Introduction
Cancer is a leading cause of death worldwide, and
metastasis accounts for most cancer-related fatalities due to the
limited efficacy of current therapies against disseminated disease
(1–3). This multistep cascade, from local
invasion to colonization, challenges cancer cells to overcome
profound metabolic, mechanical and signaling stresses. While
numerous oncogenic pathways contribute to dissemination, including
Phosphoinositide 3-kinase/Protein kinase B (PI3K/AKT),
mitogen-activated protein kinase (MAPK) signaling, and immune
evasion mechanisms, the present review focuses on two core,
interdependent systems that are fundamental to the metastatic
process which are the cytoskeletal machinery and mitochondrial
bioenergetics (4,5).
A defining feature of metastatic cells is their
capacity for dynamic cytoskeletal remodeling. This process is
driven by actin polymerization, which forms lamellipodia and
filopodia for protrusion. Simultaneously, actomyosin contractility
generates the force required for invasion. Microtubules and
intermediate filaments further coordinate cell polarity and vesicle
transport (6). These processes are
precisely governed by Rho family GTPases (RhoA, Rac1 and Cdc42),
which act as molecular switches that spatially and temporally
regulate cytoskeletal dynamics through effectors such as
Rho-associated coiled-coil containing protein kinase (ROCK) and
WAVE/Arp2/3 (6–8). Consequently, elevated Rho GTPase
activity is a recurrent feature across cancers and is strongly
associated with aggressive invasion, epithelial-to-mesenchymal
transition (EMT), and poor patient outcomes (9,10).
Simultaneously, to power these demanding mechanical
processes, metastatic cells undergo profound mitochondrial
reprogramming. Beyond their standard role in ATP synthesis,
mitochondria are dynamically trafficked to the cell's leading edge.
This positioning supplies local ATP for actin turnover and focal
adhesion dynamics. Additionally, mitochondria facilitate the
production of reactive oxygen species (ROS), which function as
essential second messengers in pro-migratory signaling pathways
(11–13). The upregulation of oxidative
phosphorylation (OXPHOS) and mitochondrial biogenesis, fine-tuned
by regulators such as peroxisome proliferator-activated receptor
gamma coactivator 1α (PGC-1α), supports invasion and distant
colonization (12,14). Critically, these adaptations are
also a feature of therapy-resistant, cancer stem-like cells (CSCs),
a population responsible for seeding metastases and driving relapse
(15).
Rather than operating independently, these two
aforementioned systems engage in a tightly coordinated
bidirectional crosstalk that forms a feedforward loop of
metabolic-mechanical coupling. In this loop, mitochondrial
metabolism directly supports cytoskeletal remodeling by providing
ATP for actin dynamics and generating ROS that modulate Rho GTPase
signaling through redox-sensitive pathways, including Sarcoma (Src)
family kinases and Rho regulatory proteins (11,16–19).
In parallel, activation of Rho GTPases drives cytoskeletal
reorganization and cell polarity, which in turn regulates
mitochondrial trafficking, positioning, and dynamics through
effectors such as Dynamin-related protein 1 (DRP1) and
mitochondrial Rho GTPase (MIRO) proteins (6–8,16,20).
This reciprocal interaction spatially couples energy production to
mechanical demand while reinforcing pro-migratory signaling.
Consequently, this self-sustaining loop integrates metabolic
adaptation with force generation to promote invasion, survival
under stress, and metastatic dissemination (18–21).
This integrated framework provides a mechanistic
explanation for the limited efficacy of single-target therapies,
since inhibition of either metabolic or cytoskeletal pathways alone
can be compensated by adaptive rewiring of the other. Furthermore,
it motivates a therapeutic strategy of dual targeting (18,22)
through two translationally tractable modalities: Small interfering
RNA (siRNA) based silencing of Rho GTPases to overcome the
historical intractability of inhibiting these protein-protein
interaction-dependent switches, and repurposing
mitochondria-targeting antibiotics (doxycycline and tigecycline) to
suppress OXPHOS and pro-tumorigenic ROS signaling (7,23,24).
Moreover, numerous advances in biological delivery platforms, such
as lipid nanoparticles, will be discussed addressing limitations
including off-target effects and poor bioavailability (25,26).
By synthesizing mechanistic insights into mitochondrial-Rho GTPase
crosstalk and integrating them with emerging therapeutic
strategies, the present review aims to establish a dual-target
framework for disrupting this feedforward loop and suppressing
metastatic progression (Fig.
1).
