Introduction
Renal cancer is the most prevalent malignancy
affecting the urinary system, with both incidence and mortality
rates rising steadily each year. In 2020, kidney cancer accounted
for an estimated 431,288 new cases and 179,368 deaths worldwide,
with ~73,600 new cases and ~25,200 deaths in China (1–3). Clear
cell renal cell carcinoma (ccRCC) is the most common subtype,
accounting for 70–80% of all renal cancer cases (4,5).
Despite improvements in the diagnosis and treatment of ccRCC, the
prognosis remains unfavorable, largely due to late-stage diagnosis,
the tendency for metastasis and limited treatment options,
particularly in advanced or refractory cases (6–8).
At the molecular level, ccRCC development and
progression are driven by complex genetic, epigenetic and signaling
alterations that collectively promote tumor growth, dissemination
and therapeutic resistance. Among the pathways implicated in ccRCC
biology, the Janus kinase-signal transducer and activator of
transcription (JAK-STAT) signaling pathway has emerged as a
critical regulator of renal tumorigenesis and disease
aggressiveness (9,10). Activated primarily by cytokines and
growth factors, the JAK-STAT pathway governs essential cellular
processes, including proliferation, survival, differentiation and
immune regulation (11).
Accumulating evidence has indicated that the aberrant activation of
JAK-STAT signaling, particularly persistent signal transducer and
activator of transcription 3 (STAT3) activation, contributes to
enhanced tumor cell growth, migration, resistance to apoptosis, and
immune evasion in multiple malignancies, including ccRCC (12–19).
In addition, dysregulated STAT3 signaling has been directly linked
to ccRCC progression, drug resistance and aggressive clinical
behavior (20–22).
Mucins are a family of high-molecular-weight
glycoproteins that are predominantly expressed on epithelial
surfaces, where they play essential roles in maintaining epithelial
integrity, barrier function, and cell-cell communication. Beyond
their physiological functions, aberrant mucin expression has been
increasingly recognized as a contributor to tumor initiation,
progression and metastasis (23,24).
Among membrane-associated mucins, mucin 3A (MUC3A) is a
transmembrane glycoprotein primarily localized to the apical
surface of epithelial cells and has been implicated in regulating
epithelial homeostasis and signaling processes (25,26).
Previous studies have identified MUC3A as an
important oncogenic regulator in several malignancies. In
colorectal cancer, MUC3A has been demonstrated to promote tumor
progression through the activation of the PI3K/AKT/mTOR signaling
pathway (27). In non-small cell
lung cancer, MUC3A was shown to contribute to tumor growth and
radioresistance through the activation of NF-κB signaling and
modulation of immune-related pathways, including programmed
death-ligand 1 expression (28,29).
In addition, emerging evidence suggests that MUC3A may participate
in inflammatory and immune-related signaling networks, further
supporting its potential role in shaping the tumor microenvironment
(30,31).
Despite these findings in other cancer types, the
expression pattern, biological function and mechanistic role of
MUC3A in ccRCC remain largely unexplored. Given its
membrane-associated structure, involvement in oncogenic signaling,
and reported links to immune and inflammatory pathways, it is
plausible that MUC3A may influence ccRCC progression through the
modulation of key signaling cascades. In particular, the functional
convergence between MUC3A-associated signaling and the JAK-STAT
pathway raises the possibility that MUC3A may act as an upstream
regulator or facilitator of JAK-STAT activation in ccRCC, a
hypothesis that has not yet been systematically investigated.
The present study, sought to comprehensively
characterize the expression and clinical relevance of MUC3A in
ccRCC and explore its potential functional association with the
JAK-STAT signaling pathway. By integrating bioinformatics analyses
based on public datasets with in vitro functional and
mechanistic experiments, the aim was to determine whether MUC3A
contributes to ccRCC progression and to clarify its possible role
in regulating JAK-STAT-mediated oncogenic processes. Through this
approach, the present study provided novel insights into
mucin-associated signaling in ccRCC and identified MUC3A as a
potential molecular target worthy of further investigation.
Materials and methods
Cell culture
Human ccRCC cell lines 786-O (cat. no. CL-0010),
OSRC-2 (cat. no. CL-0177), Caki-1 (cat. no. CL-0052) and ACHN (cat.
no. CL-0021), as well as non-malignant renal epithelial cells HK-2
(cat. no. CL-0109; all from Procell Life Science & Technology
Co., Ltd.) and RPTEC/TERT1 (cat. no. CRL-4031; American Type
Culture Collection), were used in the present study. All cell lines
were maintained in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin, at 37°C in a humidified incubator
containing 5% CO2.
Cells were routinely passaged every 2–3 days and
used for experiments during the logarithmic growth phase. To
minimize variability, all in vitro experiments were
performed under identical culture conditions, and comparative
analyses were conducted within the same cell line, with the control
and treatment groups processed in parallel.
Small interfering RNA (siRNA)
transfection
siRNAs targeting human MUC3A and a non-targeting
negative control siRNA were synthesized by Shanghai GenePharma Co.,
Ltd., consistent with previously reported renal cell carcinoma
studies (32,33). The siRNA sequences (RNA, U-bases)
were as follows: Human mucin 3A (hMUC3A) si-1 sense,
5′-ACgACCUUCCCAgCAACAUAU-3′ and antisense,
5′-AUAUgUUgCUgggAAggUCgU-3′; hMUC3A si-2 sense,
5′-CUACgUUgggUACCAUggUAA-3′ and antisense,
5′-UUACCAUggUACCCAACgUAg-3′; hMUC3A si-3 sense,
5′-ggACCAACUUUCACAAgUA-3′ and antisense, 5′-UACUUgUgAAAgUUggUCC-3′.
The non-targeting negative control siRNA sequences (RNA, U-bases)
were: Sense, 5′-UUCUCCGAACGUGUCACGUdUdU-3′ and antisense,
5′-ACGUGACACGUUCGGAGAAdUdU-3′.
For functional assays, 786-O and OSRC-2 cells were
seeded into 6-well plates at 2.5×105 cells/well 24 h
prior to transfection to reach 50–60% confluence at the time of
transfection. siRNAs were transfected at a final concentration of
50 nM using Lipofectamine™ RNAiMAX (cat. no. 13778150; Thermo
Fisher Scientific, Inc.) and Opti-MEM (Gibco; Thermo Fisher
Scientific, Inc.), according to the manufacturer's isntructions.
Briefly, siRNA and RNAiMAX (5 µl/well) were separately diluted in
Opti-MEM, combined and incubated for 10–20 min at room temperature,
and then added dropwise to the cells. A non-targeting siRNA
(si-Ctrl) was used as the transfection control in all
experiments.
After 6 h, the transfection medium was replaced with
complete growth medium. Knockdown efficiency was evaluated by
western blotting at 48 h post-transfection (relative to si-Ctrl).
Unless otherwise indicated, CCK-8 proliferation and wound-healing
assays were initiated at 24–48 h post-transfection; apoptosis
analysis by flow cytometry and apoptosis-related western blotting
were performed at 48 h post-transfection, and colony formation
assays were initiated at 24 h post-transfection and maintained for
10–14 days. All experiments were performed with at least three
independent biological replicates.
Western blotting
Total cellular proteins were extracted using RIPA
lysis buffer supplemented with protease and phosphatase inhibitors
(Beyotime Insitute of Biotechnology) on ice. Lysates were clarified
by centrifugation at 12,000 × g for 15 min at 4°C, and the
supernatants were collected. Protein concentrations were determined
using a BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Equal amounts of protein (30 µg per lane) were mixed with loading
buffer, denatured at 95°C for 5 min, separated by 8–12% SDS-PAGE
and transferred onto polyvinylidene difluoride membranes
(MilliporeSigma) using a wet transfer system at 300 mA for 90
min.
Membranes were blocked with 5% non-fat milk (Beijing
Solarbio Science & Technology) in Tris-buffered saline with
0.1% Tween-20 (TBST) for 1 h at room temperature and incubated
overnight at 4°C with primary antibodies against MUC3A (rabbit
polyclonal; cat. no. ab270247; dilution, 1:1,000; Abcam), JAK1
(rabbit monoclonal; cat. no. 3344; dilution, 1:1,000),
phosphorylated (p)-JAK1 (Tyr1034/1035; rabbit monoclonal; cat. no.
