Colorectal cancer (CRC) ranks third globally in
terms of occurrence rate among malignant tumors and is the second
leading cause of cancer-related mortality (1). Early-stage CRC is typically managed
with surgery combined with radiotherapy, whereas for patients with
advanced colon cancer, combination therapy involving
chemotherapeutic agents and targeted drugs is primarily employed,
such as bevacizumab (2). Although
these treatment plans have markedly improved the prognosis of
patients with CRC, clinical statistical analysis in the United
States has indicated that the 5-year survival rate for CRC is 65,
and 14% for distant-stage (metastatic) CRC (3). CRC is a highly heterogeneous and
complex disease, and patients with distinct metabolic
characteristics often respond differently to the same therapeutic
strategy (4,5). In particular, during cancer
progression, tumor cells frequently acquire diverse features and
generally become more heterogeneous (6). Consequently, although targeted
therapies have improved the overall survival of patients with CRC,
a substantial number of patients still lack effective targeted
drugs or develop drug resistance during treatment. Given the
prevalence of CRC and the limitations of current therapies, it is
imperative to refine existing clinical approaches and develop novel
therapeutic agents.
Tumor cells remodel their energy metabolic networks
to meet the bioenergetic and biosynthetic demands associated with
unlimited proliferation, invasion and metastasis. This metabolic
reprogramming involves multi-level restructuring of glucose, lipid
and amino acid metabolism. It not only supplies adenosine
triphosphate (ATP) to tumor cells but also utilizes metabolic
intermediates to participate in biosynthesis, counteract oxidative
stress and modulate the microenvironment, thereby shaping the tumor
microenvironment (TME) (7,8). Within the TME, tumor cells,
inflammatory cells, stromal cells and cytokines form a complex
immunosuppressive network that inhibits T-cell effector functions
and promotes T-cell exhaustion (9,10). By
remodeling the metabolic networks of glucose, lipids and glutamine
to counteract malignant biological behaviors, targeting tumor
metabolism has emerged as a novel strategy for cancer therapy
(11).
T cell-based immunotherapy has been widely applied
in the treatment of various types of cancer and has achieved
notable clinical outcomes, making it a prominent research focus in
the field of oncology (12). For
example, chimeric antigen receptor T-cell (CAR-T) therapy has
demonstrated favorable efficacy in hematological malignancies
(13). However, its effectiveness
against solid tumors, including CRC, remains limited.
CD8+ T cells are the primary effector cells of the
cellular immune response; they can specifically recognize antigens
and efficiently eliminate tumor cells, thereby serving a critical
role in antitumor immune responses (14). The differentiation status and
infiltration proportion of intratumoral CD8+ T cells are
closely associated with patient clinical prognosis, which forms an
essential basis for the development of T cell-based immunotherapies
(15,16). Currently, metabolism has been
recognized as a key mechanism regulating the antitumor activity of
T cells within the TME.
Metabolic checkpoints refer to a series of cellular
metabolic molecular switches that ensure the timely and accurate
conversion of nutrients into metabolites. Studies have shown that
metabolic checkpoints can regulate the development, differentiation
and function of T cells, as well as the competition for nutrients
in the TME between immune cells and tumor cells (17–21).
Metabolites within the TME can impose metabolic stress on
infiltrating T cells, contributing to local immunosuppression and
immune evasion (22,23). Consequently, modulating the
metabolic pathways of tumor cells and T cells via metabolic
checkpoints to enhance the antitumor efficacy of CAR-T therapy has
emerged as a promising therapeutic strategy (24). Notably, CRC exhibits tumor
heterogeneity, with substantial variations in metabolic profiles
and TME composition among different patients (5,6,25).
Current technological advances, such as single-cell metabolomics
analysis, enable the assessment of individual patient metabolic
status (25). Consequently, precise
immunometabolic therapy may represent a promising therapeutic
direction.
The present review incorporates the latest research
literature published between 2023 and 2025. Unlike previous
studies, which have focused solely on either tumor metabolism
regulation (3) or CAR-T
optimization (26), the current
review systematically elaborates on how the metabolic features of
CRC drive immunosuppression. Furthermore, it discusses the
potential strategies for reshaping the TME and enhancing the
antitumor effects of T cells by targeting metabolic checkpoints,
ranging from glycolysis and glutamine metabolism to fatty acid
oxidation (FAO). Furthermore, the perspective of tailoring
treatments based on distinct CRC metabolic genealogies is proposed,
aiming to provide insights for developing precision immunotherapies
targeting different CRC metabolic subtypes.
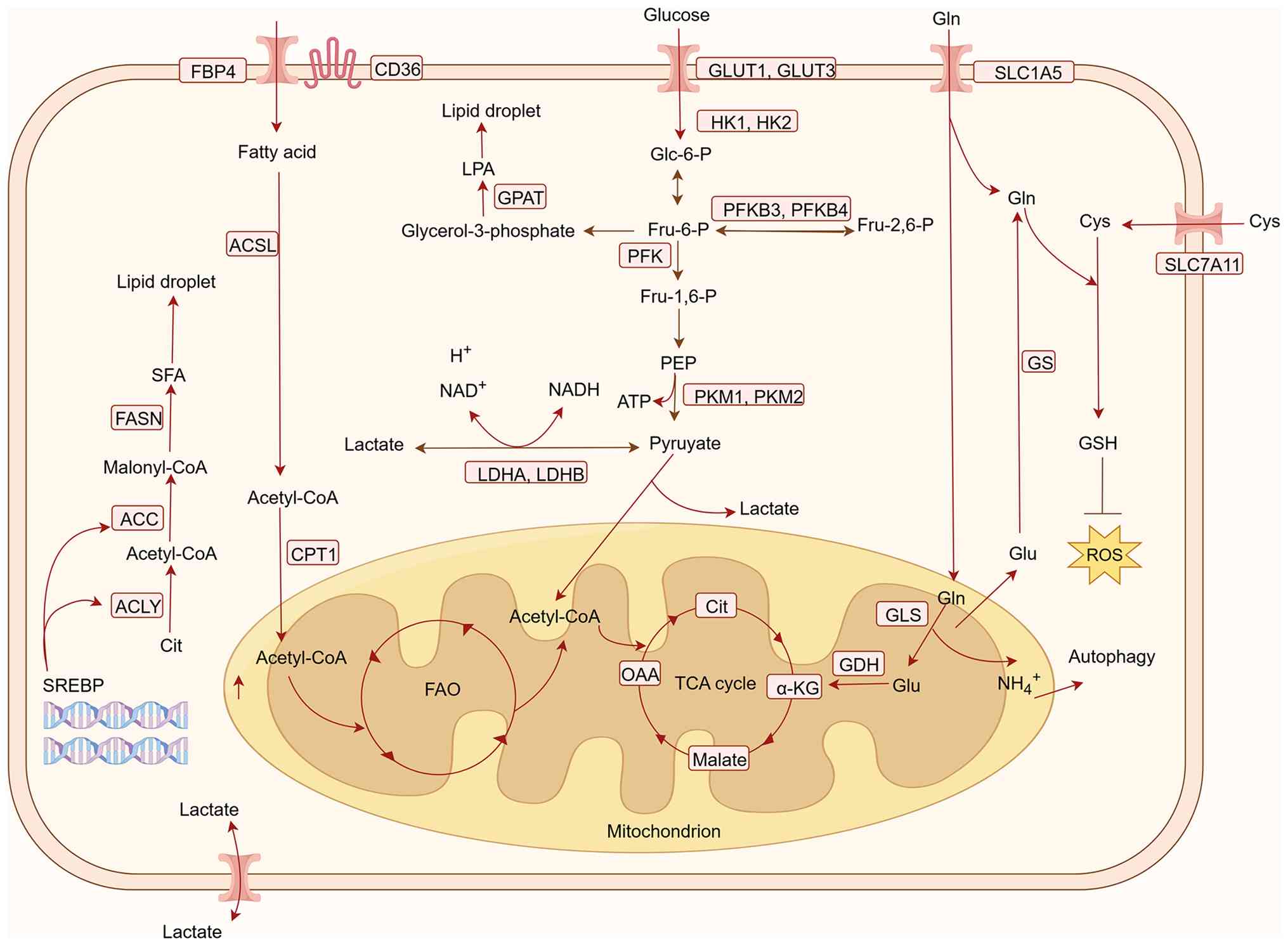
The key metabolic pathways in CRC and potential
therapeutic targets are integrated and illustrated in Fig. 1.
The Warburg effect, characterized by aerobic
glycolysis under normoxic conditions, is a distinct metabolic
feature observed in cancer cells. Despite sufficient oxygen
availability, glycolysis preferentially produces lactate, thereby
supporting rapid proliferation and meeting biosynthetic demands in
CRC (27). Glycolysis is an
inefficient energy-generating pathway, through which tumor cells
competitively consume large amounts of glucose to fuel their own
growth, while markedly impairing the antitumor response of immune
cells (28). It has been shown that
the excessive consumption of glucose by tumor cells leads to a
glucose-depleted TME, which inhibits the proliferation of
tumor-infiltrating lymphocytes, and suppresses mammalian target of
rapamycin (mTOR) activity, glycolytic capacity and the production
of effector molecules, such as interferon-γ (29). On the other hand, the enhanced
aerobic glycolysis in tumor cells generates substantial amounts of
metabolic byproducts, including lactate and CO2, leading
to lactate accumulation and acidification within the TME. This
further imposes metabolic stress on infiltrating immune cells. The
acidic extracellular microenvironment impedes lactate efflux from
cytotoxic T lymphocytes, directly affecting their proliferation and
cytokine secretion, ultimately resulting in impaired cytotoxic
function (30,31).