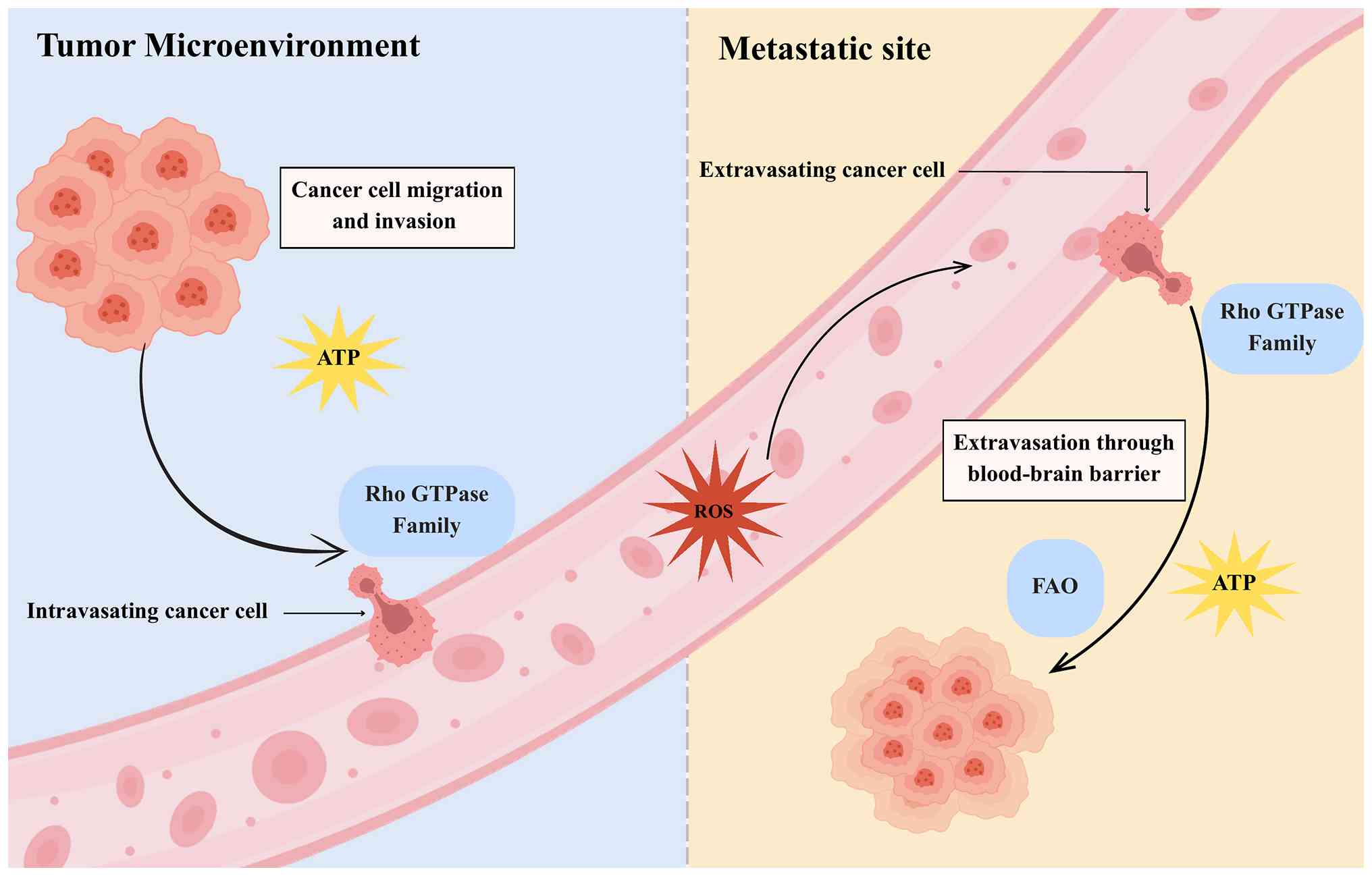 | Figure 1.Metastatic cascade with energetic and
mechanical demands. This schematic illustrates the sequential steps
of metastasis from the primary tumor microenvironment to the
metastatic site, highlighting specific energy and signaling
requirements. At the invasion and intravasation steps, cancer cells
rely on local ATP production and Rho GTPase signaling to drive
actin remodeling and cytoskeletal contractility required for matrix
degradation and endothelial penetration. During circulation,
mitochondrial-derived ROS act as adaptive signals that promote cell
survival in the bloodstream. At extravasation, Rho GTPases again
support cytoskeletal reorganization, while mitochondrial metabolism
provides bioenergetic support for barrier crossing. Finally, at
colonization in the distant site, cells depend on ATP production
and FAO to adapt to nutrient limitations and establish
proliferative growth. Overall, this process reflects a
bidirectional metabolic-mechanical feedback loop, in which
mitochondrial metabolism and Rho GTPase-driven cytoskeletal
dynamics reciprocally reinforce metastatic progression. ROS,
reactive oxygen species; FAO, fatty acid oxidation. |
Mitochondrial metabolism and cancer
metastasis
Energy demands for metastatic
cells
The metastatic cascade imposes exceptional
bioenergetic demands on disseminating tumor cells, requiring ATP
for motility, invasion and adaptation to hostile microenvironments.