74129; dilution, 1:1,000), STAT3 (rabbit monoclonal; cat. no. 9139;
dilution, 1:1,000), p-STAT3 (Tyr705) (rabbit monoclonal; cat. no.
#9145; dilution, 1:1,000), Bcl-2 (rabbit monoclonal; cat. no.
#4223; dilution, 1:1,000), and cleaved Caspase-3 (rabbit
monoclonal; cat. no. #9664; dilution, 1:1,000; all from Cell
Signaling Technology, Inc.), and GAPDH (mouse monoclonal; cat. no.
60004-1-Ig; dilution, 1:5,000; Proteintech Group, Inc.). Membranes
were washed in TBST (3 washes for 5-10 min) and incubated with
HRP-conjugated secondary antibodies (anti-rabbit IgG; cat. no.
7074; dilution, 1:5,000; anti-mouse IgG; cat. no. 7076; dilution,
1:5,000; Cell Signaling Technology, Inc.) for 1 h at room
temperature.
Protein bands were visualized using enhanced
chemiluminescence reagents (Pierce™ ECL Western Blotting Substrate;
cat. no. 32106; Thermo Fisher Scientific, Inc.) and quantified by
ImageJ software (version 1.53; National Institutes of Health)
(34,35). Densitometric values were normalized
to GAPDH and expressed relative to the corresponding control group.
All western blotting experiments were performed with at least three
independent biological replicates.
Cell migration and invasion
assays
Cell migration and invasion were assessed using
Transwell chambers with 8-µm pore size polycarbonate membranes
(cat. no. 3422, 12-well format; Corning, Inc.), as previously
described in ccRCC studies (36,37).
For migration assays, 5×104 786-O and OSRC-2 cells
suspended in 100 µl serum-free medium were seeded into the upper
chambers, while the lower chambers were filled with 800 µl complete
medium containing 10% FBS as a chemoattractant.
For invasion assays, the upper chambers were
pre-coated with Matrigel Basement Membrane Matrix (cat. no. 356234;
Corning, Inc.), diluted at 1:8 in serum-free medium, and allowed to
polymerize at 37°C for 30 min prior to cell seeding. A total of
1×105 786-O and OSRC-2 cells in 100 µl serum-free medium
were seeded per insert, and 800 µl complete medium with 10% FBS was
added to the lower chamber. Cells were incubated at 37°C for 24 h
(migration) or 36–48 h (invasion). To minimize the influence of
cell proliferation, cells were maintained under serum-free
conditions in the upper chamber, and incubation times were kept
within commonly used ranges.
Following incubation, non-migrated/non-invasive
cells on the upper surface were gently removed using a cotton swab.
Cells on the lower surface were fixed with 4% paraformaldehyde
(Beijing Solarbio Science & Technology) for 15 min at room
temperature, stained with 0.1% crystal violet (Beijing Solarbio
Science & Technology) for 15 min at room temperature, and
counted in five randomly selected, non-overlapping fields per
insert under a light microscope (scale bar, 100 µm). Each condition
was assayed in triplicate inserts and repeated in at least three
independent biological experiments.
Wound healing assay
Cell migration was assessed using a wound healing
assay. 786-O and OSRC-2 cells were seeded into 6-well plates and
cultured until reaching near confluence (90–100%). A uniform
scratch was generated across the cell monolayer using a sterile
200-µl pipette tip. Detached cells were gently removed by washing
with PBS three times, and cells were subsequently cultured in
serum-free DMEM to minimize the confounding effects of cell
proliferation.
Images of the wound area were captured at 0 and 24 h
under a light microscope at the same marked locations per well.
Wound closure was quantified using ImageJ software (version 1.53)
by measuring the wound area at each time point. The percentage of
wound closure was calculated as (Ao -
At)/Ao × 100%, where Ao is the
wound area at 0 h and At is the wound area at 24 h
(38). Each condition was assayed
in triplicate wells and repeated in at least three independent
biological experiments.
Cell proliferation assay
Cell proliferation was assessed using the Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.). 786-O
and OSRC-2 cells were seeded into 96-well plates at a density of
5×102 cells/well in 100 µl complete medium and allowed
to attach overnight. Cells were then subjected to the indicated
transfections/treatments. At 24, 48, 72 and 96 h, 10 µl CCK-8
reagent was added to each well and incubated for 2 h at 37°C.
Absorbance was measured at 450 nm using a microplate reader. Wells
containing medium plus CCK-8 but without cells were used as blanks
for background subtraction. The proliferation rate was determined
based on the optical density readings following blank correction.
Each condition was assayed in six replicate wells and repeated in
at least three independent biological experiments.
Colony formation assay
To evaluate clonogenic ability, 786-O and OSRC-2
cells were seeded into 6-well plates at a density of 500
cells/well. Cells were subsequently transfected with si-MUC3A or
the si-Ctrl as described above. Following transfection, cells were
maintained in complete medium, which was refreshed every 2 days,
and colonies were allowed to form for 10–14 days.
At the end of the incubation period, colonies were
fixed with 4% paraformaldehyde for 15 min at room temperature and
stained with 0.1% crystal violet dye for 15 min at room
temperature. Colonies containing >50 cells were counted manually
under a light microscope (Leica DMi1; Leica Microsystems GmbH).
Clonogenic efficiency was quantified as the number of colonies per
well. Each condition was assayed in triplicate wells and repeated
in at least three independent biological experiments.
Flow cytometric analysis of
apoptosis
Apoptosis was assessed by flow cytometry using
Annexin V-FITC/propidium iodide (PI) staining. 786-O and OSRC-2
cells were harvested 48 h following transfection, washed twice with
cold PBS, and resuspended in 1X binding buffer. For each sample,
1×105 cells were stained according to the manufacturer's
instructions using an Annexin V-FITC Apoptosis Detection Kit (cat.
no. 556547; BD Biosciences). Briefly, cells were incubated with
Annexin V-FITC and PI for 15 min at room temperature in the dark,
followed by the addition of binding buffer prior to
acquisition.
Flow cytometric analysis was performed on a BD
FACSCanto II flow cytometer (BD Biosciences). Unstained cells,
single-stained controls and negative controls were included for
compensation and gating. A minimum of 10,000 events per sample was
collected. Initial gating was performed on forward scatter versus
side scatter to exclude debris, followed by quadrant analysis of
Annexin V-FITC and PI to distinguish viable (Annexin
V−/PI−), early apoptotic (Annexin
V+/PI−) and late apoptotic/necrotic (Annexin
V+/PI+) cells. Total apoptosis was calculated
as the sum of early and late apoptotic populations (Annexin
V+/PI− + Annexin
V+/PI+). Data were analyzed using FlowJo
software (version 11; BD Biosciences) (39,40).
Each condition was assayed in triplicate and repeated in at least
three independent biological experiments.
In silico analysis
Publicly available transcriptomic data and
corresponding clinical information of patients with ccRCC were
obtained from The Cancer Genome Atlas (TCGA) database through the
Genomic Data Commons data portal (https://portal.gdc.cancer.gov/). RNA sequencing data
[HTSeq-transcripts per million (TPM) format] from the TCGA-kidney
renal clear cell carcinoma (KIRC) cohort were downloaded and used
for subsequent analyses. Pan-cancer expression analysis of MUC3A
was performed using the Gene Expression Profiling Interactive
Analysis 2 (GEPIA2) web server (http://gepia2.cancer-pku.cn/). MUC3A expression levels
across different tumor types were visualized using the ‘Expression
Analysis - General’ module with log2(TPM + 1) normalization.
Differential expression of MUC3A between tumor and normal tissues
in the TCGA-KIRC cohort was evaluated using the ‘Expression DIY -
Boxplot’ module, with the following parameters: |log2 fold change|
cutoff=1 and P-value cutoff=0.01, using matched TCGA normal samples
when available.
Survival analysis was conducted using GEPIA2 based
on TCGA-KIRC data. Overall survival (OS) and disease-free survival
(DFS) were analyzed by stratifying patients into high- and
low-expression groups according to MUC3A expression levels. For OS
analysis, the expression cutoff was set at the upper 80% vs. lower
20%, while for DFS analysis, the cutoff was set at 87.5% vs. 12.5%.
