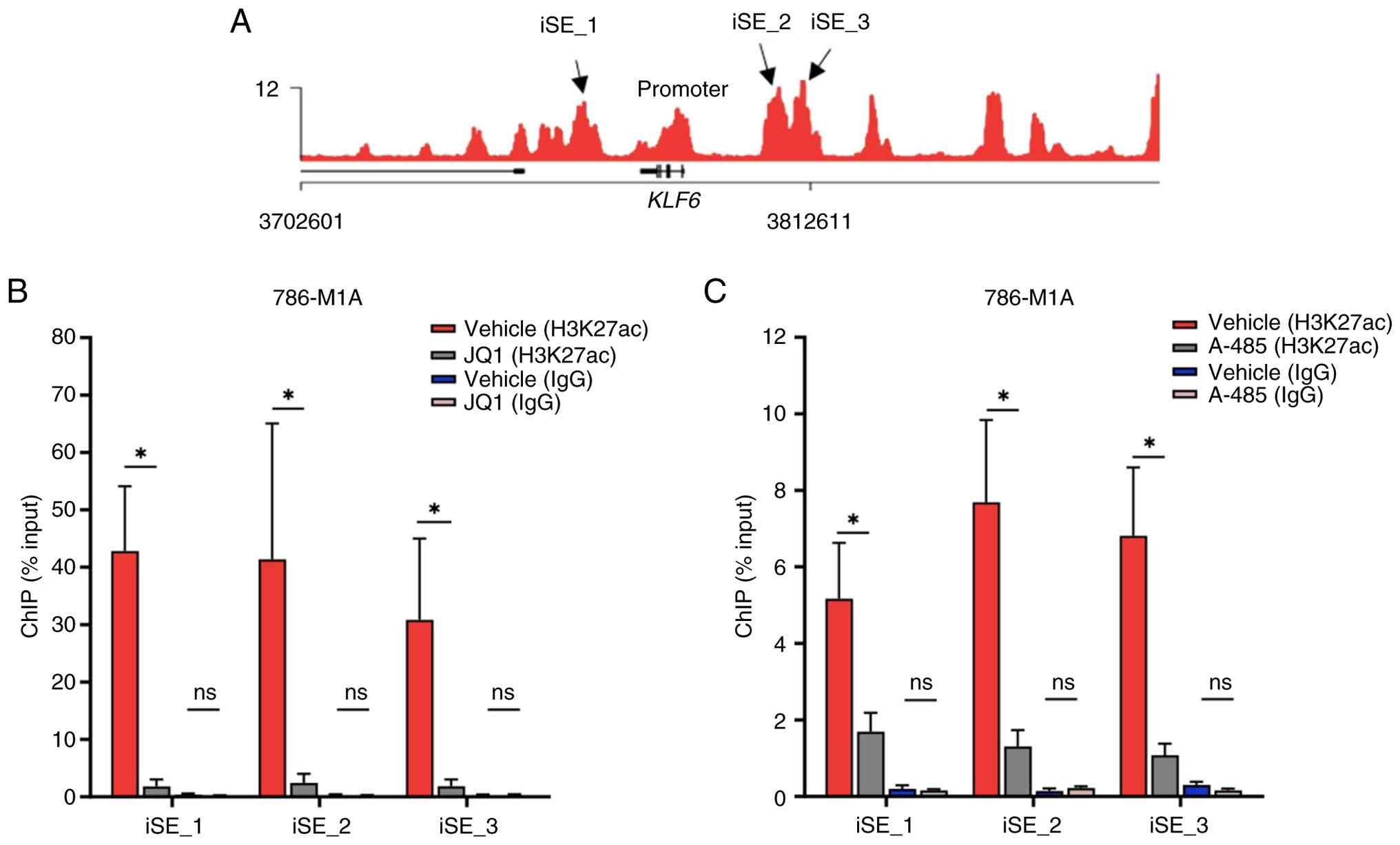
H3K27ac signals are decreased
following BRD4 and p300 inhibition
The present study investigated whether the
downregulation of KLF6 following BRD4 and p300 inhibition
was associated with changes in the chromatin landscape at the
KLF6 SE region. H3K27ac ChIP-qPCR was performed to assess
H3K27ac signal levels in JQ1- and A-485-treated 786-M1A cells.
Constituent enhancers near the KLF6 locus (SE_1, SE_2 and
SE_3) were selected from our previous H3K27ac ChIP-Seq dataset in
ccRCC (25) based on their
enrichment signal and proximity to the KLF6 promoter
(Fig. 4A). Inhibition of BRD4 with
10 µM JQ1 decreased H3K27ac signals at all three constituent
enhancers compared with those of vehicle-treated cells (Fig. 4B).