While numerous primary tumors exhibit a glycolytic phenotype
(Warburg effect), metastatic cells often show a reliance on
mitochondrial OXPHOS to meet the sustained energy requirements for
migration and colonization (14,27,28).
It is important to note that metastatic cells are metabolically
plastic, often retaining the capacity to utilize glycolysis in
hypoxic niches while upregulating OXPHOS to fuel invasion (29).
The invasive process necessitates dynamic
cytoskeletal remodeling to form protrusions such as lamellipodia
and invadopodia which degrade the extracellular matrix (ECM). These
energy-intensive processes require a high, localized supply of ATP
at the leading edge. Mitochondria are actively redistributed to
these subcellular locations through DRP1-mediated mitochondrial
fission and trafficking along microtubules, where they provide
energy for actin polymerization and focal adhesion turnover
(11,30). Beyond ATP, mitochondria generate ROS
at controlled levels, which function as essential signaling
molecules. Mitochondrial ROS can oxidize cysteine residues in key
regulatory proteins, thereby directly activating pro-migratory
signaling cascades, including those involving Rho GTPases, focal
adhesion kinase (FAK), and Src kinase (31–34).
The secretion of proteolytic enzymes, such as matrix
metalloproteinases (MMPs), for ECM remodeling is another process
heavily dependent on OXPHOS-derived ATP, underscoring the critical
role of mitochondria in invasion (12,35).
Upon intravasation, tumor cells endure extreme stresses, including
shear stress, anoikis and immune surveillance. Mitochondria
contribute to survival during this phase. Moderately elevated ROS
levels can stabilize hypoxia-inducible factor-1α (HIF-1α) through
the inhibition of prolyl hydroxylases (PHDs), promoting EMT and
anoikis resistance (33,36,37).
Concurrently, mitochondrial metabolism supports antioxidant defense
systems, such as generating nicotinamide adenine dinucleotide
phosphate (NADPH), allowing circulating tumor cells to mitigate
oxidative damage (14,38).
The last step of colonization requires adaptation to
the stringent and often nutrient-poor, microenvironment of distant
organs. Through the tricarboxylic acid (TCA) cycle and fatty acid
oxidation (FAO), mitochondria allow metastatic cells to utilize
available nutrients, such as lipids and glutamine. For instance, in
breast cancer models, enhanced FAO is critical for metastatic
outgrowth in the lipid-rich environment of the liver and lung
(39,40). Similarly, OXPHOS dependency has been
demonstrated in metastatic melanoma and pancreatic cancer (41,42).
In addition to bioenergetic and biosynthetic
functions, other mitochondrial processes support metastasis.
Mitophagy maintains a healthy pool of mitochondria to sustain
energy production during stress, and mitochondrial-ER contacts
facilitate lipid transfer and calcium signaling, both of which can
influence cell migration (43,44).
In summary, mitochondria are central, dynamic
regulators of metastatic competence, far exceeding their role as
mere powerhouses. They integrate energy production (OXPHOS, FAO),
redox signaling (ROS), and metabolic plasticity to enable every
step of dissemination. This foundational role establishes them as a
compelling therapeutic target and provides the rationale for
exploring their inhibition in combination with strategies targeting
the cytoskeletal machinery they fuel, as discussed in the following
sections.
Mitochondrial pathways in cancer
cells
Building upon the exceptional bioenergetic demands
of metastasis, this section delves into the specific mitochondrial
pathways that are co-opted to fuel dissemination. Mitochondria
occupy a central position in bioenergetic and biosynthetic
pathways, and their reprogramming is a hallmark of metastatic
efficiency. The core metabolic axes, OXPHOS, TCA cycle and FAO are
systematically rewired to maximize adaptability to the fluctuating
nutrient and oxygen conditions met during dissemination (45).
Metastatic cells exhibit pronounced metabolic
plasticity, often retaining glycolytic capacity and switching
between energy sources based on microenvironmental cues (29). While the classical Warburg effect
emphasizes glycolysis, accumulating evidence indicates that
metastatic cells frequently upregulate OXPHOS to meet the high ATP
demands of invasion and colonization. Enhanced OXPHOS supports the
intense energy requirements of actin cytoskeletal remodeling and
membrane trafficking (14,28). A key byproduct of electron transport
is the generation of ROS. While excessive ROS induces cell death,
controlled levels serve as critical signaling molecules.
Mitochondrial ROS can oxidize key cysteine residues in
phosphatases, such as PTEN, and activate kinases within the
Src/PI3K pathway, thereby promoting the activation of Rho GTPases
and FAK to drive motility (16,32,33).