Kaplan-Meier survival curves were generated, and statistical
significance was assessed using the log-rank test.
For pathway enrichment analyses, differentially
expressed genes associated with MUC3A expression were identified,
and genes with P<0.05 were selected for downstream functional
analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
enrichment analysis was performed using the clusterProfiler R
package (version 4.12.6; Bioconductor; http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
with the enrichKEGG function. Gene set enrichment analysis (GSEA)
was conducted using the GSEA function implemented in the same
package to assess pathway-level differences between high- and
low-MUC3A expression groups. Enrichment results were visualized
using standard plotting functions provided by the clusterProfiler
package.
Statistical analysis
All experiments were performed with at least three
independent biological replicates (n≥3), and data are presented as
the mean ± standard deviation (SD) unless otherwise stated.
Statistical analyses were conducted using GraphPad Prism 9
(GraphPad Software, Inc.; Dotmatics). For comparisons between two
groups, a two-tailed unpaired Student's t-test was used. For
comparisons among multiple groups, one- or two-way analysis of
variance (ANOVA) was performed as appropriate, followed by
Dunnett's post hoc test (when comparing multiple groups to a single
control) or Tukey's post hoc test (for all pairwise comparisons),
as indicated in the figure legends. Fold-change values were
calculated relative to the corresponding control group where
applicable. P<0.05 was considered to indicate a statistically
significant difference. Significance levels are denoted as
P<0.05, P<0.01 and P<0.001, unless otherwise
specified.
Results
High expression of MUC3A in ccRCC is
associated with poor prognosis
First, as shown in Fig.
1A, a pan-cancer analysis of MUC3A expression was performed and
it was found that MUC3A was significantly upregulated in multiple
tumor types, including colon adenocarcinoma (COAD),
cholangiocarcinoma (CHOL), and KIRC. Furthermore, in unpaired
samples from the TCGA-KIRC cohort (523 tumor samples vs. 72 normal
samples), MUC3A expression levels were markedly higher in tumor
tissues than adjacent normal tissues (Fig. 1B). To confirm the oncogenic role of
MUC3A in ccRCC, MUC3A expression was assessed in HK-2 and
RPTEC/TERT1 non-malignant renal epithelial cell lines, as well as
in CAKI-1, OSRC-2, 786-O and ACHN renal cancer cell lines. The
findings showed elevated protein levels of MUC3A in renal cancer
cell lines compared with non-malignant renal epithelial cell lines
(Fig. 1C). Data from the TCGA
database revealed that patients with high MUC3A expression
exhibited a significantly poorer OS and DFS than patients with low
MUC3A expression (Fig. 1E and F).
These findings indicated that MUC3A is aberrantly upregulated in
ccRCC and that elevated MUC3A expression is associated with
unfavorable clinical outcomes.
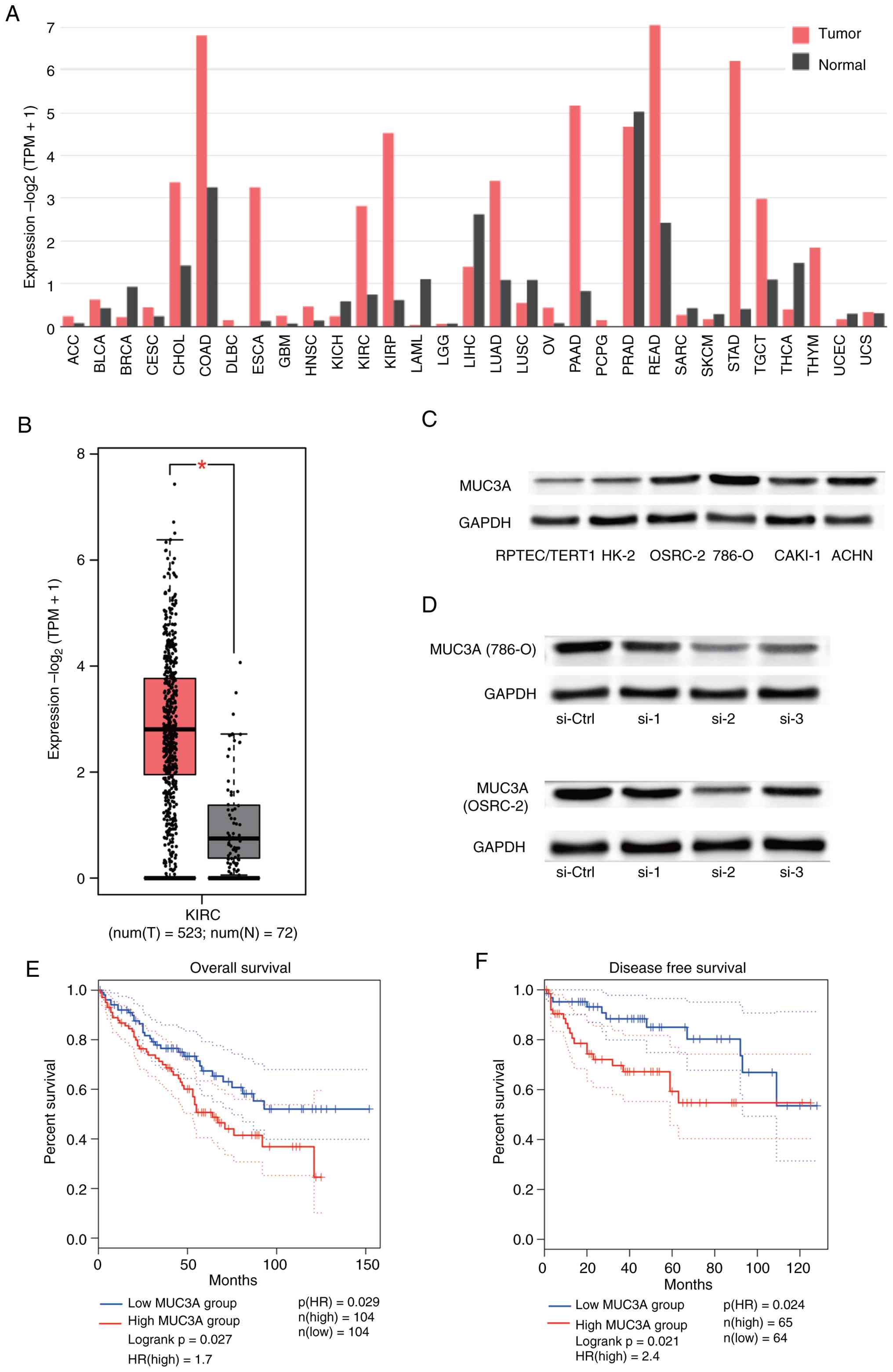 | Figure 1.MUC3A is aberrantly upregulated in
ccRCC and associated with poor prognosis. (A) Pan-cancer analysis
of MUC3A expression across multiple tumor types based on TCGA data,
generated using the GEPIA2 platform. Gene expression values are
presented as log2(TPM + 1). (B) Differential expression of MUC3A in
tumor and normal samples from the TCGA-KIRC cohort (523 tumor
samples vs. 72 normal samples). *P<0.05. (C) Western blotting of
MUC3A protein expression in HK-2 and RPTEC/TERT1 non-malignant
renal epithelial cell lines and ccRCC cell lines (CAKI-1, OSRC-2,
786-O and ACHN). GAPDH was used as a loading control. (D) Western
blotting of MUC3A knockdown efficiency in 786-O and OSRC-2 cells
following transient transfection with three independent siRNAs
targeting MUC3A. si-2 was selected for subsequent functional
experiments due to its superior knockdown efficiency. (E)
Kaplan-Meier OS analysis of ccRCC patients stratified into high-
and low-MUC3A expression groups using the GEPIA2 platform. (F)
Kaplan-Meier DFS analysis of ccRCC patients based on MUC3A
expression levels. Data are derived from TCGA unless otherwise
indicated. MUC3A, mucin 3A; ccRCC, clear cell renal cell carcinoma;
TCGA, The Cancer Genome Atlas; KIRC, kidney renal clear cell
carcinoma; GEPIA2, Gene Expression Profiling Interactive Analysis
2; TPM, transcripts per million; KIRC, kidney renal clear cell
carcinoma; OS, overall survival; DFS, disease-free survival; siRNA,
small interfering RNA; si-MUC3A, MUC3A-targeting siRNA; si-Ctrl,
non-targeting control siRNA; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase; si-1/si-2/si-3, three independent siRNAs targeting
MUC3A. |
Knockdown of MUC3A suppresses ccRCC
cell proliferation and colony-forming ability
To investigate the functional role of MUC3A in
ccRCC, siRNA targeting MUC3A was used to transiently knock down
MUC3A in two cell lines with the highest endogenous expression
(786-O and OSRC-2), and knockdown efficiency was verified. Among
the tested siRNAs, si-2 exhibited the most effective silencing
(Fig. 1D). si-2 was selected for
subsequent functional experiments due to its superior knockdown
efficiency.