Similarly, treatment with the p300 inhibitor A-485
also decreased H3K27ac signals at these enhancer regions (Fig. 4C). As a negative control, IgG
ChIP-qPCR was performed in parallel under the same conditions,
which showed minimal background enrichment across all loci,
confirming antibody specificity and negligible non-specific binding
(Fig. 4B and C). Collectively,
inhibition of BRD4 and p300 reduced H3K27ac signals, a marker of
open chromatin and active transcription (7,10), at
SE_1, SE_2 and SE_3 constituent enhancers, which potentially
contributed to KLF6 downregulation.
CRISPR-mediated deacetylation of SE_1
decreases KLF6 expression
A CRISPR-based approach was used to deacetylate the
SE_1, SE_2 and SE_3 regions and assess the effects on chromatin
accessibility and KLF6 expression. Prior to generating the
786-M1A cells that stably express dCas9-HDAC3, the plasmid was
modified to remove the stop codon between the dCas9-HDAC3 and
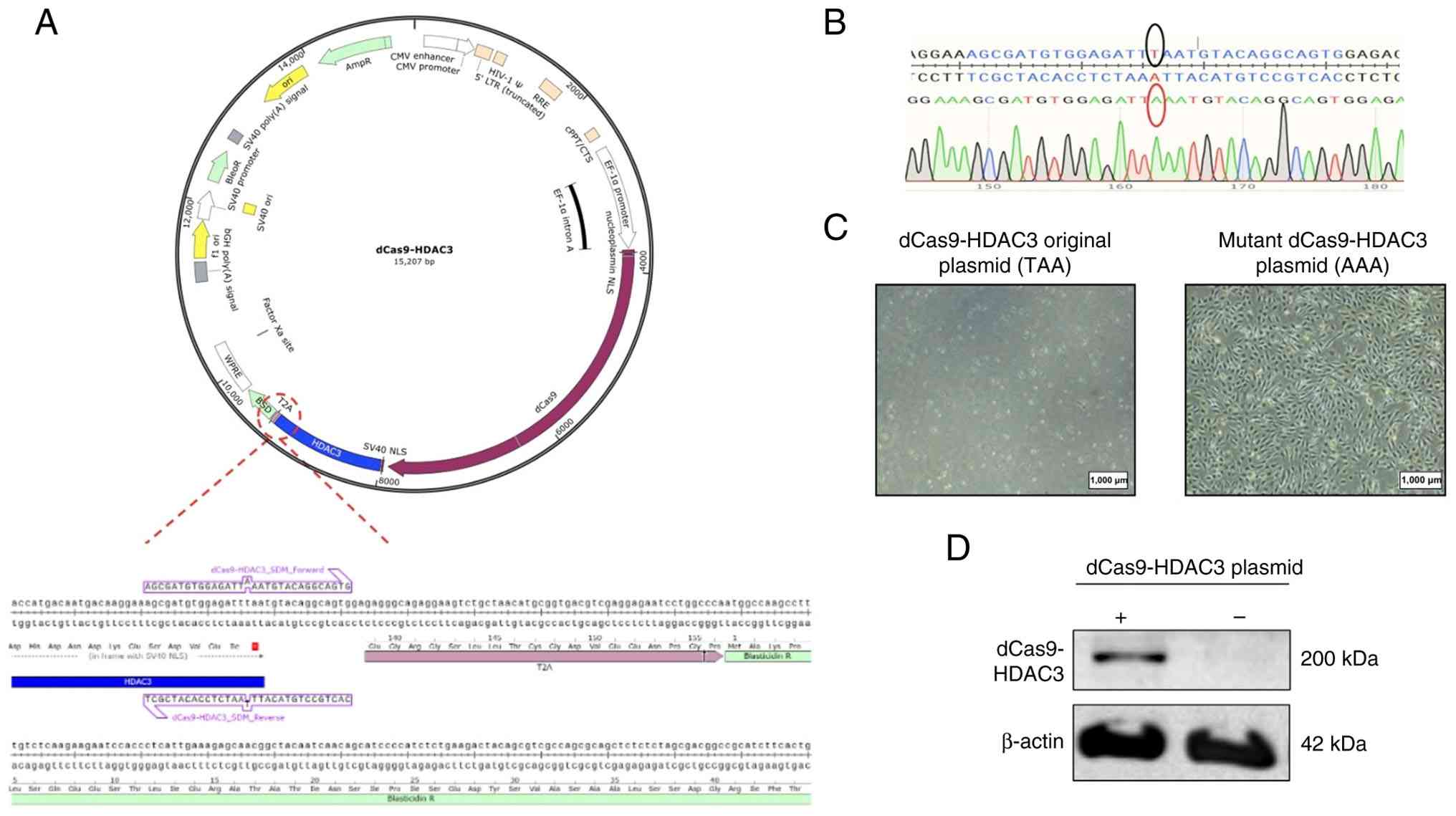
blasticidin-resistant gene reading frames (Fig. 5A). Site-directed mutagenesis was
used to replace the stop codon (TAA) with alanine (AAA), allowing
for blasticidin selection of cells that were successfully
transduced with the dCas9-HDAC3-expressing plasmid (Fig. 5B and C). dCas9-HDAC3 protein
expression in the transduced 786-M1A cells was confirmed by western
blotting (Fig. 5D). These stable
dCas9-HDAC3-expressing 786-M1A cells were transduced with either a
non-targeting sgRNA control or iSE_1, iSE_2 and iSE_3 sgRNAs, which
specifically target the SE_1, SE_2 or SE_3 enhancer regions,
respectively. All sgRNAs were cloned into an sgRNA expression
vector as described in our previous study (25). Western blot analysis confirmed
stable expression of dCas9-HDAC3 in 786-M1A cells transduced with
each sgRNA (Fig. 6A).