Under the Warburg effect, the resulting hypoxic and
acidic TME leads to the substantial accumulation of
hypoxia-inducible factor-1α, which restricts mitochondrial aerobic
activity and promotes glutaminolysis to meet the demands of
tumorigenesis and progression (32). Glutamine is the most abundant
non-essential amino acid in the human body. It enters cells via the
transporters solute carrier family 1 member 5 (SLC1A5) and solute
carrier family 7 member 5 (SLC7A5), and is converted by glutaminase
(GLS) into glutamate and ammonia. Subsequently, glutamate is
transformed into α-ketoglutarate (α-KG) through catalysis by
glutamate dehydrogenase (GDH) or transaminases, entering the
tricarboxylic acid (TCA) cycle to participate in the synthesis of
nucleotides, amino acids and fatty acids (33). Concurrently, glutamine can also be
converted into glutathione to maintain cellular redox homeostasis
and prevent damage to biological macromolecules (34,35).
Tumor cells often rely on glutamine metabolism to provide
biosynthetic precursors, energy supply and maintenance of
intracellular homeostasis for their rapid proliferation. The
enhanced glutamine metabolism in tumor cells leads to increased
ammonia release, which activates autophagy in adjacent
cancer-associated fibroblasts and promotes the release of
intracellular glutamine. This released glutamine can then be taken
up and utilized by tumor cells to sustain their proliferative
demands (36).
Early activated T cells require glutamine metabolism
to initiate proliferation and immune responses. However, glutamine
deficiency directly suppresses nucleotide synthesis and glutathione
production necessary for early T-cell activation, leading to
proliferation arrest and functional exhaustion of T cells (37). Furthermore, ammonia and α-KG
produced by tumor cells can remodel the pH and metabolite profile
of the TME, further inhibiting the function of CD8+
effector T (Teff) and T helper (Th)1 cells, while promoting the
polarization of regulatory T (Treg) cells and M2 macrophages
(38). The dependence on glutamine
metabolism varies markedly among already differentiated T-cell
subsets. Th17 cells maintain a high level of glutamine metabolism,
and their differentiation and effector functions are entirely
dependent on glutaminolysis catalyzed by GLS; notably, inhibition
of GLS rapidly impairs their proliferation and effector capacity.
By contrast, Th1 and CD8+ Teff cells gradually reduce
their reliance on glutamine metabolism during later stages of
activation, shifting toward FAO as the primary energy source. At
this stage, inhibiting GLS does not impair their function; instead,
it enhances their antitumor effects through epigenetic regulation
(39). Thus, the bidirectional
nature of glutamine modulation demonstrates that the timing and
target of intervention are critical determinants of therapeutic
efficacy.
Alterations in lipid metabolism serve a crucial role
in the occurrence and development of tumors, and are an important
characteristic of CRC. Lipid metabolism provides the necessary ATP
and macromolecules for tumor growth, division and survival, and
disordered lipid metabolic is a typical feature of CRC, with the
most notable alteration being increased de novo lipogenesis
and subsequent massive intracellular accumulation of lipids in the
form of lipid droplets (40).
Different T-cell subsets exhibit distinct lipid
metabolic demands, leading to notable divergence in their
sensitivity and response direction to lipid dysregulation within
the TME. In the early activation phase, CD8+ Teff cells
rely on de novo fatty acid synthesis to construct new cell
membranes to meet proliferation needs; however, upon entering the
TME, abnormal activation of acetyl-CoA carboxylase (ACC) and
CD36-mediated uptake of oxidized lipids result in substantial lipid
accumulation within T cells (40,41).
Concurrently, the FAO pathway in CD8+ Teff cells is
markedly suppressed and the large quantities of toxic lipids taken
up via CD36 can damage mitochondria (41). Th17 cells depend on de novo
fatty acid synthesis and glycolysis; enhanced FAO within the TME
inhibits their differentiation, whereas lipid peroxidation directly
impairs their effector functions (42,43).
Increased fatty acid content in the TME favors the generation of
Treg cells, which rely on exogenous fatty acid uptake to exert
their immunosuppressive functions (44). Based on these mechanisms, combined
strategies targeting tumor metabolic sensitization and T-cell
immune enhancement can be designed to precisely reverse T-cell
lipid metabolic disorders.
The efficacy of T cells in CRC is influenced by
metabolic competition and the metabolic microenvironment of CRC.
Theoretically, altering the competitive advantage of CRC tumor
cells for nutrients, reducing the production of immunosuppressive
substances and modifying the metabolic adaptability of T cells
themselves could all enhance the efficacy of T cells in CRC.
Nutrient competition within the TME, particularly
glucose scarcity, is a key factor impairing T-cell function
(29). Therefore, targeting the
aberrantly active glycolytic pathway in tumor cells to undermine
their metabolic advantage has emerged as a core strategy for
enhancing T-cell effector functions (45). This strategy primarily focuses on
two aspects: Inhibiting glucose transporters (GLUTs) and key
metabolic enzymes (46). GLUT1 is a
major GLUT that serves a crucial role in cancer cells; it not only
facilitates the uptake of large amounts of glucose to meet the
energy demands of rapid proliferation but also protects cancer
cells from oxidative stress induced by glucose deprivation, thereby
enhancing their anti-apoptotic capacity (47). In CRC, upregulation of GLUT1 is
strongly associated with poor patient prognosis (47). It has been demonstrated that the
expression of Hes family BHLH transcription factor 1 (HES1) is
markedly higher in CRC tissues compared with that in adjacent
normal tissues (48), and HES1
promotes CRC progression by enhancing the stability of modified
GLUT1 mRNA in an IGF2BP2-dependent manner. By contrast, knockdown
of HES1 reduces aerobic glycolysis activity and inhibits
tumorigenicity in CRC (49,50). Other research has revealed that the
upregulation of TANK-binding kinase 1 (TBK1) in CRC is associated
with disease progression. TBK1-mediated inhibition of mTOR complex
(mTORC)1 induces intracellular autophagy, subsequently reducing
GLUT1 degradation; this adaptive signaling cascade between TBK1 and
GLUT1 provides a novel strategic avenue for CRC treatment (51). Beyond modulating gene expression to
inhibit GLUT1 activity, GLUT1 inhibitors can also be employed. A
recent study showed that the GLUT1 inhibitor BAY-876 can suppress
CRC cell growth in both in vivo and ex vivo
experiments. Its primary mechanism of action involves reducing
glycolysis, thus forcing tumor cells to rely on oxidative
phosphorylation for ATP production. This leads to enhanced
mitochondrial respiration, reactive oxygen species (ROS)
accumulation, and ultimately, apoptosis (52).
Alterations in metabolic enzymes can directly
influence the glycolytic capacity of tumor cells. Current research
has identified key rate-limiting enzymes in the aerobic glycolysis
of tumor cells, such as hexokinase 2 (HK2), pyruvate kinase (PK),
lactate dehydrogenase A (LDHA) and phosphofructokinase-1 (PFK1), as
potential therapeutic targets for regulating tumor growth (53,54).
The first step of glycolysis is catalyzed by HK2, the expression of
which is markedly elevated in CRC. Notably, targeted inhibition of
HK2 can reduce the level of glycolysis in CRC cells (55). PK is another critical glycolytic
enzyme that is highly expressed in colon cancer. Changes in the
PKM1/PKM2 ratio may alter glucose metabolism; specifically, a
decrease in PKM1 expression coupled with an increase in PKM2 can
induce the Warburg effect, thereby promoting tumor proliferation
(56,57). Therefore, targeting PK expression
may be an effective strategy for modulating tumor glycolysis. LDHA
primarily acts in the final step of the glycolytic pathway,
catalyzing the conversion of pyruvate to lactate.
A recent study, through extensive examination of 40
pan-cancer single-cell RNA sequencing cohorts, established a novel
lactate metabolism-related signature, demonstrating that LDHA is a
rational therapeutic target across different cancer types and
represents a promising treatment strategy to enhance antitumor
immune responses (58). In CRC,
regenerating family member 1α (REG1α) is highly expressed and
closely associated with poor prognosis. REG1α can exert its
pro-tumorigenic function by inducing aerobic glycolysis in cancer
cells via the β-catenin/MYC proto-oncogene/LDHA axis. The use of
MYC proto-oncogene inhibitors and LDHA inhibitors can suppress the
REG1α-induced increase in glycolysis, providing a basis for
investigating feasible treatment plans for CRC (59).
PFK1 serves a crucial role in the conversion of
fructose-6-phosphate to fructose-1,6-bisphosphate during
glycolysis. Among the PFK1 family,
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3)
exhibits the strongest activity and is the most important
phosphokinase in the glycolytic process (60). PFKFB3 is highly expressed in CRC,
and is associated with disease progression and poor prognosis
(61). It has been shown that
PFKFB3 is associated with the tumor immunosuppressive
microenvironment and is related to the IL-2/STAT5 signaling pathway
(62). Previous research has
indicated that the IL-2/STAT5 pathway is essential for maintaining
the homeostasis and migration of Treg cells and the exhaustion of
CD8+ T cells (63,64).