The TCA cycle functions as a crucial biosynthetic
hub. Metastatic cells divert intermediates to support anabolic
processes. For example, citrate is exported for lipid synthesis and
histone acetylation, while α-ketoglutarate (α-KG) serves as a
cofactor for α-KG-dependent dioxygenases, including histone and DNA
demethylases. This reprogramming reinforces pro-metastatic
phenotypes, including EMT and stemness (41,46).
Furthermore, the oncometabolites succinate and fumarate, when
accumulated, inhibit PHDs, leading to HIF-1α stabilization and the
expression of pro-angiogenic and pro-invasive genes even under
normoxia (36,47).
FAO represents another critical adaptive pathway. In
nutrient-deprived niches, metastatic cells oxidize fatty acids to
generate NADH and FADH2 to fuel OXPHOS. This pathway is
particularly important for colonization of lipid-rich environments;
for example, FAO is essential for breast cancer metastasis to the
bone marrow and for ovarian cancer metastasis to the adipose-rich
omentum (14,39,48).
Additionally, FAO generates NADPH, which is essential for
maintaining redox homeostasis and mitigating oxidative stress
during circulation (38).
In addition to metabolic reprogramming, the physical
dynamics of mitochondria (fission and fusion) are precisely
regulated. DRP1-mediated fission generates fragmented mitochondria
that can be trafficked to the cell's leading edge to supply
localized ATP for protrusions and invasion. Conversely, Mitofusin 1
and Mitofusin 2 (MFN1/2)-mediated fusion maintains an
interconnected network that supports efficient OXPHOS and biomass
synthesis. The balance between these states precisely regulates
energy allocation to support migratory demands (16,30).
The coordination of these functions is managed by
key upstream regulators. The transcriptional coactivator PGC-1α,
which is upregulated in specific aggressive subtypes
(triple-negative breast cancer, BRAF-resistant melanoma), drives
mitochondrial biogenesis and enhances oxidative capacity (14,49).
While HIF-1α typically promotes glycolysis, it can also support
metastasis by maintaining pro-migratory ROS signaling (36). The nutrient sensors adenosine
monophosphate activated protein kinase (AMPK) and mechanistic
target of rapamycin (mTOR) integrate energy availability with
anabolic processes, critically influencing metastatic fitness
(45,50). Other regulators such as MYC and
Nuclear Factor Erythroid 2-Related Factor 2 (NRF2) further
contribute to this coordinated mitochondrial reprogramming,
promoting biogenesis and antioxidant responses, respectively
(51,52).
Collectively, metastatic cells exploit OXPHOS, the
TCA cycle, and FAO in a coordinated manner, supported by dynamic
organelle remodeling and primary transcriptional regulators. This
integrated rewiring ensures that tumor cells can generate energy,