In the CCK-8 assay, MUC3A knockdown resulted in a
modest but statistically significant reduction in cell
proliferation in 786-O cells (~10% decrease compared with siCtrl;
P<0.01), whereas a more pronounced inhibitory effect was
observed in OSRC-2 cells, with proliferation reduced by more than
threefold (P<0.001; Fig. 2A and
B). Consistently, the colony formation assay showed that MUC3A
knockdown significantly impaired clonogenic ability, with the
number of colonies reduced by ~30% in 786-O cells and 40% in OSRC-2
cells (both P<0.05), as compared with control groups (Fig. 2C and D).
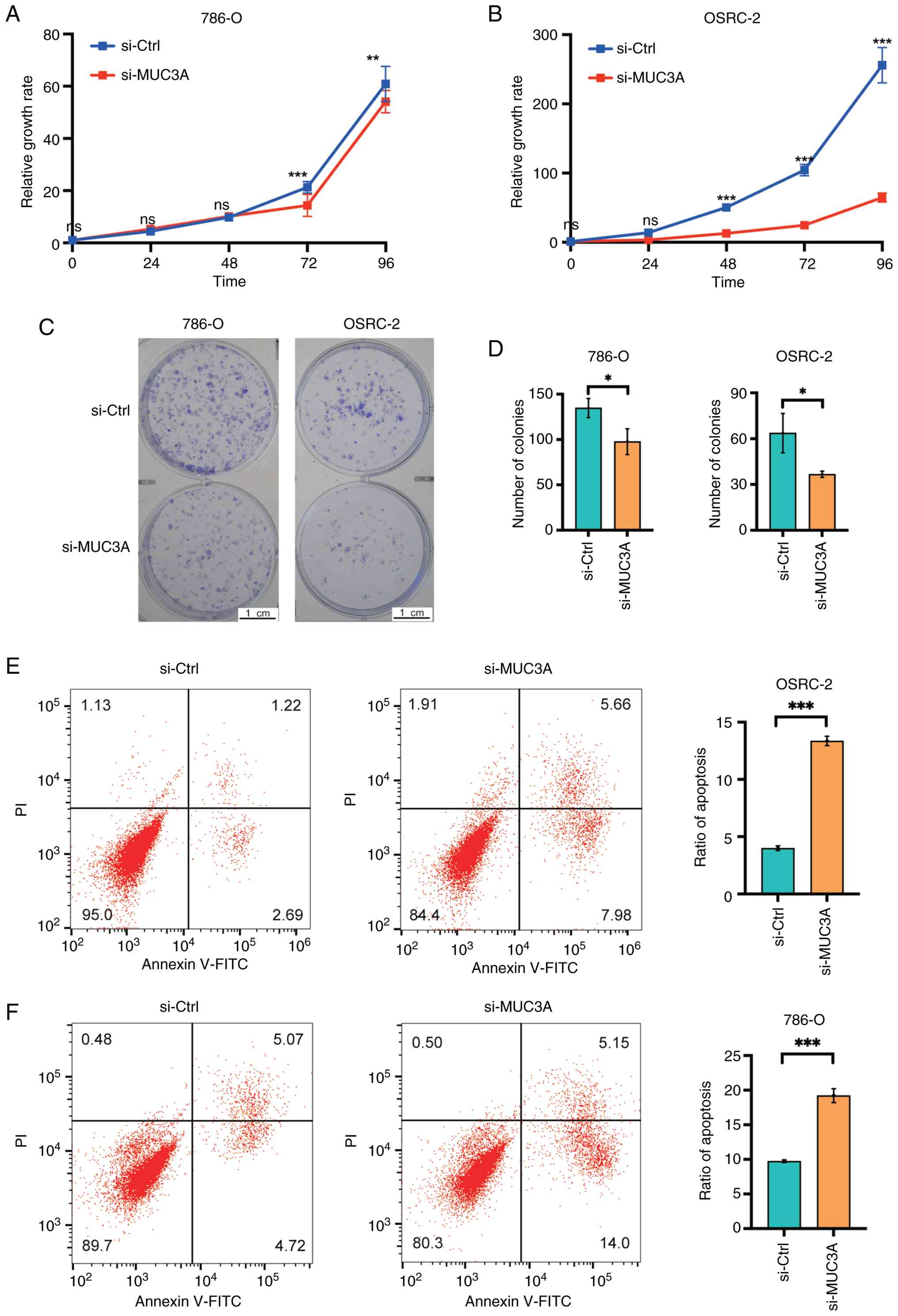 | Figure 2.MUC3A knockdown suppresses
proliferation and promotes apoptosis in ccRCC cells. (A and B) Cell
proliferation of 786-O and OSRC-2 cells following transfection with
si-MUC3A or si-Ctrl, as assessed by CCK-8 assays at the indicated
time points. (C and D) Colony formation assays showing the
clonogenic capacity of 786-O and OSRC-2 cells following MUC3A
knockdown. Representative images and quantitative analysis are
shown. (E and F) Flow cytometric analysis of apoptosis in 786-O and
OSRC-2 cells using Annexin V-FITC/PI staining following MUC3A
silencing. Representative dot plots and corresponding quantitative
results are presented. All experiments were performed with at least
three independent biological replicates (n≥3). Data are presented
as the mean ± SD. Statistical significance was determined using a
two-tailed unpaired Student's t-test. *P<0.05, **P<0.01 and
***P<0.001. MUC3A, mucin 3A; ccRCC, clear cell renal cell
carcinoma; CCK-8, Cell Counting Kit-8; si-MUC3A, MUC3A-targeting
siRNA; si-Ctrl, non-targeting control siRNA; PI, propidium iodide;
SD, standard deviation; ns, not significant. |
MUC3A knockdown promotes apoptosis in
ccRCC cells
To elucidate the effect of MUC3A on apoptosis,
Annexin V-FITC/PI staining was performed followed by flow
cytometric analysis. It was observed that MUC3A knockdown led to a
significant increase in apoptotic cell populations in both 786-O
and OSRC-2 cells (Fig. 2E and F).
Quantitative analysis showed that the apoptotic rate in OSRC-2
cells increased to approximately threefold that of the control
group, while a roughly two-fold increase was observed in 786-O
cells following MUC3A silencing (both P<0.001).