Consequently, combining PFKFB3 inhibitors with immunotherapy holds
promise for improving the response rate and prolonging overall
survival in patients with CRC subjected to immunotherapy (62).
In tumor cells, glutamine metabolism is involved in
the biosynthesis of precursors, maintains redox homeostasis and
provides energy, thereby promoting cell proliferation, migration
and invasion. Enzymes related to glutamine metabolism, such as
GLS1/2 (65), glutamine synthetase
(GS) (66), GDH (67) and transglutaminase (TG) (65), as well as protein transporters
involved in glutamine transport, such as SLC1A5, solute carrier
family 38 member 2 (SLC38A2) and solute carrier family 38 member 3,
have regulatory roles in the TME. GLS is a mitochondrial enzyme,
divided into two subtypes, GLS1 and GLS2. GLS1 exhibits high
activity in CRC and is positively associated with tumor progression
(68). The use of GLS inhibitors,
such as BPTES and CB-839, to attenuate GLS1 expression activity in
tumors can markedly inhibit tumor cell migration (69,70).
In contrast to GLS1, GLS2 is generally considered a tumor
suppressor in most contexts, with lower expression and activity in
tumors; in addition, its expression level is negatively associated
with tumor progression (71). GS
catalyzes the intracellular synthesis of glutamine from glutamate
and free ammonia, meeting the demands of rapidly proliferating
tumor cells by intracellularly synthesizing glutamine (72). A decrease in GS activity can mediate
the phenotypic transformation of tumor-associated macrophages and
inhibit tumor metastasis (73). GDH
catalyzes the conversion of glutamate to α-KG, which enters the TCA
cycle to participate in energy supply and biosynthesis. High
expression of GDH is closely associated with clinical prognosis;
GDH can promote CRC cell migration via STAT3-mediated
epithelial-mesenchymal transition (EMT) and may serve as a
prognostic marker for CRC metastasis (74). TG catalyzes the hydrolysis of
glutamine residues within proteins or polypeptide chains and the
exchange of amide groups with other amino groups. It has been
reported that TG2 expression is higher in the metastatic colon
cancer cell line SW620 than in primary colon cancer cell lines, and
is associated with EMT progression (75). Subsequently, the developed selective
small-molecule active-site-directed inhibitor of TG2, 1–155, has
been shown to attenuate the ability of TG2 to induce EMT (76).
Glutamine transporters are located on the cell
membrane, facilitating the uptake of sufficient glutamine from the
external environment by tumor cells to meet their growth and
metabolic demands. SLC1A5, also known as alanine-serine-cysteine
transporter 2, is a transporter for glutamine, serine and cysteine,
belonging to the sodium-dependent neutral amino acid transporter
family (77). SLC1A5 has high
affinity for glutamine and particularly promotes glutamine
transport in tumor cells under acidic conditions (78). SLC1A5 is highly expressed in CRC,
and targeted intervention using V-9302 can effectively inhibit CRC
growth in vitro and in murine models in vivo
(79). Solute carrier family 6
member 14 (SLC6A14) serves a crucial role in maintaining amino acid
transport and cellular metabolic balance. Its primary function is
to promote the unidirectional influx of amino acids such as
glutamate (80,81). High expression of SLC6A14 is closely
associated with metastasis and poor prognosis in patients with CRC
(82). α-methyltryptophan (α-MT) is
a tryptophan metabolism inhibitor and tryptophan is a substrate of
SLC6A14. α-MT can inhibit the proliferation of SLC6A14-positive CRC
cells but has minimal effect on SLC6A14-negative cells (83). Furthermore, compared with in primary
tumors, SLC38A2 is highly expressed in metastatic CRC tissues, and
the absence of SLC38A2 is beneficial for inhibiting tumor
progression (84). The amino acid
antiporter SLC7A5 is also crucial for tumor metastasis and
development in advanced CRC; the absence of SLC7A5 can notably
prolong the survival of model mice and reduce tumor metastasis
(85).
Lipid metabolic reprogramming in the TME increases
the nutrient supply for tumor cells. In CRC, lipid metabolism
mainly involves changes in several key genes and metabolic enzymes,
including sterol regulatory element-binding proteins (SREBPs), ATP
citrate lyase (ACLY) and adipose triglyceride lipase (ATGL). SREBPs
are crucial transcription factors in lipid metabolism; the
activation of SREBPs and their target genes, such as fatty acid
synthase (FASN) and stearoyl-CoA desaturase, markedly alters
cellular metabolic pathways. Knockdown of SREBP-1 or SREBP-2 in CRC
cell lines leads to reduced cell numbers, decreased proliferation
rates and diminished capacity to form tumor spheroids (86,87).
Therefore, targeting SREBP-1 and SREBP-2 serves an important role
in lipid metabolism-mediated tumor proliferation in CRC. A previous
study revealed that ACLY knockdown results in a notable decrease in
SREBP-1 levels in LoVo cells, leading to increased apoptosis rates.
Furthermore, the apoptosis rate in cells with SREBP-1 knockdown has
been shown to be higher than that in the ACLY knockdown group
(88).
ACLY, the first rate-limiting enzyme in fatty acid
synthesis, is associated with the metastatic potential of CRC.
Knockdown of ACLY reduces the activity of β-catenin, inhibits its
transcriptional activity, and consequently decreases the migration
and invasion of CRC cells (89). It
has been reported that zinc finger DHHC-type palmitoyltransferase
6, a palmitoyltransferase regulating fatty acid synthesis, directly
palmitoylates and stabilizes peroxisome proliferator-activated
receptor (PPAR)γ. This stabilization, in turn, activates ACLY
transcription-related metabolic pathways, stimulates fatty acid
production and is associated with CRC severity (90). Carnitine palmitoyltransferase 1A
(CPT1A) is highly expressed in CRC. High CPT1A expression enhances
FAO, promotes high survival rates of CRC cells and facilitates
metastasis. Notably, CPT1A is not only associated with FAO
activation but also interacts with the B-cell lymphoma 2 family,
directly influencing metastasis (91–93).
Furthermore, it has been demonstrated that ATGL is highly expressed
in CRC; upregulation of ATGL promotes CRC cell proliferation and is
negatively associated with patient survival (94). Glycerol-3-phosphate acyltransferase
3 (GPAT3) has been reported to promote lipid droplet generation and
confer chemoresistance in CRC. Following oxaliplatin treatment,
high GPAT3 expression transforms CRC cells into non-immunogenic
cells, attributed to reduced cytotoxic interferon-γ release and
CD8+ T-cell exhaustion (95).
The fatty acid-binding protein (FABP) family serves
an important role in lipid metabolism in CRC. A previous study
indicated that FABP6 expression is negatively associated with
immune infiltration in CRC; knockdown of FABP6 increases the
expression of major histocompatibility complex class I molecules
and promotes the secretion of immune-related chemoattractants,
suggesting enhanced immunogenicity of tumor cells (96). Upregulation of FABP4 promotes
phosphorylated AKT expression to facilitate the EMT process,
thereby enhancing CRC migration and invasion. Conversely, the FABP4
inhibitor BMS309403 yields opposite effects (97,98).
FABP5, a transporter involved in fatty acid uptake, increases CRC
cell numbers in a manner independent of the PPARβ/PPARδ signaling
pathway (99). FASN is responsible
for converting acetyl-CoA and malonyl-CoA into long-chain fatty
acids (such as palmitic acid), and high FASN expression is
negatively associated with CRC survival. Utilizing FASN inhibitors,
such as cerulenin, TVB-3166, TVB-3664 and TVB-369380, can modulate
the AKT and adenosine 5′-monophosphate-activated protein kinase
(AMPK) pathways to inhibit CRC progression, although treatment
safety must be considered (100,101). Furthermore, FASN drives cancer
cell proliferation, metastasis and phosphatidylcholine metabolism
via the specificity protein 1/phospholipase A2 group IVB axis.
Knockdown of FASN can markedly inhibit tumor growth and the spread
of CRC cells to the lungs (102).
Alterations in lipid metabolism have become a key driving factor in
the progression of CRC, involving multiple key molecules as
aforementioned. Targeting these key genes or alterations in
critical metabolic enzymes can induce apoptosis and reduce invasive
capacity, with value as therapeutic targets for CRC.