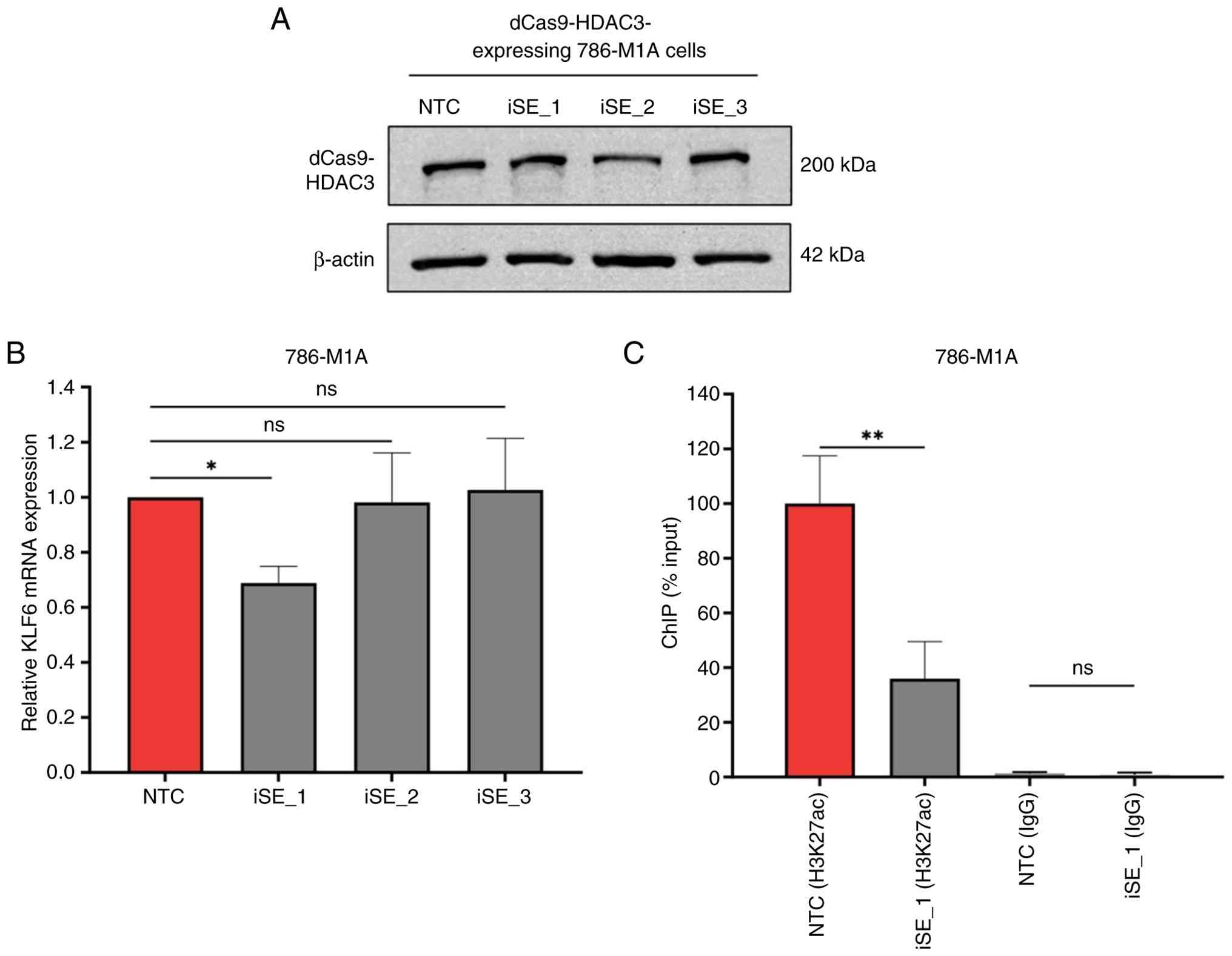 | Figure 6.CRISPR-mediated deacetylation of SE_1
decreases KLF6 expression. (A) Western blot analysis
confirmed stable dCas9-HDAC3 expression in 786-M1A cells transduced
with single guide RNAs. (B) qPCR analysis showed reduced
KLF6 expression in iSE_1-transduced cells compared with NTC
cells. P-values were determined by one-way ANOVA followed by
Dunnett's multiple-comparison post hoc test. (C) ChIP-qPCR showed
decreased H3K27ac enrichment at SE_1 in iSE_1-transduced cells
compared with NTC cells. IgG ChIP-qPCR showed minimal enrichment at
SE_1 in both NTC and iSE_1 transduced cells, confirming low
background signal and antibody specificity. P-values were
determined using an unpaired t-test with Welch's correction.