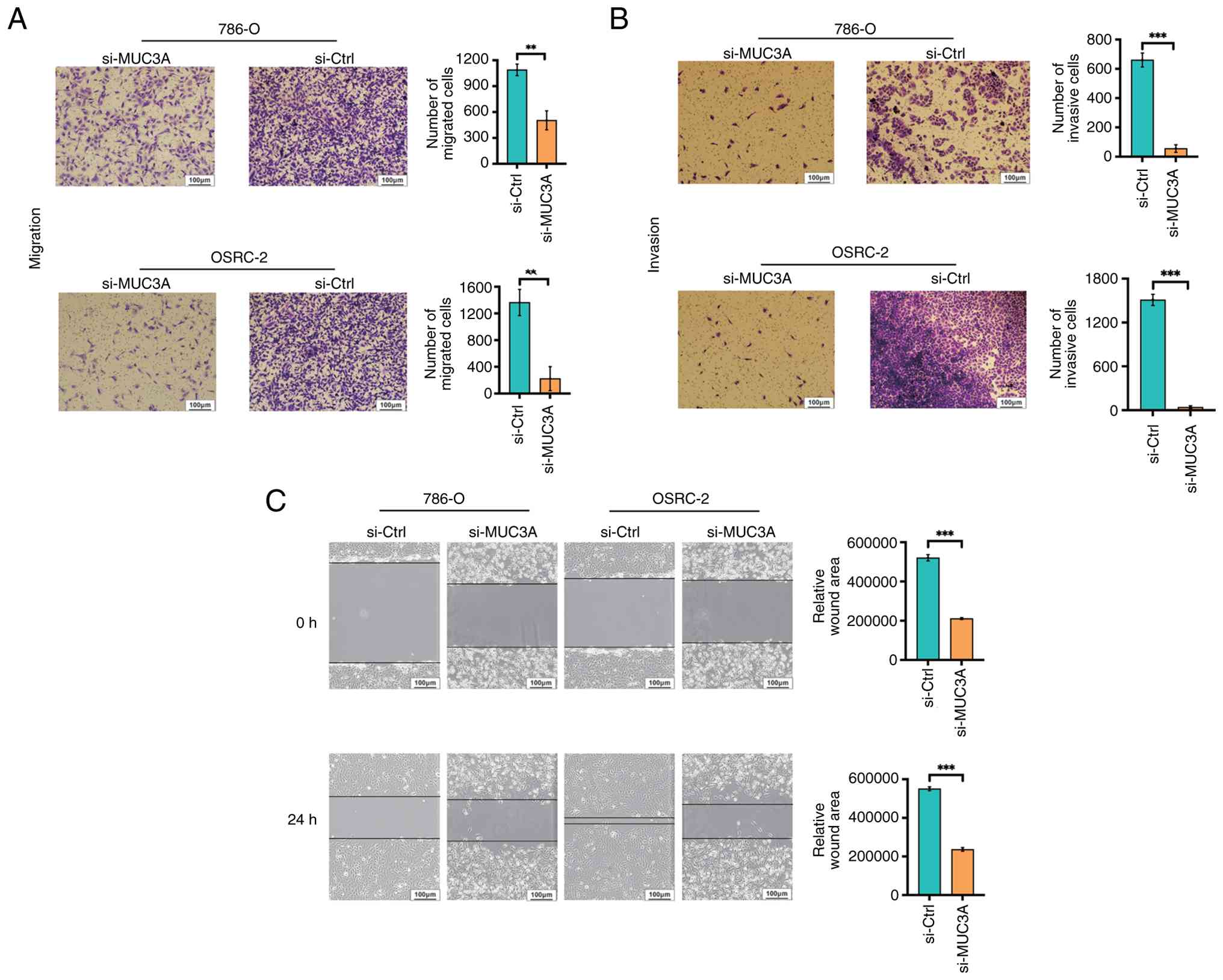
MUC3A knockdown inhibits ccRCC cell
migration and invasion
Transwell migration and Transwell invasion assays,
as well as wound healing assays, were performed to investigate the
role of MUC3A in ccRCC cell motility. Transwell migration assays
demonstrated a marked suppression of migratory capacity following
MUC3A silencing. Quantitative analysis showed that the migratory
rate of 786-O cells was reduced to ~50% of control levels
(P<0.01), whereas a more pronounced inhibitory effect was
observed in OSRC-2 cells, with migration reduced to ~1/7 of the
control levels (P<0.01; Fig.
3A).
Furthermore, Transwell invasion assays revealed a
marked inhibitory effect of MUC3A knockdown on invasive capacity
(Fig. 3B). Compared with control
cells, the number of invasive 786-O cells was reduced by more than
one order of magnitude, whereas OSRC-2 cells exhibited an even more
pronounced reduction, approaching two orders of magnitude,
following MUC3A silencing. Collectively, these results indicated
that MUC3A plays an important role in promoting the migratory and
invasive phenotypes of ccRCC cells.
Consistently, in the wound healing assay, MUC3A
knockdown significantly delayed wound closure in both 786-O and
OSRC-2 cells, with the migration rate reduced by ~50% compared with
control cells (P<0.001; Fig.
3C).
MUC3A knockdown downregulates the
JAK-STAT signaling pathway in ccRCC
To further explore the potential mechanisms through
which MUC3A contributes to ccRCC progression, KEGG pathway
enrichment analysis was performed based on MUC3A expression levels.
The results indicated that MUC3A expression was associated with
multiple signaling pathways, including ‘primary immunodeficiency’,
‘oxidative phosphorylation’, ‘Th17 cell differentiation’, ‘NF-κB
signaling’ and ‘cytokine-cytokine receptor interaction’ (Fig. 4A).
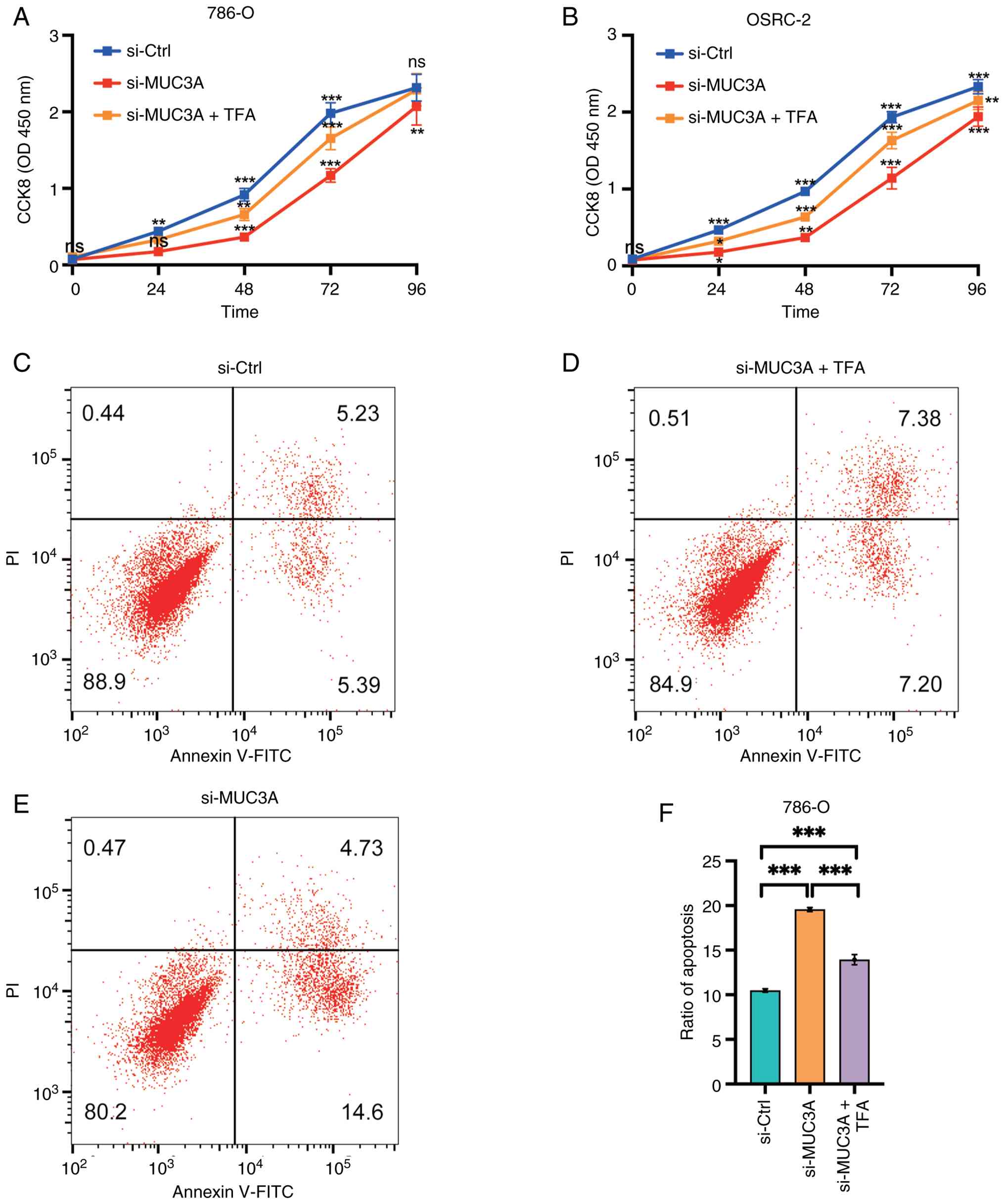
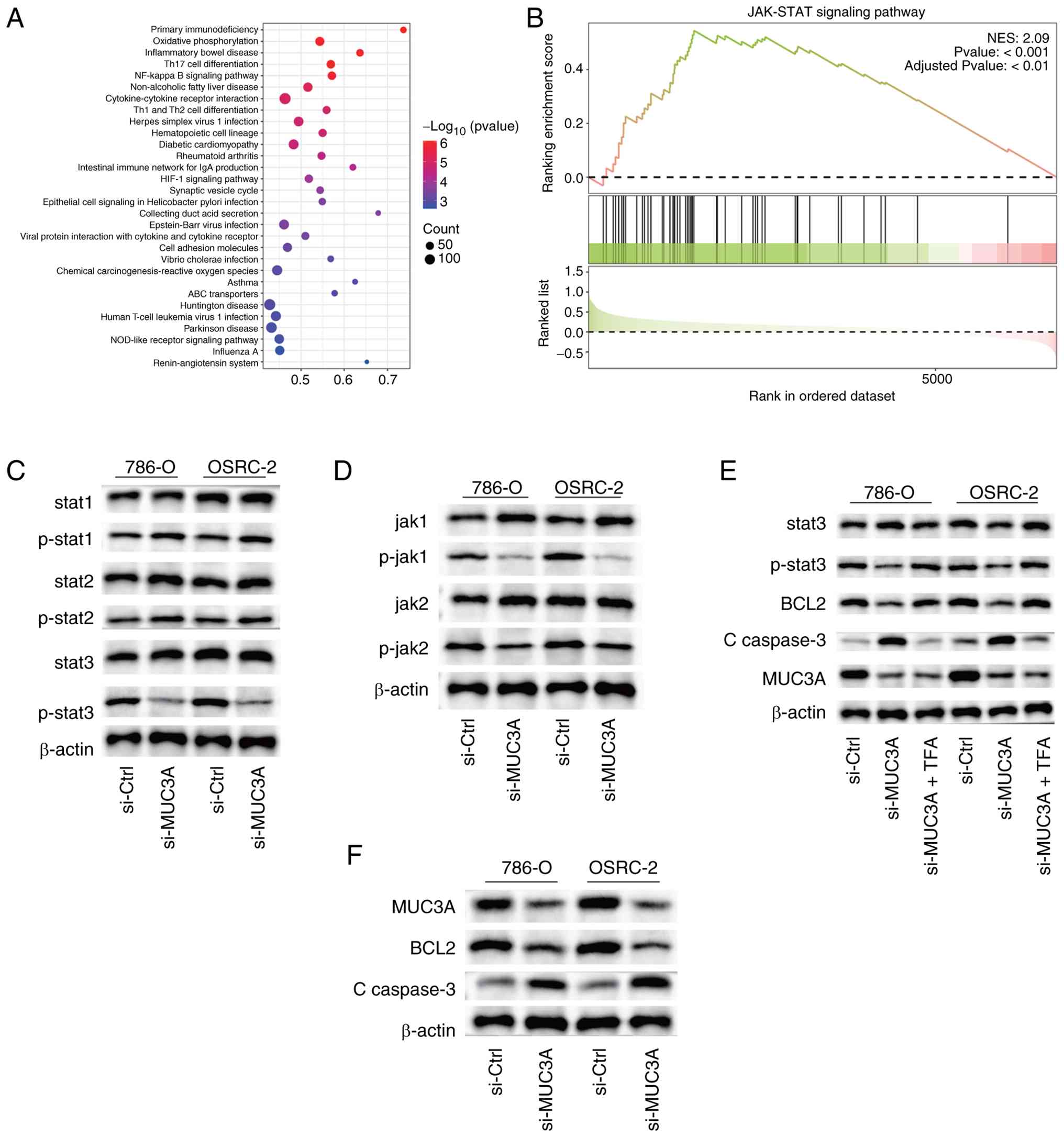 | Figure 4.MUC3A is associated with the
activation of the JAK-STAT signaling pathway in ccRCC. (A) KEGG
pathway enrichment analysis of genes associated with MUC3A
expression based on TCGA-KIRC transcriptomic data. (B) GSEA showing
significant enrichment of the JAK-STAT signaling pathway in ccRCC
samples with a high MUC3A expression. (C and D) Western blotting of
total and phosphorylated JAK1, JAK2 and STAT3 in 786-O and OSRC-2
cells following MUC3A knockdown. (E) Western blotting showing that
STAT3 activation by Colivelin TFA restores p-STAT3 levels in
si-MUC3A-transfected 786-O and OSRC-2 cells, accompanied by
increased Bcl-2 and decreased cleaved caspase-3 expression. (F)
Western blotting of apoptosis-related proteins Bcl-2 and cleaved
caspase-3 following MUC3A silencing. GAPDH served as a loading
control. All western blotting experiments were repeated
independently at least three times. MUC3A, mucin 3A; JAK, Janus
kinase; STAT, signal transducer and activator of transcription;
ccRCC, clear cell renal cell carcinoma; KEGG, Kyoto Encyclopedia of
Genes and Genomes; TCGA, The Cancer Genome Atlas; KIRC, kidney
renal clear cell carcinoma; GSEA, gene set enrichment analysis;
TFA, trifluoroacetate; p-, phosphorylated. |