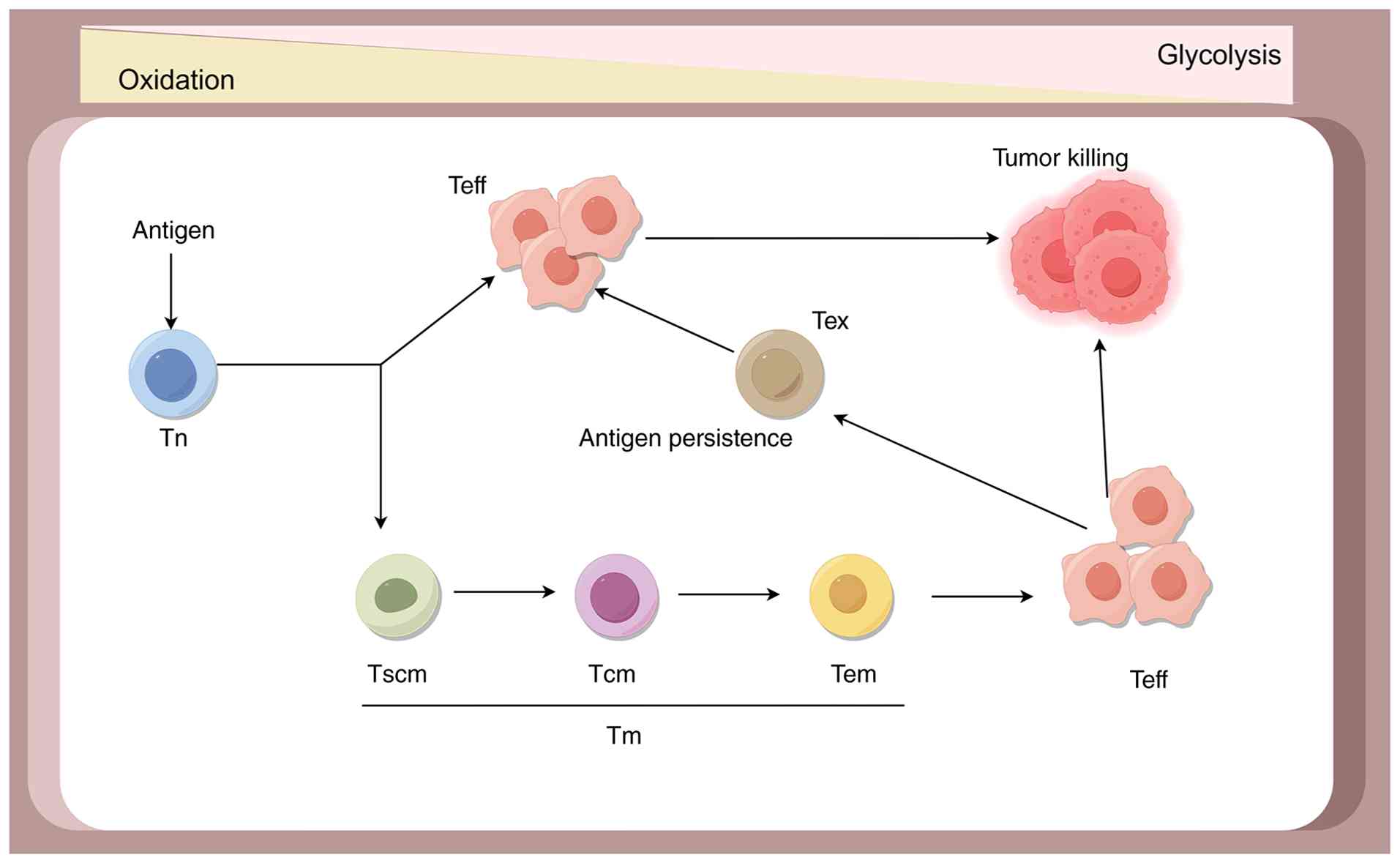
T cells are key components of the immune system and
their metabolic status influences their functional performance
(103,104). T cells in different states, such
as naïve T (Tn), Teff, memory T (Tm) and exhausted T (Tex) cells,
each possess distinct metabolic requirements. Tn cells exhibit a
relatively low metabolic rate, primarily relying on low-level
oxidative phosphorylation fueled by glucose-derived pyruvate or
fatty acids to meet their energy demands (105). Upon activation of the T-cell
receptor (TCR) in Tn cells, concomitant co-stimulatory signals
received by receptors such as CD28 enhance TCR signaling. This
triggers metabolic changes to support the anabolic processes
necessary for cell proliferation and the differentiation of Teff
cells, thereby enhancing effector functions (106–108). Following activation through
interactions with antigens, receptors, co-stimulatory ligands and
cytokine signaling, Teff cells enter a metabolically active state
characterized by increased glycolytic and oxidative phosphorylation
activity. They predominantly utilize aerobic glycolysis to provide
energy for their rapid proliferation and functional execution
(109,110). If the antigen is cleared, Teff
cells can transition into a long-lived memory state with stem
cell-like properties. Their metabolic profile reverts to a less
active state, primarily depending on FAO and oxidative
phosphorylation pathways to sustain long-term cellular survival and
energy needs. Upon re-encountering their cognate antigen, Tm cells
mount a rapid response, swiftly reactivating aerobic glycolysis to
facilitate robust effector function and proliferation (111). In cases of persistent antigen
exposure, inhibitory receptors such as PD-1 and CTLA-4, can remodel
T-cell metabolism, leading to the transition of T cells towards an
exhausted state. Tex cells exhibit reduced glucose and glutamine
metabolism, dysfunctional mitochondria and an increased reliance on
FAO (112,113). Within the TME, there exists a
complex metabolic network with interactions; tumor cells engage in
almost parasitic competition for nutrients and the metabolism of T
cells also interacts with each other. Through regulating the
metabolism of T cells, it may be possible to enhance their
antitumor ability (Fig. 2).
Fine-tuning the glycolytic pathway within T cells
themselves can optimize their differentiation fate and functional
state. mTOR is a shared serine/threonine kinase that exists in two
distinct complexes, mTORC1 and mTORC2. Of these, mTORC1 acts as a
sensor for nutrients in the microenvironment, capable of perceiving
cellular nutrient status and specific needs to coordinate cellular
functions, serving as a classic metabolic checkpoint molecule
(114,115). Studies have shown that mTORC1 can
promote glycolysis by upregulating the expression of GLUT1, GLUT3
and LDHA through upregulating hypoxia-inducible factor-1α
expression in CD8+ T cells, thereby promoting their
effector functions (116,117). Inhibition of mTORC1 with rapamycin
reduces the overall glycolytic flux in CD8+ T cells,
thereby increasing memory phenotype differentiation and enhancing
antitumor capacity (118).
Genetic editing techniques or small-molecule
inhibitors can be employed to intervene in the expression or
activity of specific proteins. For example, CD38 is a single-chain
transmembrane glycoprotein; knockdown of the CD38 gene or the use
of small-molecule inhibitors targeting CD38 enzymatic activity can
inhibit glycolysis by downregulating the CD38-cyclic adenosine
diphosphate ribose-calcium ion signaling pathway and activating the
CD38/nicotinamide adenine dinucleotide/sirtuin 1 axis. This, in
turn, promotes memory phenotype differentiation in CAR-T cells and
enhances their in vivo antitumor capacity and persistence
(119). Mitochondrial isocitrate
dehydrogenase 2 primarily mediates oxidative decarboxylation
reactions. Inhibition of this enzyme in CAR-T cells with the
clinical drug enasidenib shifts cellular metabolism from glycolysis
to the pentose phosphate pathway, reduces ROS levels, and enhances
the persistence and antitumor function of CAR-T cells (120). Suppressing overactive glycolysis
can promote the formation of memory-like T cells and enhance their
persistence in vivo. Conversely, during stages requiring
rapid expansion and potent cytotoxic activity, timely promotion of
glycolysis in CAR-T cells may be beneficial. This indicates that
metabolic regulation of CAR-T cells to meet the functional
requirements at different stages is the key to enhancing their
therapeutic efficacy.
The uptake of lipids from the TME is crucial for
sustaining the effector function of CD8+ T cells, as
tumor cells are often deficient in glucose as an energy source. To
adapt to the TME, CD8+ T cells upregulate lipid
transporters to acquire lipids as an energy source under nutrient
stress, leading to excessive lipid accumulation within these cells,
which contributes to their dysfunction and exhaustion (121). Most tumor-infiltrating lymphocytes
exhibit suppressed mitochondrial biogenesis and a limited capacity
to utilize FAO, further inhibiting T-cell effector function
(122). Therefore, regulating
T-cell lipid transporters to reduce excessive intracellular lipid
accumulation may be an effective strategy to enhance the antitumor
capacity of T cells.
CD36, a scavenger receptor responsible for
transporting fatty acids and oxidized lipids, is highly expressed
in tumor-infiltrating CD8+ T cells and impairs their
function (123). Knockout of the
CD36 gene in CD8+ T cells has been shown to markedly
inhibit tumor growth in CRC (41,124).
Notably, anti-CD36 blocking antibodies can also target
metastasis-initiating cells, inhibiting metastasis in both
immunodeficient and immunocompetent mouse models without marked
side effects (125). Consequently,
CD36 represents a promising drug target, and the development of
novel CD36 inhibitors, particularly in combination with CAR-T
therapy, holds potential for advancing cancer therapies.
Tumor-infiltrating CD8+ T cells exhibit increased lipid
storage due to elevated ACC activity. Restriction of ACC activity
can reprogram T-cell metabolism, enhance survival and
polyfunctionality, and suppress tumor growth (126). FASN, a critical enzyme in fatty
acid synthesis, serves a marked role in the function and survival
of memory CD8+ T cells. These cells rely on fatty acid
synthesis to achieve long-term persistence, and inhibition of FASN
leads to decreased survival of memory CD8+ T cells,
whereas effector CD8+ T cells remain largely unaffected
(127). However, the specific role
of FASN in tumor-infiltrating CD8+ T cells remains
unclear, and further investigation is required to determine its
impact on antitumor function.
The present review systematically elucidates the
complex mechanisms underlying the interplay between immunity and
metabolism in CRC. Tumor cells actively shape an immunosuppressive
TME through metabolic reprogramming, thereby compromising the
efficacy of T-cell therapy. Through an in-depth analysis of these
mechanisms, it may be hypothesized that dual breakthroughs are
required at the fundamental conceptual and technical levels.