biomass, and signaling molecules to complete the metastatic
cascade. The centrality of these mitochondrial functions
establishes them as a compelling therapeutic vulnerability, a
premise that will be explored in the following sections on
targeting strategies (Fig. 2).
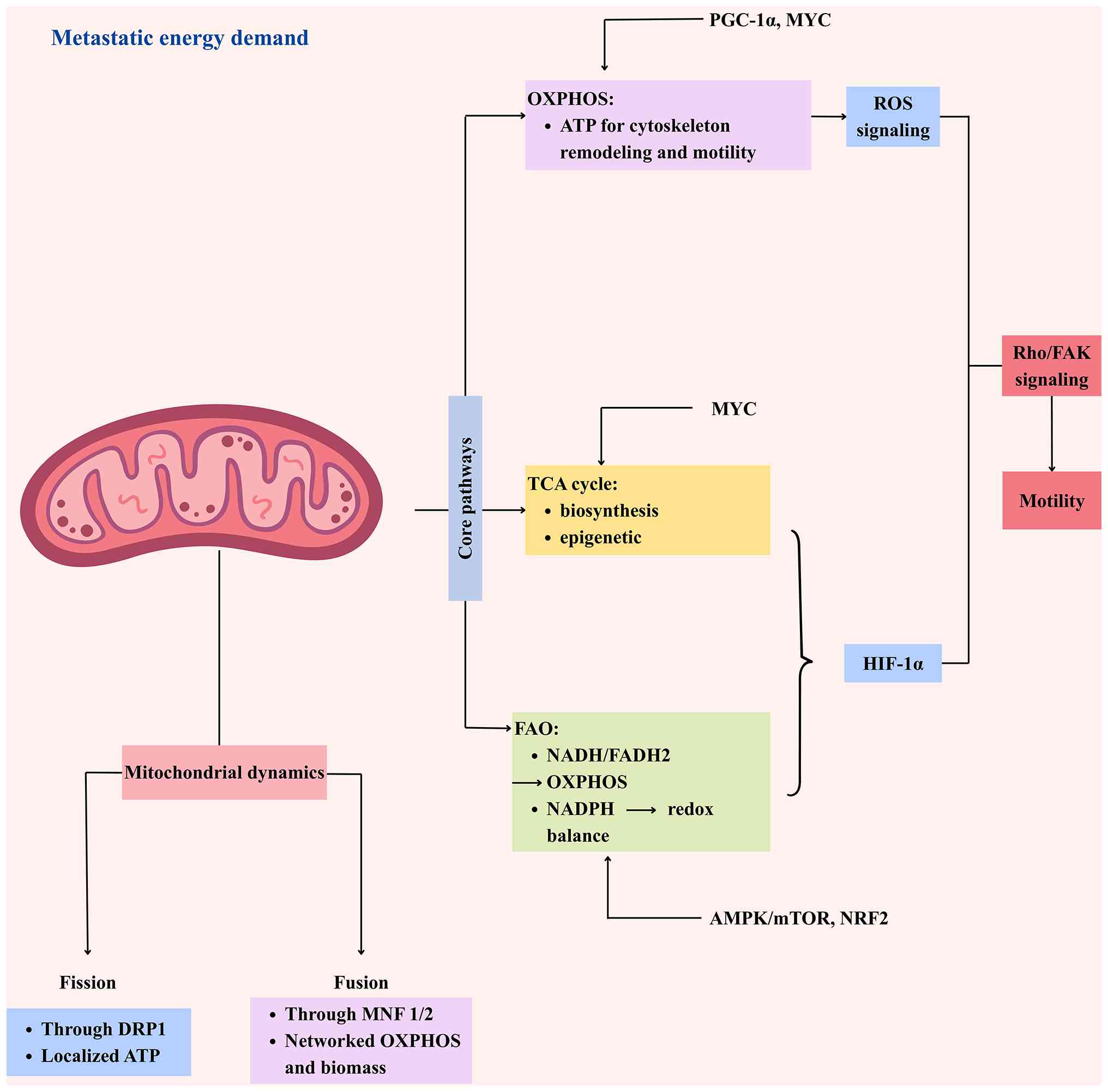 | Figure 2.Mitochondrial coordination of
metastatic energy metabolism and motility. Metastatic cancer cells
rewire core mitochondrial pathways (OXPHOS, TCA, FAO) to produce
ATP, biosynthetic precursors, and redox signals that support
motility. ROS and HIF-1α activate Rho/FAK signaling, driving
cytoskeletal remodeling. Mitochondrial dynamics (DRP1-mediated
fission and MFN1/2-mediated fusion) distribute ATP and maintain
networked metabolism to meet energetic and biosynthetic demands.
Key upstream regulators (PGC-1α, MYC, AMPK/mTOR, NRF2) orchestrate
metabolic rewiring, dynamics, and redox balance. The integrated
output enhances energy availability, biomass synthesis, ROS
signaling, and promotes cancer cell migration, invasion, and
colonization. OXPHOS, oxidative phosphorylation; TCA, tricarboxylic
acid; FAO, fatty acid oxidation; ROS, reactive oxygen species. |
Mitochondria as therapeutic
targets
Mitochondrial metabolism plays a leading role in
powering metastasis. Consequently, numerous strategies have been
developed to target mitochondrial function, ranging from direct
inhibition of respiration to interference with biogenesis and
dynamics.
Direct pharmacological inhibition of OXPHOS impairs
ATP production and attenuates essential ROS signaling, disrupting
the cytoskeletal dynamics required for invasion. Small-molecule
inhibitors targeting electron transport chain complexes, such as
complex I (IACS-010759) and complex III, have proven efficacy in
preclinical models by reducing metastatic burden (24). For instance, the complex I inhibitor
IACS-010759 has demonstrated potent antitumor activity in acute
myeloid leukemia (AML) and solid tumor xenograft models, achieving
significant tumor growth inhibition at doses of 5–10 mg/kg
(53). The complex I inhibitor
IACS-010759 has progressed to early-phase clinical evaluation in
advanced solid tumors and hematological malignancies. Notably, the
Phase I clinical trial NCT03291938 (2017–2020; n=29) assessed the
safety and feasibility of this approach (54). However, peer-reviewed results from
this trial have not yet been formally published, and currently
available data are limited to clinical trial registry reports.
Despite initial promise, dose-limiting toxicities, including
cardiac and neurological adverse effects, as well as metabolic
adaptation through glycolytic compensation, have limited its
clinical efficacy (53,55). These limitations highlight the
current gap between promising preclinical findings and clinically
validated mitochondrial-targeted therapies.