Of note, GSEA revealed a significant enrichment of
the JAK-STAT signaling pathway in samples with high MUC3A
expression (Fig. 4B). Given the
established role of the JAK-STAT pathway in regulating key
biological processes such as cell proliferation, apoptosis and
immune responses, these findings suggested a potential link between
MUC3A and JAK-STAT pathway activation in ccRCC.
To further validate this association at the protein
level, western blotting was performed, which demonstrated that
MUC3A knockdown resulted in reduced phosphorylation levels of JAK1,
JAK2 and STAT3, while total protein levels remained largely
unchanged (Fig. 4C and D). To
further validate the involvement of the JAK-STAT pathway, 786-O and
OSRC-2 cells were treated with Colivelin trifluoroacetate (TFA), a
specific STAT3 agonist, following MUC3A knockdown. At the protein
level, p-STAT3 expression was restored in the si-MUC3A + Colivelin
TFA group, accompanied by increased Bcl-2 and decreased cleaved
caspase-3 levels (Fig. 4E).
Furthermore, consistent with the flow cytometric findings
aforementioned, western blotting demonstrated a marked decrease in
the expression of the anti-apoptotic protein Bcl-2, accompanied by
increased levels of cleaved caspase-3 upon MUC3A knockdown
(Fig. 4F). Collectively, these
results supported the notion that MUC3A may promote ccRCC
progression, at least in part, through modulation of the JAK-STAT
signaling pathway.
MUC3A regulates ccRCC proliferation
and apoptosis via the JAK-STAT pathway
Building on the protein-level validation of JAK-STAT
activation, the functional consequences of restoring this pathway
were next examined. Treatment with Colivelin TFA rescued the
proliferation inhibition caused by si-MUC3A, restoring cell growth
to levels comparable to those of the controls (Fig. 5A and B). Similarly, the
apoptosis-promoting effect of MUC3A knockdown was reversed upon
Colivelin TFA treatment (Fig.
5C-F). These findings indicated that MUC3A promotes ccRCC
proliferation and suppresses apoptosis through the activation of
the JAK-STAT signaling pathway.
Discussion
In the present study, the expression, functional
relevance and potential signaling mechanisms of MUC3A were
systematically investigated in ccRCC. The findings demonstrated
that MUC3A is aberrantly upregulated in ccRCC and that its
silencing leads to a significant suppression of tumor-associated
phenotypes, including cell proliferation, clonogenic capacity,
migration, invasion and resistance to apoptosis. Collectively,
these results supported the role for MUC3A as a contributor to
ccRCC progression rather than a passive bystander.
Mucins have long been recognized as important
regulators of epithelial biology, and accumulating evidence
suggests that dysregulated mucin expression contributes to
tumorigenesis through mechanisms involving cell adhesion, immune
evasion and intracellular signaling modulation. Previous studies on
mucins, such as MUC1 and MUC4, have established their oncogenic
roles in multiple malignancies, including colorectal, pancreatic
and lung cancer, where they participate in signal transduction and
promote aggressive tumor behavior (23,24).