*P<0.05, **P<0.005. dCas9-HDAC3, dead Cas9-histone
deacetylase 3; qPCR, quantitative PCR; KLF6, Kruppel-like factor 6;
iSE, CRISPRi-targeted super enhancer; NTC, non-targeting control;
ChIP, chromatin immunoprecipitation; H3K27ac, acetylation at lysine
27 of histone H3; ns, not significant. |
qPCR demonstrated ~25% KLF6 expression
downregulation in dCas9-HDAC3-expressing 786-M1A cells transduced
with iSE_1 sgRNA. However, there was no significant decrease in
KLF6 expression in the iSE_2- and iSE_3-transduced cells
(Fig. 6B). The downregulation of
KLF6 in the iSE_1-transduced cells suggested that the
decreased KLF6 expression may result from
dCas9-HDAC3-mediated deacetylation of the SE_1 region. To assess
the deacetylation efficiency, H3K27ac ChIP-qPCR was performed on
the iSE_1 sgRNA-transduced cells. Decreased H3K27ac signals at the
iSE_1-targeted enhancer indicated deacetylation of this region,
which consequently altered chromatin accessibility and impaired
KLF6 transcription (Fig.
6C). As a negative control, parallel IgG ChIP-qPCR demonstrated
low background signal at this locus, confirming the specificity of
the antibody and minimal non-specific binding.
Discussion
The present findings revealed that BRD4 and p300,
key epigenetic modifiers, serve integral roles in the regulation of
chromatin accessibility within the KLF6 SE region and drive
high expression of this gene in ccRCC. These results align with
previous research highlighting the key roles of BRD4 and p300 in
sustaining SE activity and promoting the transcription of oncogenes
in numerous types of cancer including breast and lung cancer and
neuroblastoma (10,31,32).
However, although inhibition of BRD4 or p300 significantly reduced
KLF6 expression in ccRCC cells, the magnitude of this reduction was
moderate (40–50%). KLF6 expression in ccRCC is driven by a
large and robust SE region. Therefore, we hypothesized that
redundancy mechanisms or the involvement of other epigenetic and
transcriptional regulators may compensate for the loss of BRD4 or
p300 activity, allowing the transcription of KLF6 to persist
despite inhibition of these SE regulators.
The modest downregulation of KLF6 observed
following JQ1 treatment may also reflect the intrinsic robustness
of the large KLF6 SE. Given its extensive size and modular
architecture, partial inhibition of BRD4 may be insufficient to
fully disrupt the transcriptional machinery assembled across all
constituent enhancers (2). When
BRD4 activity is decreased, residual occupancy of BRD4 or
compensation by other bromodomain and extraterminal domain family
members together with redundant enhancer modules within the SE may
sustain sufficient chromatin accessibility and co-activator
recruitment to maintain KLF6 transcription (33). This is consistent with our previous
study showing that simultaneous suppression of multiple constituent
enhancers, or deletion of a large genomic region encompassing these
multiple constituent enhancers, was required to achieve a more
pronounced KLF6 downregulation in ccRCC compared with
individual enhancer perturbations (25). Additionally, it is possible that the
JQ1 and A-485 treatment conditions did not result in complete
inhibition of BRD4 and p300, respectively.