Targeting the upstream drivers of mitochondrial mass
represents another potential strategy. Metastatic and
therapy-resistant subpopulations often depend on PGC-1α-driven
mitochondrial biogenesis. While directly inhibiting PGC-1α remains
challenging, targeting its upstream regulators or synthetic lethal
partners shows preclinical promise in suppressing oxidative
capacity and metastasis (14,56).
A particularly tractable approach is the repurposing
of antibiotics that inhibit mitochondrial translation, exploiting
the bacterial ancestry of mitochondrial ribosomes. Tigecycline and
doxycycline collapse OXPHOS capacity and have shown selective
efficacy against metastatic and cancer stem cells in models of
breast, ovarian and blood cancers (23,46).
For example, tigecycline has been shown to significantly reduce
leukemia burden and target leukemia stem cells in AML xenograft
models (57). Similarly,
doxycycline reduced metastatic tumor burden by ~70% in an
intracardiac MDA-MB-231 breast cancer model in nude mice (58). This selectivity arises from the
heightened mitochondrial dependency of these aggressive cells.
Unfortunately, this therapeutic window remains context-dependent,
as normal tissues with high mitochondrial activity may also be
affected, limiting clinical applicability. Moreover, the clinical
translation of long-term, high-dose antibiotic regimens is
complicated by systemic toxicity, microbiome disruption, and the
emergence of drug-resistant bacterial strains (57,59).
Beyond metabolism, targeting the machinery that
controls mitochondrial dynamics is gaining interest. Inhibiting
DRP1-mediated fission (with mdivi-1) prevents the mitochondrial
fragmentation and redistribution needed for invasion, thereby
impairing cell motility. Consistently, inhibition of DRP1 has been
reported to reduce migration and invasion phenotypes in cancer cell
models, supporting its role in metastatic progression (60). It is important to note that
compounds such as mdivi-1 may have off-target effects, underscoring
the need for more specific inhibitors (61,62).
Conversely, disrupting fusion through MFN inhibition compromises
metabolic efficiency and sensitizes cells to stress.
ROS-modulating therapies present a complex, but
context-dependent, therapeutic opportunity due to the dual role of
ROS in both pro-migratory signaling and oxidative cell death. The
strategy involves either elevating ROS to toxic levels to induce
apoptosis or scavenging mitochondrial ROS to blunt pro-invasive
signaling. However, the clinical application of ROS modulators has
been challenging due to their context-dependent effects and the
risk of harming normal tissues (36,63).
A major limitation across these strategies is the
inherent metabolic plasticity of tumor cells. Inhibition of OXPHOS
or mitochondrial function often triggers adaptive responses,
including a shift to glycolysis, increased glutaminolysis, or
upregulation of alternative nutrient salvage pathways, leading to
therapeutic resistance (29,55).
In addition, toxicity to normal tissues with high metabolic demands
(cardiac muscle, neurons) remains a primary barrier.
In summary, targeting mitochondrial metabolism
offers a rational, multi-pronged approach to disrupt the energetic
and signaling foundations of metastasis. The combinatory use of
these strategies, or their integration with agents targeting
compensatory pathways (glycolysis inhibitors), may help overcome
the limitations of monotherapy. The translational feasibility of
these approaches, particularly with advanced delivery systems to
improve selectivity, will be explored in the context of targeting
the cytoskeletal machinery in Section 3 (Table I).
 | Table I.Experimental evidence supporting
mitochondrial-targeting agents in metastatic cancer models. |
Table I.
Experimental evidence supporting
mitochondrial-targeting agents in metastatic cancer models.
| First author/s,
year | Drug | Mechanism | Model system | Dose | Duration | Quantitative
outcome | Statistical
analysis | (Refs.) |
|---|
| Duivenvoorden et
al, 2002 | Doxycycline | Mitochondrial
translation inhibitor | Female nude mice
(BALB/c nu/nu, ~5 weeks), intracardiac injection of MDA-MB-231
breast cancer cells; n=9-14/group | 10 mg slow-release
pellet (equivalent systemic exposure) | 21 days | ~70% reduction in
bone metastatic tumor burden (histomorphometric analysis) | Student's t-test,
P<0.01 | (58) |
| Škrtić et
al, 2011 | Tigecycline | Mitochondrial
translation inhibitor | NOD/SCID mice
xenografted with human AML (OCI-AML2 cells); n ≈ 8–10/group | 50 mg/kg
(intraperitoneal injection) | ~14 days | ~70–77% reduction
in leukemia burden and impaired leukemia stem cell function | Student's t-test,
P<0.05 | (57) |
| Molina et
al, 2018; Yap et al, 2023 | IACS-010759 | Complex I (OXPHOS)
inhibitor | NSG mice
xenografted with human AML and brain tumor cells; n ≈
8–10/group | 5–10 mg/kg (oral
administration) | 14–21 days | 50–70% reduction in
tumor growth and evidence of OXPHOS-dependent tumors | ANOVA,
P<0.01 | (53,54) |
| Kashatus et
al, 2015 | Midvi-1 | DRP1-mediated
mitochondrial fission inhibitor | Breast cancer cell
lines (MDA-MB-231) migration/invasion assays (in vitro); n≥3
independent experiments | 25–50 µM | 24–72 h | ~40–60% reduction
in migration and invasion capacity | Student's t-test,
P<0.05 | (60) |
Advantages and limitations of
targeting mitochondria
While the preceding section outlined various
strategies to target mitochondrial function, a critical assessment
of their advantages and limitations is essential for evaluating
their therapeutic potential. The approach offers distinct benefits
rooted in the biology of metastasis but also faces significant
challenges due to the indispensable role of mitochondria in normal
physiology.