While traditional research has predominantly focused
on the roles of cytokines and immune checkpoint molecules within
the TME, the current review highlights the pivotal role of
metabolic regulation in fostering immunosuppression (22,23,132).
The interaction between tumor cells and T cells extends beyond
competition for physical space and signaling pathways to encompass
the regulation of nutrient acquisition and metabolic pathways.
Lactate, the end product of glycolysis, not only contributes to an
acidic milieu but can also directly regulate the expression of
immune-related genes (such as Foxp3) by inducing histone
lactylation, thereby stabilizing the functional state of
immunosuppressive cells, such as Treg cells (31,32).
Conversely, aberrant lipid metabolism leads to the accumulation of
oxidized lipids within the TME, which can directly impair
mitochondrial function in CD8+ T cells and promote the
development of an exhausted phenotype (41,124).
Consequently, effective therapeutic strategies must address two key
aspects: Improving nutrient availability within the TME and
actively clearing or neutralizing these bioactive immunosuppressive
metabolites.
Notable metabolic heterogeneity exists among
patients with CRC, forming a crucial basis for differential
therapeutic responses (4,24). For example, intervention strategies
targeting glutamine metabolism are effective only in tumors with
high GLS1 expression (68,69), showing limited efficacy against
tumor subtypes with metabolic compensation mechanisms and
potentially affecting normal T-cell function, which also relies on
glutamine (39,133). Therefore, future clinical practice
necessitates the establishment of a patient stratification system
based on metabolic profiles. By integrating metabolic imaging
techniques with single-cell metabolomic analyses (4,134,135), the metabolic landscape of a
patient can be systematically assessed prior to treatment,
facilitating the development of individualized combination
strategies. The precise identification of tumor-specific metabolic
dependencies is a prerequisite for achieving precision
immunometabolic therapy.
A reason for the limited functionality of current T
cells within the TME is their insufficient metabolic adaptability
(29,136). Future technological developments
should focus on enhancing the metabolic adaptability of CAR-T
cells, improving metabolic plasticity, regulating key metabolic
regulatory factors through genetic engineering, or introducing
heterologous metabolic enzymes to enable CAR-T cells to utilize
multiple energy substrates and adapt to nutrient-limited conditions
within the TME (17,131). The construction of metabolic
resistance can be achieved by utilizing gene editing technologies
to knock out specific receptors, thereby reducing the sensitivity
of CAR-T cells to inhibitory metabolic signals within the TME
(121,122,137). Furthermore, by using exogenous
small molecules to regulate metabolic pathways, researchers can
precisely control when and where the CAR-T cells change their
metabolic state. For example, glycolytic capacity can be enhanced
during the expansion phase, whereas a shift toward oxidative
phosphorylation can be promoted during the effector phase to
facilitate the formation and maintenance of memory subsets
(107,131).
The simple combination of metabolic modulators with
T cells is unlikely to achieve optimal synergistic effects
(54,72). A sequential treatment strategy is
therefore proposed: Prior to T-cell infusion, the TME could be
pre-conditioned with metabolic modulators to improve the metabolic
milieu; following CAR-T cell infusion, a metabolic support regimen
centered on sustaining T-cell function could be implemented
(62,72). Furthermore, the combined application
of metabolic interventions with epigenetic regulators may induce a
deeper synergistic antitumor effect by remodeling the
transcriptional states of both tumor and immune cells (73,103).
The application bottleneck of T-cell therapy in CRC
is fundamentally a problem of mismatch between therapeutic cells
and the metabolic characteristics of the TME. The solution should
shift from mere targeted killing to the systemic remodeling of the
TME and the precise optimization of the metabolic functions of
CAR-T cells. This necessitates the establishment of a patient
stratification system based on multi-omics analysis and the
development of next-generation CAR-T cells with metabolic
adaptability. This research direction represents a notable
developmental trend in cancer immunotherapy, namely immunometabolic
precision therapy aimed at breaking metabolic constraints. Its
advancement will provide a novel breakthrough pathway for the
treatment of solid tumors such as CRC.
Not applicable.
This work was supported by the Yantai Science and Technology
Program Development Project (grant no. 2024YD014).
Not applicable.
QY contributed to conceptualization, project
administration, writing the original draft, reviewing and editing.
PL contributed to investigation, supervision, reviewing and
editing. JL contributed to investigation, reviewing and editing. LW
contributed to conceptualization, supervision, reviewing and
editing. Data authentication is not applicable. All authors read
and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
2
|
Wolpin BM and Mayer RJ: Systemic treatment
of colorectal cancer. Gastroenterology. 134:1296–1310. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Kratzer TB, Wagle NS, Sung H
and Jemal A: Cancer statistics, 2026. CA Cancer J Clin.
76:e700432026.PubMed/NCBI
|
|
4
|
Wang Y, Qiu X, Li Q, Qin J, Ye L, Zhang X,
Huang X, Wen X, Wang Z, He W, et al: Single-cell and
spatial-resolved profiling reveals cancer-associated fibroblast
heterogeneity in colorectal cancer metabolic subtypes. J Transl
Med. 23:1752025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valdeolivas A, Amberg B, Giroud N,
Richardson M, Gálvez EJC, Badillo S, Julien-Laferrière A, Túrós D,
Voith von Voithenberg L, Wells I, et al: Profiling the
heterogeneity of colorectal cancer consensus molecular subtypes
using spatial transcriptomics. NPJ Precis Oncol. 8:102024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ogden S, Metic N, Leylek O, Smith EA,
Berner AM, Baker AM, Uddin I, Buzzetti M, Gerlinger M; Cancer
Tissue Bank, ; et al: Phenotypic heterogeneity and plasticity in
colorectal cancer metastasis. Cell Genom. 5:1008812025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Biswas SK: Metabolic reprogramming of
immune cells in cancer progression. Immunity. 43:435–449. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koundouros N and Poulogiannis P:
Reprogramming of fatty acid metabolism in cancer. Br J Cancer.
122:4–22. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hwang HS, Kim D and Choi J: Distinct
mutational profile and immune microenvironment in
microsatellite-unstable and POLE-mutated tumors. J Immunother
Cancer. 9:e0027972021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin B, Du L, Li H, Zhu X, Cui L and Li X:
Tumor-infiltrating lymphocytes: Warriors fight against tumors
powerfully. Biomed Pharmacother. 132:1108732020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu H, Wang S, Wang J, Guo X, Song Y, Fu
K, Gao Z, Liu D, He W and Yang LL: Energy metabolism in health and
diseases. Signal Transduct Target Ther. 10:692025. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brudno JN, Maus MV and Hinrichs CS: CAR T
cells and T-cell therapies for cancer: A translational science
review. JAMA. 332:1924–1935. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kantarjian H, Stein A, Gökbuget N,
Fielding AK, Schuh AC, Ribera JM, Wei A, Dombret H, Foà R, Bassan
R, et al: Blinatumomab versus chemotherapy for advanced acute
lymphoblastic leukemia. N Engl J Med. 376:836–847. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giles JR, Globig AM, Kaech SM and Wherry
EJ: CD8+ T cells in the cancer-immunity cycle. Immunity.
56:2231–2253. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lerner EC, Woroniecka KI, D'Anniballe VM,
Wilkinson DS, Mohan AA, Lorrey SJ, Waibl-Polania J, Wachsmuth LP,
Miggelbrink AM, Jackson JD, et al: CD8+ T cells maintain
killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL
axis. Nat Cancer. 4:1258–1272. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
MacNabb BW, Chen X, Tumuluru S, Godfrey J,
Kasal DN, Yu J, Jongsma MLM, Spaapen RM, Kline DE and Kline J:
Dendritic cells can prime anti-tumor CD8+ T cell
responses through major histocompatibility complex cross-dressing.
Immunity. 55:2206–2208. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Frisch AT, Wang Y, Xie B, Yang A, Ford BR,
Joshi S, Kedziora KM, Peralta R, Wilfahrt D, Mullett SJ, et al:
Redirecting glucose flux during in vitro expansion generates
epigenetically and metabolically superior T cells for cancer
immunotherapy. Cell Metab. 37:870–885.e8. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Markowitz GJ, Ban Y, Tavarez DA, Yoffe L,
Podaza E, He Y, Martin MT, Crowley MJP, Sandoval TA, Gao D, et al:
Deficiency of metabolic regulator PKM2 activates the pentose
phosphate pathway and generates TCF1+ progenitor
CD8+ T cells to improve immunotherapy. Nat Immunol.
25:1884–1899. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mortazavi Farsani SS, Soni J, Jin L, Yadav
AK, Bansal S, Mi T, Hilakivi-Clarke L, Clarke R, Youngblood B,
Cheema A and Verma V: Pyruvate kinase M2 activation reprograms
mitochondria in CD8 T cells, enhancing effector functions and
efficacy of anti-PD1 therapy. Cell Metab. 37:1294–1310.e7. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qiu Y, Su Y, Xie E, Cheng H, Du J, Xu Y,
Pan X, Wang Z, Chen DG, Zhu H, et al: Mannose metabolism reshapes T
cell differentiation to enhance anti-tumor immunity. Cancer Cell.