Our previous genome-wide H3K27ac ChIP-Seq profiling
and CRISPR/Cas9 deletion of enhancer clusters upstream of
KLF6 provided direct evidence that this SE is functionally
required for KLF6 transcription in ccRCC (25). While genome-wide H3K27ac profiling
was not performed in the present study, the targeted ChIP-qPCR and
transcriptional assays in the present study focused specifically on
previously validated SE constituents (25). The present study assessed H3K27ac
signals in only three constituent enhancers via ChIP-qPCR. Although
significant decreases in H3K27ac signals were observed at SE_1,
SE_2 and SE_3 regions upon BRD4 and p300 inhibition, future studies
should investigate the effect of these inhibitions on H3K27ac
signals not only across the entire KLF6 SE locus but also at
the genome-wide level using ChIP-Seq. This comprehensive analysis
may allow for a deeper understanding of the global chromatin
modifications induced by these inhibitors and their broader impact
on gene expression regulation.
The present study deepens understanding of the KLF6
transcriptional networks in ccRCC by elucidating the roles of the
epigenetic regulators BRD4 and p300 in governing this process.
These insights may pave the way for the development of novel
diagnostic and therapeutic strategies for ccRCC. Although
inhibition of BRD4 and p300 decreased H3K27ac signals and
KLF6 expression, it remains uncertain whether the decreased
viability in BRD4- or p300-inhibited cells is directly linked to
perturbation in the KLF6 transcriptional network. As BRD4 and p300
are essential epigenetic and transcriptional regulators, targeting
these proteins affects genome-wide transcriptional activity,
suggesting that the phenotypic impacts of these inhibitors may
extend beyond the direct regulation of KLF6 expression. As
shown in our previous study, KLF6 knockdown impairs ccRCC
cell proliferation and decreases PDGFβ transcription,
supporting its key role in cell viability and downstream signaling
(25). These data support KLF6 as a
key effector of BRD4/p300-regulated phenotypes. In line with this,
BRD4 inhibition suppresses ccRCC cell proliferation and
epithelial-to-mesenchymal transition by activating the NF-κB/NLR
family pyrin domain containing 3/caspase-1 pyroptosis signaling
pathway (34). JQ1-mediated BRD4
inhibition exerts anticancer effects in ccRCC by downregulating the
expression of numerous oncogenes, including MYC (35). On the other hand, Dong et al
(36) used a computational approach
to link normal developmental programs to cancer-specific driver
mutations. The aforementioned study identified a regulatory network
associated with a poor prognosis subtype, classified as the C0
group, that was maintained by p300. Analysis of drug response data
from ccRCC cell lines predicted to be in the C0 group revealed that
8 out of 15 cell lines were sensitive to the p300 inhibitor A-485
(36).
Although several studies, including the present
study, have demonstrated the inhibitory effects of JQ1 and A-485 on
ccRCC cell viability (34–37), their impact on normal cells and
potential genome-wide consequences warrant further investigation. A
challenge in targeting SEs for cancer treatment is specificity.
This is because the main SEs and transcription regulatory proteins
such as BRD4 and p300 also serve key roles in modulating normal
cell and physiological processes (1,2),
making it difficult to selectively target cancer cells without
affecting normal cell function. Therefore, it is crucial to
determine the SE-driven transcriptional networks that are governed
by these epigenetic regulators. By identifying other candidate
proteins or pathways that are specifically active in cancer cells,
it may be possible to develop more targeted therapies with minimal
off-target effects on normal cells. For example, our previous study
identified a cell signaling loop that links SE-driven KLF6 to
mTORC1 signaling hyperactivation and lipid metabolism, which
support ccRCC cell proliferation and progression (25). KLF6 serves a central role in
regulating this cellular network by transactivating the expression
of pro-angiogenic PDGFβ, a key agonist of the mTORC1
signaling pathway (25,38). The upregulation of angiogenesis and
hyperactivation of the mTORC1 signaling pathway are ccRCC hallmark
features; a number of clinically approved therapies for ccRCC have
been designed to target these phenotypes, including the
angiogenesis inhibitors sunitinib and axitinib as well as the
mTORC1 pathway inhibitors everolimus and temsirolimus (39). Collectively, the aforementioned
findings on KLF6 underscore the importance of a comprehensive
investigation and understanding of SE regulation and activity, as
well as downstream cellular networks, to uncover cancer-specific
drivers and identify novel therapeutic targets.
Advancements in CRISPR technology have led to the
evolution of its applications beyond traditional gene editing. The
present study used the CRISPR-mediated histone deacetylation tool
to assess its efficiency in perturbing KLF6 SE activity and
the transcription of this gene. Of the three constituent enhancers,
modest downregulation of KLF6 expression was observed only
in SE_1-targeted cells. This corroborates our previous study on the
robustness of this SE region and its modular function in driving
KLF6 expression in ccRCC (25). In our previous study, a marked
reduction in KLF6 expression was only achieved when multiple
constituent enhancers were concurrently suppressed using CRISPRi or
by deleting large genomic region containing these multiple clusters
of enhancer using traditional CRISPR/Cas9 (25). These findings contrast with numerous
studies that have shown that cancer-associated SEs are sensitive to
perturbations of constituent enhancers or the components
responsible for regulating the SE landscape (38–40).