These findings extend this concept by identifying MUC3A as a
functionally relevant mucin in ccRCC, a cancer type in which the
role of membrane-related mucins has been comparatively
underexplored.
A key observation of the present study is the
association between MUC3A expression and activation of the JAK-STAT
signaling pathway in ccRCC. Through integrated bioinformatics
analyses and experimental validation, it was found that MUC3A
knockdown resulted in the reduced phosphorylation of JAK1, JAK2 and
STAT3, without substantially affecting total protein levels. These
findings suggested that MUC3A may influence ccRCC progression, at
least in part, by modulating the activity of the JAK-STAT pathway
rather than altering its basal expression. Of note, the present
results do not imply that MUC3A is the sole upstream regulator of
JAK-STAT signaling; instead, they indicate that MUC3A may function
as a facilitator or enhancer within a broader oncogenic signaling
network.
The biological relevance of JAK-STAT signaling in
ccRCC has been well documented. Persistent activation of STAT3 has
been shown to drive tumor cell proliferation, enhance migratory and
invasive potential, promote resistance to apoptosis and contribute
to immune evasion in renal cancer (20–22).
The present data were consistent with these observations and
further suggested that MUC3A may represent one of the upstream
molecular factors capable of shaping STAT3 activation status in
ccRCC cells. Of note, rescue experiments using a STAT3 agonist
partially reversed the anti-proliferative and pro-apoptotic effects
induced by MUC3A silencing, providing functional support for the
involvement of JAK-STAT signaling downstream of MUC3A. Consistent
with this mechanism-oriented interpretation, a recent ccRCC study
reported that apolipoprotein C2 promotes malignant phenotypes at
least partly through the activation of the JAK-STAT pathway,
supporting the concept that distinct upstream molecules may
converge on JAK/STAT signaling to drive ccRCC progression (41). In addition, a recent systematic
review highlighted JAK/STAT as a major signaling axis in renal
carcinoma and summarized its therapeutic potential, which further
supported the translational relevance of the present findings
linking MUC3A to JAK/STAT activation (42).
From a translational perspective, the JAK-STAT
pathway has emerged as a promising therapeutic target in multiple
malignancies, including renal cancer. Pharmacological inhibitors of
the JAK or STAT signaling, such as ruxolitinib, are currently being
evaluated in preclinical and clinical settings, either as
monotherapies or in combination with other agents (9). In this context, the present findings
raise the possibility that MUC3A expression status may help
identify a subset of patients with ccRCC who are more likely to
benefit from therapies targeting the JAK-STAT axis. In addition,
therapeutic strategies aimed at reducing MUC3A expression or
disrupting its functional interactions could potentially enhance
the efficacy of JAK-STAT-directed treatments.
Despite the strengths of the present study, several
limitations should be acknowledged. First, although TCGA-based
analyses provide valuable insights into gene expression patterns
and clinical associations at the tissue level, such data reflect
bulk tumor samples and cannot fully capture intratumoral
heterogeneity or microenvironmental influences. Secondly, these
functional experiments were conducted in immortalized ccRCC cell
lines cultured under standardized in vitro conditions. While
all comparisons were performed under identical conditions, the use
of DMEM rather than RPMI-1640 may influence cellular behavior and
signaling responses, and this potential confounding factor cannot
be completely excluded. Thirdly, the absence of in vivo
validation limits the direct translational applicability of the
present findings.
Future studies should focus on elucidating the
precise molecular mechanisms through which MUC3A interfaces with
JAK-STAT signaling, including whether this regulation occurs
through direct protein interactions or via intermediary signaling
molecules. In addition, given the central role of JAK-STAT
signaling in immune regulation, it will be important to explore the
contribution of MUC3A to immune modulation and immune checkpoint
responsiveness in ccRCC. Finally, in vivo studies using
xenograft or patient-derived xenograft models will be essential to
validate the oncogenic role of MUC3A and to assess its potential as
a therapeutic target in clinically relevant settings.
To the best of our knowledge, the present study is
the first to systematically characterize the expression, functional
significance and mechanistic role of MUC3A in ccRCC. While the
JAK-STAT pathway has been extensively implicated in ccRCC, the
involvement of MUC3A as a modulator of this signaling axis has not
been previously reported. By integrating bioinformatics analyses
with functional and rescue experiments, the present study
identified MUC3A as a previously underappreciated contributor to
ccRCC progression.
In conclusion, MUC3A was identified herein as a
previously underappreciated contributor to ccRCC progression and
provided evidence linking its oncogenic effects to the modulation
of the JAK-STAT signaling pathway. These findings expanded the
current understanding of mucin biology in renal cancer and
highlighted MUC3A as a potential biomarker and therapeutic target
worthy of further investigation.
In conclusion, the present study provided novel
insights into the oncogenic role of MUC3A in ccRCC and highlighted
its potential as a therapeutic target. By elucidating the
mechanistic relationship between MUC3A and the JAK/STAT signaling
pathway, the groundwork was laid for future studies aimed at
developing targeted therapies that can improve the prognosis of
ccRCC patients.
Acknowledgements
The authors would like to thank the staff of the
Department of Urology, Peking University First Hospital (Beijing,
China), for their technical support and insightful discussions.
Funding
The present study was supported by the Beijing Natural Science
Foundation (grant no. 7232179).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YY conceived and designed the study, performed the
major experiments, analyzed and interpreted the data, and drafted
the manuscript. XJ contributed to the study design, performed key
experiments, and participated in data analysis and manuscript
revision. SH contributed to experimental work and data collection,
and participated in data analysis and manuscript revision. TW, YL,
ZL, ZS, PZ and LY provided technical assistance and helped with
data collection and/or verification. WY conceived and supervised
the study, contributed to the study design and data interpretation,
and critically revised the manuscript. YY and WY confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yao L, Du Y, Dong W, Gao X, Guo J, Qiu J,
Wei Q, Wu S, Ye D, Yu W, et al: Chinese quality control indices for
standardized diagnosis and treatment of renal cancer (2022
edition). J Natl Cancer Cent. 4:6–13. 2024.PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
3
|
Capitanio U, Bensalah K, Bex A, Boorjian
SA, Bray F, Coleman J, Gore JL, Sun M, Wood C and Russo P:
Epidemiology of renal cell carcinoma. Eur Urol. 75:74–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33.
2022.PubMed/NCBI
|
|
5
|
Lv XQ, Zhang KB, Guo X, Pei L and Li F:
Higher TYROBP and lower SOX6 as predictive biomarkers for poor
prognosis of clear cell renal cell carcinoma: A pilot study.
Medicine (Baltimore). 101:e306582022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Colombo PE, Quenet F, Alric P, Mourregot
A, Neron M, Portales F, Rouanet P and Carrier G: Distal
pancreatectomy with celiac axis resection (Modified Appleby
Procedure) and arterial reconstruction for locally advanced
pancreatic adenocarcinoma after FOLFIRINOX chemotherapy and
chemoradiation therapy. Ann Surg Oncol. 28:1106–1108. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Q, Ren H, Ge L, Zhang W, Song F and
Huang P: A review on the role of long non-coding RNA and microRNA
network in clear cell renal cell carcinoma and its tumor
microenvironment. Cancer Cell Int. 23:162023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song Z, Guan C, Li T, Li C, Zhang N, Liu
K, Yang C, Zhu Y and Xu Y: Vaporization phosphorization-mediated
synthesis of phosphorus-doped TiO2 nanocomposites for
combined photodynamic and photothermal therapy of renal cell
carcinoma. J Mater Chem B. 12:4039–4052. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meng K, Li YY, Liu DY, Hu LL, Pan YL,
Zhang CZ and He QY: A five-protein prognostic signature with GBP2
functioning in immune cell infiltration of clear cell renal cell
carcinoma. Comput Struct Biotechnol J. 21:2621–2630. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao Y, Qi JC, Li X, Sun JP, Ji H and Li
QH: Decreased expression of TXNIP predicts poor prognosis in
patients with clear cell renal cell carcinoma. Oncol Lett.
19:763–770. 2020.PubMed/NCBI
|
|
11
|
Sánchez A and Fabregat I: Growth factor-
and cytokine-driven pathways governing liver stemness and
differentiation. World J Gastroenterol. 16:5148–5161. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sabaawy HE, Ryan BM, Khiabanian H and Pine
SR: JAK/STAT of all trades: linking inflammation with cancer
development, tumor progression and therapy resistance.