43:103–121.e8. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patel CH and Powell JD: Targeting T cell
metabolism to regulate T cell activation, differentiation and
function in disease. Curr Opin Immunol. 46:82–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Notarangelo G, Spinelli JB, Perez EM,
Baker GJ, Kurmi K, Elia I, Stopka SA, Baquer G, Lin JR, Golby AJ,
et al: Oncometabolite d-2HG alters T cell metabolism to impair
CD8+ T cell function. Science. 377:1519–1529. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Willson J: Tumour-derived D-2HG blocks T
cell cytotoxicity. Nat Rev Cancer. 22:6572022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trefny MP, Kroemer G, Zitvogel L and
Kobold S: Metabolites as agents and targets for cancer
immunotherapy. Nat Rev Drug Discov. 24:764–784. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chu X, Li X, Zhang Y, Dang G, Miao Y, Xu
W, Wang J, Zhang Z and Cheng S: Integrative single-cell analysis of
human colorectal cancer reveals patient stratification with
distinct immune evasion mechanisms. Nat Cancer. 5:1409–1426. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sterner RC and Sterner RM: CAR-T cell
therapy: Current limitations and potential strategies. Blood Cancer
J. 11:692021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liberti MV and Locasale JW: The Warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leone RD and Powell JD: Metabolism of
immune cells in cancer. Nat Rev Cancer. 9:516–531. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang CH, Qiu J, O'Sullivan D, Buck MD,
Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ,
et al: Metabolic competition in the tumor microenvironment is a
driver of cancer progression. Cell. 162:1229–1241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brand A, Singer K, Koehl GE, Kolitzus M,
Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et
al: LDHA-associated lactic acid production blunts tumor
immunosurveillance by T and NK cells. Cell Metab. 24:657–671. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu Y, Zhang Q, Xing B, Luo N, Gao R, Yu
K, Hu X, Bu Z, Peng J, Ren X and Zhang Z: Immune phenotypic linkage
between colorectal cancer and liver metastasis. Cancer Cell.
40:424–437.e5. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Damiani C, Colombo R, Gaglio D,
Mastroianni F, Pescini D, Westerhoff HV, Mauri G, Vanoni M and
Alberghina L: A metabolic core model elucidates how enhanced
utilization of glucose and glutamine, with enhanced
glutamine-dependent lactate production, promotes cancer cell
growth: The WarburQ effect. PLoS Comput Biol. 13:e10057582017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yoo HC, Yu YC, Sung Y and Han JM:
Glutamine reliance in cell metabolism. Exp Mol Med. 52:1496–1516.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen TH, Wang HC, Chang CJ and Lee SY:
Mitochondrial glutathione in cellular redox homeostasis and disease
manifestation. Int J Mol Sci. 25:13142024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bai C, Hua J, Meng D, Xu Y, Zhong B, Liu
M, Wang Z, Zhou W, Liu L, Wang H, et al: Glutaminase-1 mediated
glutaminolysis to glutathione synthesis maintains redox homeostasis
and modulates ferroptosis sensitivity in cancer cells. Cell Prolif.
11:e700362025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ko YH, Lin Z, Flomenberg N, Pestell RG,
Howell A, Sotgia F, Lisanti MP and Martinez-Outschoorn UE:
Glutamine fuels a vicious cycle of autophagy in the tumor stroma
and oxidative mitochondrial metabolism in epithelial cancer cells:
Implications for preventing chemotherapy resistance. Cancer Biol
Ther. 12:1085–1097. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Edwards DN, Ngwa VM, Raybuck AL, Wang S,
Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S, et al: Selective
glutamine metabolism inhibition in tumor cells improves antitumor T
lymphocyte activity in triple-negative breast cancer. J Clin
Invest. 131:e1401002021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matias MI, Yong CS, Foroushani A,
Goldsmith C, Mongellaz C, Sezgin E, Levental KR, Talebi A, Perrault
J, Rivière A, et al: Regulatory T cell differentiation is
controlled by αKG-induced alterations in mitochondrial metabolism
and lipid homeostasis. Cell Rep. 37:1099112021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Johnson MO, Wolf MM, Madden MZ, Andrejeva
G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton
RJ, et al: Distinct regulation of Th17 and Th1 cell differentiation
by glutaminase-dependent metabolism. Cell. 175:1780–1795.e19. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cao J, Qin S, Li B, Zhang Z, Miao P, Yan
H, Duan J, He B, He K, Peng P, et al: Extracellular vesicle-induced
lipid dysregulation drives liver premetastatic niche formation in
colorectal cancer. Gut. 74:2012–2023. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E,
Wang Q, Yang M, Qian J and Yi Q: CD36-mediated ferroptosis dampens
intratumoral CD8+ T cell effector function and impairs
their antitumor ability. Cell Metab. 33:1001–1012.e5. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xing J, Man C, Liu Y, Zhang Z and Peng H:
Factors impacting the benefits and pathogenicity of Th17 cells in
the tumor microenvironment. Front Immunol. 14:12242692023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cerboni S, Gehrmann U, Preite S and Mitra
S: Cytokine-regulated Th17 plasticity in human health and diseases.
Immunology. 163:3–18. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pacella I, Procaccini C, Focaccetti C,
Miacci S, Timperi E, Faicchia D, Severa M, Rizzo F, Coccia EM,
Bonacina F, et al: Fatty acid metabolism complements glycolysis in
the selective regulatory T cell expansion during tumor growth. Proc
Natl Acad Sci USA. 115:E6546–E6555. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cascone T, McKenzie JA, Mbofung RM, Punt
S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al:
Increased tumor glycolysis characterizes immune resistance to
adoptive T cell therapy. Cell Metab. 27:977–987.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qiao Q, Hu S and Wang X: The regulatory
roles and clinical significance of glycolysis in tumor. Cancer
Commun (Lond). 44:761–786. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shen YM, Arbman G, Olsson B and Sun XF:
Overexpression of GLUT1 in colorectal cancer is independently
associated with poor prognosis. Int J Biol Markers. 26:166–172.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yuan R, Ke J, Sun L, He Z, Zou Y, He X,
Chen Y, Wu X, Cai Z, Wang L, et al: HES1 promotes metastasis and
predicts poor survival in patients with colorectal cancer. Clin Exp
Metastasis. 32:169–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Weng MT, Tsao PN, Lin HL, Tung CC, Change
MC, Chang YT, Wong JM and Wei SC: Hes1 increases the invasion
ability of colorectal cancer cells via the STAT3-MMP14 pathway.
PLoS One. 10:e01443222015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang J, Zhu M, Zhu J, Li J, Zhu X, Wang K,
Shen K, Yang K, Ni X, Liu X, et al: HES1 promotes aerobic
glycolysis and cancer progression of colorectal cancer via
IGF2BP2-mediated GLUT1 m6A modification. Cell Death Discov.
9:4112023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou D, Yao Y, Zong L, Zhou G, Feng M,
Chen J, Liu G, Chen G, Sun K, Yao H, et al: TBK1 facilitates
GLUT1-dependent glucose consumption by suppressing mTORC1 signaling
in colorectal cancer progression. Int J Biol Sci. 18:3374–3389.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hayashi M, Nakamura K, Harada S, Tanaka M,
Kobayashi A, Saito H, Tsuji T, Yamamoto D, Moriyama H, Kinoshita J
and Inaki N: GLUT1 inhibition by BAY-876 induces metabolic changes
and cell death in human colorectal cancer cells. BMC Cancer.
25:7162025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sun X, Peng Y, Zhao J, Xie Z, Lei X and
Tang G: Discovery and development of tumor glycolysis rate-limiting
enzyme inhibitors. Bioorg Chem. 112:1048912021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sawant Dessai A, Kalhotra P, Novickis AT
and Dasgupta S: Regulation of tumor metabolism by post
translational modifications on metabolic enzymes. Cancer Gene Ther.
30:548–558. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhong J, Lu S, Jia X, Li Q, Liu L, Xie P,
Wang G, Lu M, Gao W, Zhao T, et al: Role of endoplasmic reticulum
stress in apoptosis induced by HK2 inhibitor and its potential as a
new drug combination strategy. Cell Stress Chaperones. 27:273–283.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ma L, Zhang X, Liu Y, Jin H, Li D, Zhang
H, Feng L, Zuo J, Wang Y, Liu J and Han J: The ratio of PKM1/PKM2
is the key factor affecting the glucose metabolism and biological
function of colorectal cancer cells. Transl Cancer Res.
13:3522–3535. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhao J, Li J, Hassan W, Xu D, Wang X and
Huang Z: Sam68 promotes aerobic glycolysis in colorectal cancer by
regulating PKM2 alternative splicing. Ann Transl Med. 8:4592020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen D, Liu P, Lu X, Li J, Qi D, Zang L,
Lin J, Liu Y, Zhai S, Fu D, et al: Pan-cancer analysis implicates
novel insights of lactate metabolism into immunotherapy response
prediction and survival prognostication. J Exp Clin Cancer Res.