Nonetheless, the redundancy and robustness in the regulation of
KLF6 SE activity and transcription are aligned with the role
of KLF6 in driving ccRCC phenotypes (25). Most key biological processes and
developmental transcriptional programs are relatively insensitive
to genetic changes or environmental perturbations, reflecting
regulatory robustness (42).
The HDAC family comprises >10 members, including
HDAC3 (43). Given the diverse
regulatory functions and substrates of HDACs (43), dCas9-HDAC3 alone may not be
sufficient to efficiently deacetylate the targeted constituent
enhancers. A more comprehensive approach, potentially involving
deacetylation by multiple HDACs, warrants further investigation to
evaluate the efficiency of the CRISPR approach in modifying the
chromatin landscape and modulating transcriptional regulation.
Future studies should evaluate dCas9 fusions with alternative HDAC
isoforms to determine whether specific HDACs display higher
efficiency at renal SE regions.
A limitation of this study is the absence of
non-tumorigenic renal epithelial as well as non-renal cancer
models, precluding definitive assessment of whether
BRD4/p300-dependent KLF6 super-enhancer regulation is
ccRCC-specific. Future studies including such models will be
necessary to establish lineage specificity. This is particularly
relevant because renal enhancer identity and transcriptional
programs in ccRCC are shaped by renal lineage factors such as
paired box 8 (44). JQ1 and A-485
exert broad epigenetic effects and KLF6 downregulation may
not be exclusively due to direct SE disruption. However, our
previous study demonstrated that genetic perturbation of
KLF6 or its upstream enhancer cluster result in similar
phenotypes, including suppression of mTORC1-associated signaling
and decreased ccRCC cell viability (20). While the present study did not
perform KLF6 rescue experiments following BRD4 or p300
inhibition, the consistent decrease in KLF6 expression and
associated phenotypes support its functional contribution. Future
studies incorporating KLF6 rescue or epistasis analyses
following pharmacological perturbation would be valuable to further
clarify this causal association. Additionally, genome-wide
approaches such as RNA-sequencing and H3K27ac ChIP-seq following
BRD4 or p300 inhibition would help distinguish KLF6-dependent
effects from broader transcriptional and chromatin changes.
In conclusion, the present study revealed that BRD4
and p300 are key regulators of KLF6 super-enhancer activity
in ccRCC. Together with our previous findings demonstrating the
super enhancer activity in driving high KLF6 expression
(25), these results further
highlight BRD4 and p300 as potential therapeutic targets in this
cancer. Additionally, targeted deacetylation of KLF6
enhancer regions using dCas9-HDAC3 demonstrated that deacetylation
of an individual region was insufficient to fully suppress
KLF6 expression. This observation aligns with our previous
study, which highlighted the robustness of the KLF6 SE and
the existence of regulatory redundancy or compensatory mechanisms
within this SE landscape (25).
Future research employing combinatorial epigenetic perturbation
strategies may provide valuable insights into the complex
regulatory networks of SEs in cancer, potentially leading to the
development of more effective and targeted therapeutic
approaches.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Dr Joan
Massagué, Memorial Sloan Kettering Cancer Center, NY, USA, for
providing the 786-M1A and OS-LM1B ccRCC cell lines. We also thank
Dr Marston Linehan, National Cancer Institute, MD, USA, for
providing the UOK101 ccRCC cell line.
Funding
The present study was supported by Geran Universiti
Penyelidikan, Universiti Kebangsaan Malaysia (grant no.
GUP-2023-005), the Dr Ranjeet Bhagwan Singh Medical Research Grant
(grant no. JJ-2020-005) and the Ministry of Higher Education
Fundamental Research Grant Scheme (grant no.
FRGS/1/2020/SKK0/UKM/03/3).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SES and MAM conceived and designed the study and
edited the manuscript. SES, NNMZ, AYA, NQAAR and SNHMY performed
experiments. SES and NNMZ analyzed the data and wrote the
manuscript. SES, NNMZ and NQAAR confirm the authenticity of all the
raw data. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Whyte WA, Orlando DA, Hnisz D, Abraham BJ,
Lin CY, Kagey MH, Rahl PB, Lee TI and Young RA: Master
transcription factors and mediator establish super-enhancers at key
cell identity genes. Cell. 153:307–319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hnisz D, Abraham BJ, Lee TI, Lau A,
Saint-André V, Sigova AA, Hoke HA and Young RA: Super-enhancers in
the control of cell identity and disease. Cell. 155:934–947. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou RW and Parsons RE: Etiology of
super-enhancer reprogramming and activation in cancer. Epigenetics
Chromatin. 16:292023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sengupta S and George RE:
Super-enhancer-driven transcriptional dependencies in cancer.