Carcinogenesis. 42:1411–1419. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Owen KL, Brockwell NK and Parker BS:
JAK-STAT signaling: A double-edged sword of immune regulation and
cancer progression. Cancers (Basel). 11:20022019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rah B, Rather RA, Bhat GR, Baba AB,
Mushtaq I, Farooq M, Yousuf T, Dar SB, Parveen S, Hassan R, et al:
JAK/STAT signaling: molecular targets, therapeutic opportunities,
and limitations of targeted inhibitions in solid malignancies.
Front Pharmacol. 13:8213442022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pang S, Zhao S, Dongye Y, Fan Y and Liu J:
Identification and validation of m6A-associated ferroptosis genes
in renal clear cell carcinoma. Cell Biol Int. 48:777–794. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiao W, Wang X, Wang T and Xing J:
Overexpression of BMP1 reflects poor prognosis in clear cell renal
cell carcinoma. Cancer Gene Ther. 27:330–340. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun J, Du Y, Zhang X, Wang Z, Lin Y, Song
Q, Wang X, Guo J, Li S, Nan J and Yang J: Discovery and evaluation
of Atopaxar hydrobromide, a novel JAK1 and JAK2 inhibitor,
selectively induces apoptosis of cancer cells with constitutively
activated STAT3. Invest New Drugs. 38:1003–1011. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dinakar YH, Kumar H, Mudavath SL, Jain R,
Ajmeer R and Jain V: Role of STAT3 in the initiation, progression,
proliferation and metastasis of breast cancer and strategies to
deliver JAK and STAT3 inhibitors. Life Sci. 309:1209962022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Groner B and von Manstein V: Jak Stat
signaling and cancer: Opportunities, benefits and side effects of
targeted inhibition. Mol Cell Endocrinol. 451:1–14. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang C, Wang Y, Hong T, Cheng B, Gan S,
Chen L, Zhang J, Zuo L, Li J and Cui X: Blocking the autocrine
regulatory loop of Gankyrin/STAT3/CCL24/CCR3 impairs the
progression and pazopanib resistance of clear cell renal cell
carcinoma. Cell Death Dis. 11:1172020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song J, Xu S, Zhang ZH, Chen YH, Gao L,
Xie DD, Sun GP, Yu DX and Xu DX: The correlation between low
vitamin D status and renal interleukin-6/STAT3 hyper-activation in
patients with clear cell renal cell carcinoma. Steroids.
150:1084452019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arévalo J, Campoy I, Durán M, Nemours S,
Areny A, Vall-Palomar M, Martínez C, Cantero-Recasens G and
Meseguer A: STAT3 phosphorylation at serine 727 activates specific
genetic programs and promotes clear cell renal cell carcinoma
(ccRCC) aggressiveness. Sci Rep. 13:195522023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ganguly K, Rauth S, Marimuthu S, Kumar S
and Batra SK: Unraveling mucin domains in cancer and metastasis:
when protectors become predators. Cancer Metastasis Rev.
39:647–659. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang Y, Liu Z, Hu Y, Gao T, Wen T and An
G: Correlation of Tn antigen expression with mucins in Chinese
patients with colorectal cancer. Int J Clin Exp Pathol.
11:1562–1568. 2018.PubMed/NCBI
|
|
25
|
Miura K, Kinouchi M, Ishida K, Fujibuchi
W, Naitoh T, Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, et
al: 5-fu metabolism in cancer and orally-administrable 5-fu drugs.
Cancers (Basel). 2:1717–1730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Korbut E, Janmaat VT, Wierdak M, Hankus J,
Wójcik D, Surmiak M, Magierowska K, Brzozowski T, Peppelenbosch MP
and Magierowski M: Molecular profile of barrett's esophagus and
gastroesophageal reflux disease in the development of translational
physiological and pharmacological studies. Int J Mol Sci.
21:64362020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Su W, Feng B, Hu L, Guo X and Yu M: MUC3A
promotes the progression of colorectal cancer through the
PI3K/Akt/mTOR pathway. BMC Cancer. 22:6022022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Y, Sun X, You C, Ma S, Luo Y, Peng S,
Tang F, Tian X, Wang F, Huang Z, et al: MUC3A promotes non-small
cell lung cancer progression via activating the NFkappaB pathway
and attenuates radiosensitivity. Int J Biol Sci. 17:2523–2536.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo Y, Ma S, Sun Y, Peng S, Zeng Z, Han L,
Li S, Sun W, Xu J, Tian X, et al: MUC3A induces PD-L1 and reduces
tyrosine kinase inhibitors effects in EGFR-mutant non-small cell
lung cancer. Int J Biol Sci. 17:1671–1681. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu S, Zhang RF, You Y, You W, Ruan GC,
Liu YP, Zhang SY, Li Y, Feng YL, Yan XM, et al: The genomic
landscape of Cronkhite-Canada syndrome: Possible clues for
pathogenesis. J Dig Dis. 23:288–294. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hammoudeh SM, Hammoudeh AM, Bhamidimarri
PM, Al Safar H, Mahboub B, Künstner A, Busch H, Halwani R, Hamid Q,
Rahmani M and Hamoudi R: Systems immunology analysis reveals the
contribution of pulmonary and extrapulmonary tissues to the
immunopathogenesis of severe COVID-19 patients. Front Immunol.
12:5951502021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen M, Wei X, Shi X, Lu L, Zhang G, Huang
Y and Hou J: LncRNA HIF1A-AS2 accelerates malignant phenotypes of
renal carcinoma by modulating miR-30a-5p/SOX4 axis as a ceRNA.
Cancer Biol Med. 18:587–603. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeng K, Song G, Chen B, Gao X, Liu C, Miao
J, Ruan Y, Luan Y, Chen X, Liu J, et al: Comprehensive analysis to
identify the RP11-478C19.2/E2F7 axis as a novel biomarker for
treatment decisions in clear cell renal cell carcinoma. Transl
Oncol. 25:1015252022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ye S, Li S, Qin L, Zheng W, Liu B, Li X,
Ren Z, Zhao H, Hu X, Ye N and Li G: GBP2 promotes clear cell renal
cell carcinoma progression through immune infiltration and
regulation of PD-L1 expression via STAT1 signaling. Oncol Rep.
49:492023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie Z, Feng C, Hong Y, Chen L, Li M and
Deng W: Identification of key genes CCL5, PLG, LOX and C3 in clear
cell renal cell carcinoma through integrated bioinformatics
analysis. Front Mol Biosci. 12:15871962025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luo Q, Wang Q, Shi J, Lv Q, Dong Z, Li W,
Xia Y, Liu J and Yang H: PUMA reduces FASN ubiquitination to
promote lipid accumulation and tumor progression in human clear
cell renal cell carcinoma. Cell Death Dis. 16:4602025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miao D, Shi J, Xiong Z, Xiao W, Meng X, Lv
Q, Xie K, Yang H and Zhang X: As a prognostic biomarker of clear
cell renal cell carcinoma RUFY4 predicts immunotherapy
responsiveness in a PDL1-related manner. Cancer Cell Int.
22:662022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng M, Zhang S, Zhou J, Lin M and Liao
Y: ACAT1 suppresses clear cell renal cell carcinoma progression by
AMPK mediated fatty acid metabolism. Transl Oncol. 47:1020432024.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang X, Shi A, Liu J, Kong W, Huang Y, Xue
W, Yang F and Huang J: CDCA5-EEF1A1 interaction promotes
progression of clear cell renal cell carcinoma by regulating mTOR
signaling. Cancer Cell Int. 24:1472024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yun Y, Ji X, Wu T, Liu Y, Li Z, Sun Z,
Zhou P, Yang L and Yu W: APOC2 promotes clear cell renal cell
carcinoma progression via activation of the JAK-STAT signaling
pathway. Curr Issues Mol Biol. 47:9362025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shabib S, Rahnama A, Sakhtemanpour Bolouki
S, Hosseini SA and Satarzadeh M: JAK/STAT is a main signaling
pathway in renal carcinoma: A systematic review of therapeutic
potential. J Inflamm Dis. 29:e1601712025.
|