A key advantage is the disproportionate dependence
of metastatic and stem-like tumor cells on OXPHOS compared with
bulk tumor populations. This dependency can create a therapeutic
window, allowing mitochondrial inhibition to selectively impair
metastatic competence while sparing more glycolytic cells (14,64).
Furthermore, mitochondrial inhibitors exert a dual impact; they
deprive cells of ATP while simultaneously suppressing ROS-driven
signaling cascades that activate critical drivers of invasion, such
as Rho GTPases and focal adhesion kinases (63). Another promising advantage is the
opportunity to repurpose antibiotics including doxycycline and
tigecycline, which have known pharmacokinetic and safety profiles
from decades of clinical use (23).
However, long-term high-dose regimens required for antitumor
efficacy can lead to off-target effects, including microbiome
disruption and cumulative toxicity in normal tissues. Mitochondrial
inhibition also shows efficacy against numerous therapy-resistant
CSCs, potentially reducing long-term recurrence. However, it is
crucial to acknowledge that not all CSCs rely on OXPHOS; some
utilize glycolysis, highlighting the need for patient
stratification based on metabolic phenotypes (36,65).
Despite these strengths, some limitations complicate
clinical translation. These limitations reflect the same adaptive
features that enable metastatic success, underscoring the challenge
of targeting mitochondrial metabolism without affecting normal
cellular function. The indispensability of mitochondria for
high-demand normal tissues (heart, brain, nerves) poses a risk of
dose-limiting toxicities, such as cardiomyopathy and neuropathy
(66). Moreover, tumor cells
exhibit pronounced metabolic plasticity as a key resistance
mechanism. When OXPHOS is suppressed, cells can rapidly switch to
glycolysis (often driven by HIF-1α stabilization), glutaminolysis
(via MYC upregulation), or fatty acid metabolism, thereby
maintaining energy production and viability (12,29).
Both inter- and intra-tumoral heterogeneity further complicate
treatment. For example, hypoxic cells within a tumor or in certain
metastatic niches, such as the bone marrow may be inherently
glycolytic and thus resistant to OXPHOS inhibition alone (67). Finally, sustained inhibition can
select for clones with adaptive resistance mechanisms, including
altered mitochondrial dynamics or amplified compensatory glycolytic
flux (10).
Ultimately, mitochondria represent a metabolic
vulnerability that can be exploited for therapy, but they also
confer resilience through their plasticity and role in stress
adaptation. Overcoming the challenges of toxicity, heterogeneity,
and resistance will require innovative strategies. These include
developing targeted delivery systems (nanoparticles) to improve
selectivity, combining mitochondrial inhibitors with agents that
block compensatory pathways (glycolysis inhibitors), and basing
therapeutic decisions on metabolic biomarkers. The potential of
such rational combination strategies, particularly in the context
of disrupting metabolic-mechanical crosstalk, will be explored in
the closing section of this review.
Rho GTPase signaling and siRNA in cancer
metastasis
Why cancer cells depend on Rho GTPase
signaling
Having established mitochondria as the metabolic
engine of metastasis, the cytoskeletal machinery that this engine
powers is then discussed. The metastatic cascade demands that tumor
cells dynamically alter their shape and adhesion to detach, invade
the ECM, intravasate, survive circulation, and colonize distant
sites. Rho family GTPases serve as master molecular switches that
regulate these essential processes by cycling between active
GTP-bound and inactive GDP-bound states (7,9,68).
These GTPases govern distinct, well-characterized
aspects of cytoskeletal dynamics. RhoA signals through its effector
ROCK to promote actomyosin contractility and stress fiber
formation, facilitating the rounded, amoeboid migration mode that
is effective in dense tissues. Rac1 activates the WAVE/Arp2/3
complex to drive the formation of broad, sheet-like lamellipodia
for protrusive mesenchymal migration. Cdc42, via effectors like
N-WASP, stimulates the formation of finger-like filopodia for
environmental sensing and establishes front-rear cell polarity,
guiding directional movement (6,69,70).