Trends Cancer. 3:269–281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang F, Yang Z, Tan Y and Li Y:
Super-enhancer function and its application in cancer targeted
therapy. NPJ Precis Oncol. 4:22020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Filippakopoulos P, Qi J, Picaud S, Shen Y,
Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et
al: Selective inhibition of BET bromodomains. Nature.
468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chiang CM: Brd4 engagement from chromatin
targeting to transcriptional regulation: selective contact with
acetylated histone H3 and H4. F1000 Biol Rep. 1:982009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qian H, Zhu M, Tan X, Zhang Y, Liu X and
Yang L: Super-enhancers and the super-enhancer reader BRD4:
tumorigenic factors and therapeutic targets. Cell Death Discov.
9:1–11. 2023. View Article : Google Scholar
|
|
9
|
Donati B, Lorenzini E and Ciarrocchi A:
BRD4 and cancer: Going beyond transcriptional regulation. Mol
Cancer. 17:1642018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang M, Chen Q, Wang S, Xie H, Liu J,
Huang R, Xiang Y, Jiang Y, Tian D and Bian E: Super-enhancers
complexes zoom in transcription in cancer. J Exp Clin Cancer Res.
42:1832023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Strachowska M and Robaszkiewicz A:
Characteristics of anticancer activity of CBP/p300
inhibitors-Features of their classes, intracellular targets and
future perspectives of their application in cancer treatment.
Pharmacol Ther. 257:1086362024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Q, Yang B, Liu X, Zhang XD, Zhang L
and Liu T: Histone acetyltransferases CBP/p300 in tumorigenesis and
CBP/p300 inhibitors as promising novel anticancer agents.
Theranostics. 12:4935–4948. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Adli M: The CRISPR tool kit for genome
editing and beyond. Nat Commun. 9:19112018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zamberi NN, Abuhamad AY, Low TY, Mohtar MA
and Syafruddin SE: dCas9 tells tales: Probing gene function and
transcription regulation in cancer. CRISPR J. 7:73–87. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Q, Ma C, Mao H and Wang J: Epigenome
editing by a CRISPR-Cas9-based acetyltransferase activates the
ZNF334 gene to inhibit the growth of colorectal cancer. Int J Biol
Macromol. 277:1345802024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang L, Wang E, Balcazar JP, Wu Z, Xiang
K, Wang Y, Huang Q, Negrete M, Chen KY, Li W, et al: Chromatin
remodeling of colorectal cancer liver metastasis is mediated by an
HGF-PU.1-DPP4 axis. Adv Sci (Weinh). 8:e20046732021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu J, Sun M, Cho KB, Gao X and Guo B: A
CRISPR-Cas9 repressor for epigenetic silencing of KRAS. Pharmacol
Res. 164:1053042021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsieh JJ, Purdue MP, Signoretti S, Swanton
C, Albiges L, Schmidinger M, Heng DY, Larkin J and Ficarra V: Renal
cell carcinoma. Nat Rev Dis Primers. 3:170092017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barragan-Carrillo R, Saad E, Saliby RM,
Sun M, Albiges L, Bex A, Heng D, Mejean A, Motzer RJ, Plimack ER,
et al: First and second-line treatments in metastatic renal cell
carcinoma. Eur Urol. 87:143–154. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mattila KE, Vainio P and Jaakkola PM:
Prognostic factors for localized clear cell renal cell carcinoma
and their application in adjuvant therapy. Cancers (Basel).
14:2392022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Turajlic S, Xu H, Litchfield K, Rowan A,
Horswell S, Chambers T, O'Brien T, Lopez JI, Watkins TBK, Nicol D,
et al: Deterministic evolutionary trajectories influence primary
tumor growth: TRACERx renal. Cell. 173:595–610. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aweys H, Lewis D, Sheriff M, Rabbani RD,
Lapitan P, Sanchez E, Papadopoulos V, Ghose A and Boussios S: Renal
cell cancer-insights in drug resistance mechanisms. Anticancer Res.
43:4781–4792. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Makhov P, Joshi S, Ghatalia P, Kutikov A,
Uzzo RG and Kolenko VM: Resistance to systemic therapies in clear
cell renal cell carcinoma: mechanisms and management strategies.