43:1252024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhou M, He J, Li Y, Jiang L, Ran J, Wang
C, Ju C, Du D, Xu X, Wang X, et al: N6-methyladenosine
modification of REG1α facilitates colorectal cancer progression via
β-catenin/MYC/LDHA axis mediated glycolytic reprogramming. Cell
Death Dis. 14:5572023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang R, Xu F, Yang Z, Cao J, Hu L and She
Y: The mechanism of PFK-1 in the occurrence and development of
bladder cancer by regulating ZEB1 lactylation. BMC Urol. 24:592024.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yan S, Li Q, Li S, Ai Z and Yuan D: The
role of PFKFB3 in maintaining colorectal cancer cell proliferation
and stemness. Mol Biol Rep. 49:9877–9891. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lu S, Zhao R, Han Y, Shao S, Ji Y, Zhang
J, Pan H, Sun J and Feng Y: Identification of PFKFB3 as a key
factor in the development of colorectal cancer and immunotherapy
resistance. Clin Exp Med. 24:2192024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Song M, Sandoval TA, Chae CS, Chopra S,
Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, et
al: IRE1α-XBP1 controls T cell function in ovarian cancer by
regulating mitochondrial activity. Nature. 562:423–428. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tay C, Tanaka A and Sakaguchi S:
Tumor-infiltrating regulatory T cells as targets of cancer
immunotherapy. Cancer Cell. 41:450–465. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jiang B, Zhang J, Zhao G, Liu M, Hu J, Lin
F, Wang J, Zhao W, Ma H, Zhang C, et al: Filamentous GLS1 promotes
ROS-induced apoptosis upon glutamine deprivation via insufficient
asparagine synthesis. Mol Cell. 82:1821–1835.e6. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Villar VH, Allega MF, Deshmukh R,
Ackermann T, Nakasone MA, Vande Voorde J, Drake TM, Oetjen J, Bloom
A, Nixon C, et al: Hepatic glutamine synthetase controls
N5-methylglutamine in homeostasis and cancer. Nat Chem
Biol. 19:292–300. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shang M, Cappellesso F, Amorim R, Serneels
J, Virga F, Eelen G, Carobbio S, Rincon MY, Maechler P, De Bock K,
et al: Macrophage-derived glutamine boosts satellite cells and
muscle regeneration. Nature. 587:626–631. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen Y, Tan L, Gao J, Lin C, Wu F, Li Y
and Zhang J: Targeting glutaminase 1 (GLS1) by small molecules for
anticancer therapeutics. Eur J Med Chem. 252:1153062023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu HY, Zhang HS, Liu MY, Li HM, Wang XY
and Wang M: GLS1 depletion inhibited colorectal cancer
proliferation and migration via redox/Nrf2/autophagy-dependent
pathway. Arch Biochem Biophys. 708:1089642021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Miyamoto R, Takigawa H, Yuge R, Shimizu D,
Ariyoshi M, Otani R, Tsuboi A, Tanaka H, Yamashita K, Hiyama Y, et
al: Analysis of anti-tumor effect and mechanism of GLS1 inhibitor
CB-839 in colorectal cancer using a stroma-abundant tumor model.
Exp Mol Pathol. 137:1048962024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lukey MJ, Cluntun AA, Katt WP, Lin MJ,
Druso JE, Ramachandran S, Erickson JW, Le HH, Wang ZE, Blank B, et
al: Liver-type glutaminase GLS2 is a druggable metabolic node in
luminal-subtype breast cancer. Cell Rep. 29:76–88.e7. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kim GW, Lee DH, Jeon YH, Yoo J, Kim SY,
Lee SW, Cho HY and Kwon SH: Glutamine synthetase as a therapeutic
target for cancer treatment. Int J Mol Sci. 22:17012021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Palmieri EM, Menga A, Martín-Pérez R,
Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH,
Ghesquière B, McVicar DW, et al: Pharmacologic or genetic targeting
of glutamine synthetase skews macrophages toward an M1-like
phenotype and inhibits tumor metastasis. Cell Rep. 20:1654–1666.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu G, Zhu J, Yu M, Cai C, Zhou Y, Yu M,
Fu Z, Gong Y, Yang B, Li Y, et al: Glutamate dehydrogenase is a
novel prognostic marker and predicts metastases in colorectal
cancer patients. J Transl Med. 13:1442015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ayinde O, Wang Z and Griffin M: Tissue
transglutaminase induces epithelial-mesenchymal-transition and the
acquisition of stem cell like characteristics in colorectal cancer
cells. Oncotarget. 8:20025–20041. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ayinde O, Wang Z, Pinton G, Moro L and
Griffin M: Transglutaminase 2 maintains a colorectal cancer stem
phenotype by regulating epithelial-mesenchymal transition.
Oncotarget. 10:4556–4569. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Scalise M, Pochini L, Console L, Losso MA
and Indiveri C: The human SLC1A5 (ASCT2) amino acid transporter:
From function to structure and role in cell biology. Front Cell Dev
Biol. 6:962018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bröer A, Rahimi F and Bröer S: Deletion of
amino acid transporter ASCT2 (SLC1A5) reveals an essential role for
transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain
glutaminolysis in cancer cells. J Biol Chem. 291:13194–13205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Schulte ML, Fu A, Zhao P, Li J, Geng L,
Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, et al:
Pharmacological blockade of ASCT2-dependent glutamine transport
leads to antitumor efficacy in preclinical models. Nat Med.
24:194–202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dejure FR, Butzer J, Lindemann RK and
Mardin BR: Exploiting the metabolic dependencies of the broad amino
acid transporter SLC6A14. Oncotarget. 11:4490–4503. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sniegowski T, Korac K, Bhutia YD and
Ganapathy V: SLC6A14 and SLC38A5 drive the glutaminolysis and
serine-glycine-one-carbon pathways in cancer. Pharmaceuticals
(Basel). 14:2162021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Nałęcz KA: Amino acid transporter SLC6A14
(ATB0,+)-a target in combined anti-cancer therapy. Front
Cell Dev Biol. 8:5944642020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lu Y, Jiang Z, Wang K, Yu S, Hao C, Ma Z,
Fu X, Qin MQ, Xu Z and Fan L: Blockade of the amino acid
transporter SLC6A14 suppresses tumor growth in colorectal cancer.
BMC Cancer. 22:8332022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Peng W and Zeng Z: Epigenetic activation
of PTCD3 Promotes CRC glutamine metabolism and metastasis via
IGF2BP2-mediated SLC38A2 m6A modification. FASEB J. 39:e705582025.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Najumudeen AK, Ceteci F, Fey SK, Hamm G,
Steven RT, Hall H, Nikula CJ, Dexter A, Murta T, Race AM, et al:
The amino acid transporter SLC7A5 is required for efficient growth
of KRAS-mutant colorectal cancer. Nat Genet. 53:16–26. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Shen W, Xu T, Chen D and Tan X: Targeting
SREBP1 chemosensitizes colorectal cancer cells to gemcitabine by
caspase-7 upregulation. Bioengineered. 10:459–468. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wen YA, Xiong X, Zaytseva YY, Napier DL,
Vallee E, Li AT, Wang C, Weiss HL, Evers BM and Gao T:
Downregulation of SREBP inhibits tumor growth and initiation by
altering cellular metabolism in colon cancer. Cell Death Dis.
9:2652018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Qiu Z, Deng W, Hong Y, Zhao L, Li M, Guan
Y, Su Y, Chen C, Shi Q, Yu J and Wang W: Biological behavior and
lipid metabolism of colon cancer cells are regulated by a
combination of sterol regulatory element-binding protein 1 and ATP
citrate lyase. Onco Targets Ther. 14:1531–1542. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wen J, Min X, Shen M, Hua Q, Han Y, Zhao
L, Liu L, Huang G, Liu J and Zhao X: ACLY facilitates colon cancer
cell metastasis by CTNNB1. J Exp Clin Cancer Res. 38:4012019.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Shan J, Li X, Sun R, Yao Y and Sun Y,
Kuang Q, Dai X and Sun Y: Palmitoyltransferase ZDHHC6 promotes
colon tumorigenesis by targeting PPARγ-driven lipid biosynthesis
via regulating lipidome metabolic reprogramming. J Exp Clin Cancer
Res. 43:2272024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Xiong X, Wen YA, Fairchild R, Zaytseva YY,
Weiss HL, Evers BM and Gao T: Upregulation of CPT1A is essential
for the tumor-promoting effect of adipocytes in colon cancer. Cell
Death Dis. 11:7362020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He
MM, Zhao Q, Wang ZX, Li T, Lu YX, et al: CPT1A-mediated fatty acid
oxidation promotes colorectal cancer cell metastasis by inhibiting
anoikis. Oncogene. 37:6025–6040. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Paumen MB, Ishida Y, Han H, Muramatsu M,
Eguchi Y, Tsujimoto Y and Honjo T: Direct interaction of the
mitochondrial membrane protein carnitine palmitoyltransferase I
with Bcl-2. Biochem Biophys Res Commun. 231:523–525. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Yin H, Li W, Mo L, Deng S, Lin W, Ma C,
Luo Z, Luo C and Hong H: Adipose triglyceride lipase promotes the
proliferation of colorectal cancer cells via enhancing the
lipolytic pathway. J Cell Mol Med. 25:3963–3975. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang Y, Xu C, Yang X, Liu X, Guo Z, Lin X,
Li L and Huang Z: Glycerol-3-phosphate acyltransferase 3-mediated
lipid droplets accumulation confers chemoresistance of colorectal
cancer. MedComm (2020). 5:e4862024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lian W, Wang Z, Ma Y, Tong Y, Zhang X, Jin
H, Zhao S, Yu R, Ju S, Zhang X, et al: FABP6 expression correlates
with immune infiltration and immunogenicity in colorectal cancer
cells. J Immunol Res. 2022:31297652022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hua W, Ten Dijke P, Kostidis S, Giera M
and Hornsveld M: TGFβ-induced metabolic reprogramming during
epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci.