Mol Cancer Ther. 17:1355–1364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lovén J, Hoke HA, Lin CY, Lau A, Orlando
DA, Vakoc CR, Bradner JE, Lee TI and Young RA: Selective inhibition
of tumor oncogenes by disruption of super-enhancers. Cell.
153:320–334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Syafruddin SE, Rodrigues P, Vojtasova E,
Patel SA, Zaini MN, Burge J, Warren AY, Stewart GD, Eisen T, Bihary
D, et al: A KLF6-driven transcriptional network links lipid
homeostasis and tumour growth in renal carcinoma. Nat Commun.
10:1–13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vanharanta S, Shu W, Brenet F, Hakimi AA,
Heguy A, Viale A, Reuter VE, Hsieh JJ, Scandura JM and Massagué J:
Epigenetic expansion of VHL-HIF signal output drives multiorgan
metastasis in renal cancer. Nat Med. 19:50–56. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kwon DY, Zhao YT, Lamonica JM and Zhou Z:
Locus-specific histone deacetylation using a synthetic
CRISPR-Cas9-based HDAC. Nat Commun. 8:153152017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative CT method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qi LS, Larson MH, Gilbert LA, Doudna JA,
Weissman JS, Arkin AP and Lim WA: Repurposing CRISPR as an
RNA-guided platform for sequence-specific control of gene
expression. Cell. 152:1173–1183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haring M, Offermann S, Danker T, Horst I,
Peterhansel C and Stam M: Chromatin immunoprecipitation:
Optimization, quantitative analysis and data normalization. Plant
Methods. 3:112007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jia Q, Chen S, Tan Y, Li Y and Tang F:
Oncogenic super-enhancer formation in tumorigenesis and its
molecular mechanisms. Exp Mol Med. 52:713–723. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thandapani P: Super-enhancers in cancer.
Pharmacol Ther. 199:129–138. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarnik J, Popławski T and Tokarz P: BET
proteins as attractive targets for cancer therapeutics. Int J Mol
Sci. 22:111022021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tan YF, Wang M, Chen ZY, Wang L and Liu
XH: Inhibition of BRD4 prevents proliferation and
epithelial-mesenchymal transition in renal cell carcinoma via NLRP3
inflammasome-induced pyroptosis. Cell Death Dis. 11:2392020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sakaguchi T, Yoshino H, Sugita S, Miyamoto
K, Yonemori M, Osako Y, Meguro-Horike M, Horike SI, Nakagawa M and
Enokida H: Bromodomain protein BRD4 inhibitor JQ1 regulates
potential prognostic molecules in advanced renal cell carcinoma.
Oncotarget. 9:23003–23017. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dong X, Zhang D, Zhang X and Liu Y and Liu
Y: Network modeling links kidney developmental programs and the
cancer type-specificity of VHL mutations. NPJ Syst Biol Appl.
10:1142024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kuang Z, Guo K, Cao Y, Jiang M, Wang C, Wu
Q, Hu G, Ao M, Huang M, Qin J, et al: The novel CDK9 inhibitor,
XPW1, alone and in combination with BRD4 inhibitor JQ1, for the
treatment of clear cell renal cell carcinoma. Br J Cancer.
129:1915–1929. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Abuhamad AY, Zamberi NN, Vanharanta S,
Yusuf SNH, Mohtar MA and Syafruddin SE: Cancer cell-derived PDGFB
stimulates mTORC1 activation in renal carcinoma. Int J Mol Sci.
24:64472023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choueiri TK and Motzer RJ: Systemic
therapy for metastatic renal-cell carcinoma. N Engl J Med.
376:354–366. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chipumuro E, Marco E, Christensen CL,
Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C,
Altabef A, et al: CDK7 inhibition suppresses super-enhancer-linked
oncogenic transcription in MYCN-driven cancer. Cell. 159:1126–1139.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bhagwat AS, Roe J-S, Mok BYL, Hohmann AF,
Shi J and Vakoc CR: BET bromodomain inhibition releases the
mediator complex from select cis-regulatory elements. Cell Rep.
15:519–530. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Félix MA and Barkoulas M: Pervasive
robustness in biological systems. Nat Rev Genet. 16:483–496. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Seto E and Yoshida M: Erasers of histone
acetylation: The histone deacetylase enzymes. Cold Spring Harb
Perspect Biol. 6:a0187132014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Patel SA, Hirosue S, Rodrigues P,
Vojtasova E, Richardson EK, Ge J, Syafruddin SE, Speed A,
Papachristou EK, Baker D, et al: The renal lineage factor PAX8
controls oncogenic signalling in kidney cancer. Nature.
606:999–1006. 2022. View Article : Google Scholar : PubMed/NCBI
|