77:2103–2123. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Tian W, Zhang W, Zhang Y, Zhu T, Hua Y, Li
H, Zhang Q and Xia M: FABP4 promotes invasion and metastasis of
colon cancer by regulating fatty acid transport. Cancer Cell Int.
20:5122020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kawaguchi K, Senga S, Kubota C, Kawamura
Y, Ke Y and Fujii H: High expression of fatty acid-binding protein
5 promotes cell growth and metastatic potential of colorectal
cancer cells. FEBS Open Bio. 6:190–199. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lu T, Sun L, Wang Z, Zhang Y, He Z and Xu
C: Fatty acid synthase enhances colorectal cancer cell
proliferation and metastasis via regulating AMPK/mTOR pathway. Onco
Targets Ther. 12:3339–3347. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zaytseva YY, Rychahou PG, Le AT, Scott TL,
Flight RM, Kim JT, Harris J, Liu J, Wang C, Morris AJ, et al:
Preclinical evaluation of novel fatty acid synthase inhibitors in
primary colorectal cancer cells and a patient-derived xenograft
model of colorectal cancer. Oncotarget. 9:24787–24800. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Liu X, Lu J, Ni X, He Y, Wang J, Deng Z,
Zhang G, Shi T and Chen W: FASN promotes lipid metabolism and
progression in colorectal cancer via the SP1/PLA2G4B axis. Cell
Death Discov. 11:1222025. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Soriano-Baguet L and Brenner D: Metabolism
and epigenetics at the heart of T cell function. Trends Immun.
44:231–244. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
McGuire PJ: Chemical individuality in T
cells: A Garrodian view of immunometabolism. Immunol Rev.
295:82–100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chapman NM, Boothby MR and Chi H:
Metabolic coordination of T cell quiescence and activation. Nat Rev
Immunol. 20:55–70. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Esensten JH, Helou YA, Chopra G, Weiss A
and Bluestone JA: CD28 costimulation: from mechanism to therapy.
Immunity. 44:973–988. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Frauwirth KA, Riley JL, Harris MH, Parry
RV, Rathmell JC, Plas DR, Elstrom RL, June CH and Thompson CB: The
CD28 signaling pathway regulates glucose metabolism. Immunity.
16:769–777. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Mendoza A, Fang V, Chen C, Serasinghe M,
Verma A, Muller J, Chaluvadi VS, Dustin ML, Hla T, Elemento O, et
al: Lymphatic endothelial S1P promotes mitochondrial function and
survival in naive T cells. Nature. 546:158–161. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
MacIver NJ, Michalek RD and Rathmell JC:
Metabolic regulation of T lymphocytes. Annu Rev Immunol.
31:259–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
O'Neill LA, Kishton RJ and Rathmell J: A
guide to immunometabolism for immunologists. Nat Rev Immunol.
16:553–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Bantug GR, Fischer M, Grählert J, Balmer
ML, Unterstab G, Develioglu L, Steiner R, Zhang L, Costa ASH,
Gubser PM, et al: Mitochondria-endoplasmic reticulum contact sites
function as immunometabolic hubs that orchestrate the rapid recall
response of memory CD8+ T cells. Immunity.
48:542–555.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Blank CU, Haining WN, Held W, Hogan PG,
Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al:
Defining ‘T cell exhaustion’. Nat Rev Immunol. 19:665–674. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bengsch B, Johnson AL, Kurachi M, Odorizzi
PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA,
Delgoffe GM and Wherry EJ: Bioenergetic insufficiencies due to
metabolic alterations regulated by the inhibitory receptor PD-1 are
an early driver of CD8(+) T cell exhaustion. Immunity. 45:358–373.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dibble CC and Manning BD: Signal
integration by mTORC1 coordinates nutrient input with biosynthetic
output. Nat Cell Biol. 15:555–564. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Finlay DK, Rosenzweig E, Sinclair LV,
Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K
and Cantrell DA: PDK1 regulation of mTOR and hypoxia-inducible
factor 1 integrate metabolism and migration of CD8+ T cells. J Exp
Med. 209:2441–2453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Rao RR, Li Q, Odunsi K and Shrikant PA:
The mTOR kinase determines effector versus memory CD8+ T cell fate
by regulating the expression of transcription factors T-bet and
Eomesodermin. Immunity. 32:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Sukumar M, Liu J, Ji Y, Subramanian M,
Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly
ED, et al: Inhibiting glycolytic metabolism enhances CD8+ T cell
memory and antitumor function. J Clin Invest. 123:4479–4488. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhang M, Hu Y, Qian P and Huang H:
Inhibition of CD38 enzymatic activity enhances CAR-T cell
immune-therapeutic efficacy by repressing glycolytic metabolism.
Cell Rep Med. 5:1014002024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhu M, Han Y, Gu T, Wang R, Si X, Kong D,
Zhao P, Wang X, Li J, Zhai X, et al: Class I HDAC inhibitors
enhance antitumor efficacy and persistence of CAR-T cells by
activation of the Wnt pathway. Cell Rep. 43:1140652024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang
G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et
al: Enhancing CD8+ T cell fatty acid catabolism within a
metabolically challenging tumor microenvironment increases the
efficacy of melanoma immunotherapy. Cancer Cell. 32:377–391.e9.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Chamoto K, Chowdhury PS, Kumar A, Sonomura
K, Matsuda F, Fagarasan S and Honjo T: Mitochondrial activation
chemicals synergize with surface receptor PD-1 blockade for T
cell-dependent antitumor activity. Proc Natl Acad Sci USA.
114:E761–E770. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Silverstein RL and Febbraio M: CD36, a
scavenger receptor involved in immunity, metabolism, angiogenesis,
and behavior. Sci Signal. 72:re32009.PubMed/NCBI
|
|
124
|
Xu S, Chaudhary O, Rodríguez-Morales P,
Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I,
et al: Uptake of oxidized lipids by the scavenger receptor CD36
promotes lipid peroxidation and dysfunction in CD8+ T
cells in tumors. Immunity. 54:1561–1577.e7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Hueto JA, Bescós C, Di Croce L and Benitah
SA: Targeting metastasis-initiating cells through the fatty acid
receptor CD36. Nature. 541:41–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Hunt EG, Hurst KE, Riesenberg BP, Kennedy
AS, Gandy EJ, Andrews AM, Del Mar Alicea Pauneto C, Ball LE,
Wallace ED, Gao P, et al: Acetyl-CoA carboxylase obstructs
CD8+ T cell lipid utilization in the tumor
microenvironment. Cell Metab. 36:969–983.e10. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
O'Sullivan D, van der Windt GJ, Huang SC,
Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM,
et al: Memory CD8(+) T cells use cell-intrinsic lipolysis to
support the metabolic programming necessary for development.
Immunity. 41:75–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Hong M, Clubb JD and Chen YY: Engineering
CAR-T cells for next-generation cancer therapy. Cancer Cell.
38:473–488. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Timpanaro A, Piccand C, Dzhumashev D,
Anton-Joseph S, Robbi A, Moser J, Rössler J and Bernasconi M:
CD276-CAR T cells and Dual-CAR T cells targeting CD276/FGFR4
promote rhabdomyosarcoma clearance in orthotopic mouse models. J
Exp Clin Cancer Res. 42:2932023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Sanomachi T, Katsuya Y, Nakatsura T and
Koyama T: Next-generation CAR-T and TCR-T cell therapies for solid
tumors: Innovations, challenges, and global development trends.
Cancers (Basel). 17:19452025. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Choi BK, Lee DY, Lee DG, Kim YH, Kim SH,
Oh HS, Han C and Kwon BS: 4-1BB signaling activates glucose and
fatty acid metabolism to enhance CD8+ T cell proliferation. Cell
Mol Immunol. 14:748–757. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
De Martino M, Rathmell JC, Galluzzi L and
Vanpouille-Box C: Cancer cell metabolism and antitumour immunity.
Nat Rev Immunol. 24:654–669. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Carr EL, Kelman A, Wu GS, Gopaul R,
Senkevitch E, Aghvanyan A, Turay AM and Frauwirth KA: Glutamine
uptake and metabolism are coordinately regulated by ERK/MAPK during
T lymphocyte activation. J Immunol. 185:1037–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ruan T and Keshari KR: Imaging tumor
metabolism. Cold Spring Harb Perspect Med. 15:a0415512025.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Xu L, Huang G, Wang Y, Huang G, Liu J and
Chen R: 2-[18F]FDG PET-based quantification of lymph
node metabolic heterogeneity for predicting lymph node metastasis
in patients with colorectal cancer. Eur J Nucl Med Mol Imaging.
51:1729–1740. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Shi Y, Kotchetkov IS, Dobrin A, Hanina SA,
Rajasekhar VK, Healey JH and Sadelain M: GLUT1 overexpression
enhances CAR T cell metabolic fitness and anti-tumor efficacy. Mol
Ther. 32:2393–2405. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Giuffrida L, Sek K, Henderson MA, Lai J,
Chen AXY, Meyran D, Todd KL, Petley EV, Mardiana S, Mølck C, et al:
CRISPR/Cas9 mediated deletion of the adenosine A2A receptor
enhances CAR T cell efficacy. Nat Commun. 12:32362021. View Article : Google Scholar : PubMed/NCBI
|