Aberrant activation of these pathways, whether through
overexpression of the GTPases themselves, amplification of upstream
activators such as guanine nucleotide exchange factor (GEFs), or
loss of inhibitors such as GTPase-activating protein (GAPs),
confers a potent invasive advantage, identifying them as additional
critical vulnerabilities in metastatic cancers (71).
The pathway hyperactivity is often driven by
sustained upstream signaling from receptor tyrosine kinases,
integrins and G-protein-coupled receptors within the tumor
microenvironment (TME) (68). The
resulting persistent Rho GTPase enhances metastatic fitness in two
key ways. First, it enables dynamic plasticity, allowing cells to
switch between mesenchymal and amoeboid migration in response to
ECM density and confinement. Second, it promotes immune evasion;
for instance, the rapid, rounded amoeboid movement minimizes cell
surface exposure and adhesive interactions, potentially reducing
recognition by immune cells (9,72,73).
The clinical relevance is underscored by tumor-type-specific
dependencies; specifically, RhoA is frequently amplified in
melanoma and glioblastoma, while Cdc42 activity is critical for
invasion and metastasis in breast and pancreatic cancers (70,74).
Basically, Rho GTPases function as central signaling
hubs, integrating extrinsic cues from the TME with intrinsic
oncogenic drives to orchestrate dissemination. However, their
dependence on protein-protein interactions and the absence of deep
catalytic pockets have rendered them notoriously difficult to
target with conventional small molecules, leading to their
classification as ‘undruggable’ (71). This fundamental challenge motivates
the exploration of alternative therapeutic strategies, most notably
RNA interference (RNAi) to achieve precise silencing, as discussed
in the following subsection.
Rho GTPases as molecular switches and
their regulators
Building on their established role as master
regulators of metastasis, the precise molecular control of Rho
GTPase activity is fundamental to their function. Rather than
acting as simple binary switches, their activity is tightly
coordinated by a regulatory network of guanine nucleotide exchange
factors (GEFs: Vav, Tiam1 and Trio), GTPase-activating proteins
(GAPs: p190RhoGAP) and guanine nucleotide dissociation inhibitors
(GDIs), which collectively ensure the spatial and temporal
activation required for efficient cell migration (7,9,68).
In cancer, direct mutations in Rho GTPases are rare.
Instead, dysregulation most commonly occurs through overexpression
or hyperactivation of oncogenic GEFs, or the loss of
tumor-suppressive GAPs. This imbalance results in sustained and
spatially deregulated Rho GTPase signaling, a key driver of
metastatic progression across multiple cancer types (69,71).
This sustained signaling drives invasion through
distinct downstream effector pathways. Active RhoA signals through
ROCK, which phosphorylates LIM kinase and myosin light chain (MLC)
to drive stress fiber formation and actomyosin contractility,
facilitating amoeboid movement. Rac1 stimulates lamellipodia
formation via the WAVE/Arp2/3 complex and can also activate NADPH
oxidase (NOX) complexes to generate localized ROS that amplify
pro-migratory signals (8,33). Cdc42 coordinates filopodia formation
and establishes cell polarity through effectors such as N-WASP.
Furthermore, Cdc42 regulates vesicle trafficking, guiding the
exocytosis of MMPs and the endocytosis of integrins, thereby
directly facilitating ECM degradation and directional migration
(6,70).
These pathways are not isolated but are highly
interdependent, allowing for adaptive migration. A key regulatory
node is the antagonism between RhoA and Rac1, which controls the
switch between contractile amoeboid and protrusive mesenchymal
migration in response to ECM physical constraints. Furthermore,
Cdc42 often cooperates with Rac1 at the leading edge to reinforce
protrusive activity while simultaneously inhibiting RhoA-mediated
contractility at the cell rear. This dynamic, spatially-controlled
balance enables tumor cells to efficiently navigate the complex and
heterogeneous microenvironments encountered during dissemination
(75).
Importantly, the tight spatial and temporal
regulation of Rho GTPase signaling imposes significant energetic
demands, particularly for actin remodeling, vesicle trafficking and
contractility. This functional dependence on energy supply suggests
a close integration with mitochondrial metabolism, providing a
mechanistic basis for the metabolic-mechanical crosstalk explored
in Section 4.
The precise and complex nature of this regulatory
network is what makes Rho GTPases so difficult to target with
conventional small-molecule inhibitors. The lack of deep catalytic
pockets and their reliance on protein-protein interactions and they
have made Rho GTPase challenging to pursue therapeutically
(71). This challenge underscores
the need for alternative intervention strategies, such as RNAi,
which can selectively silence key nodes within this signaling
network, as discussed in the following subsection (Fig. 3).
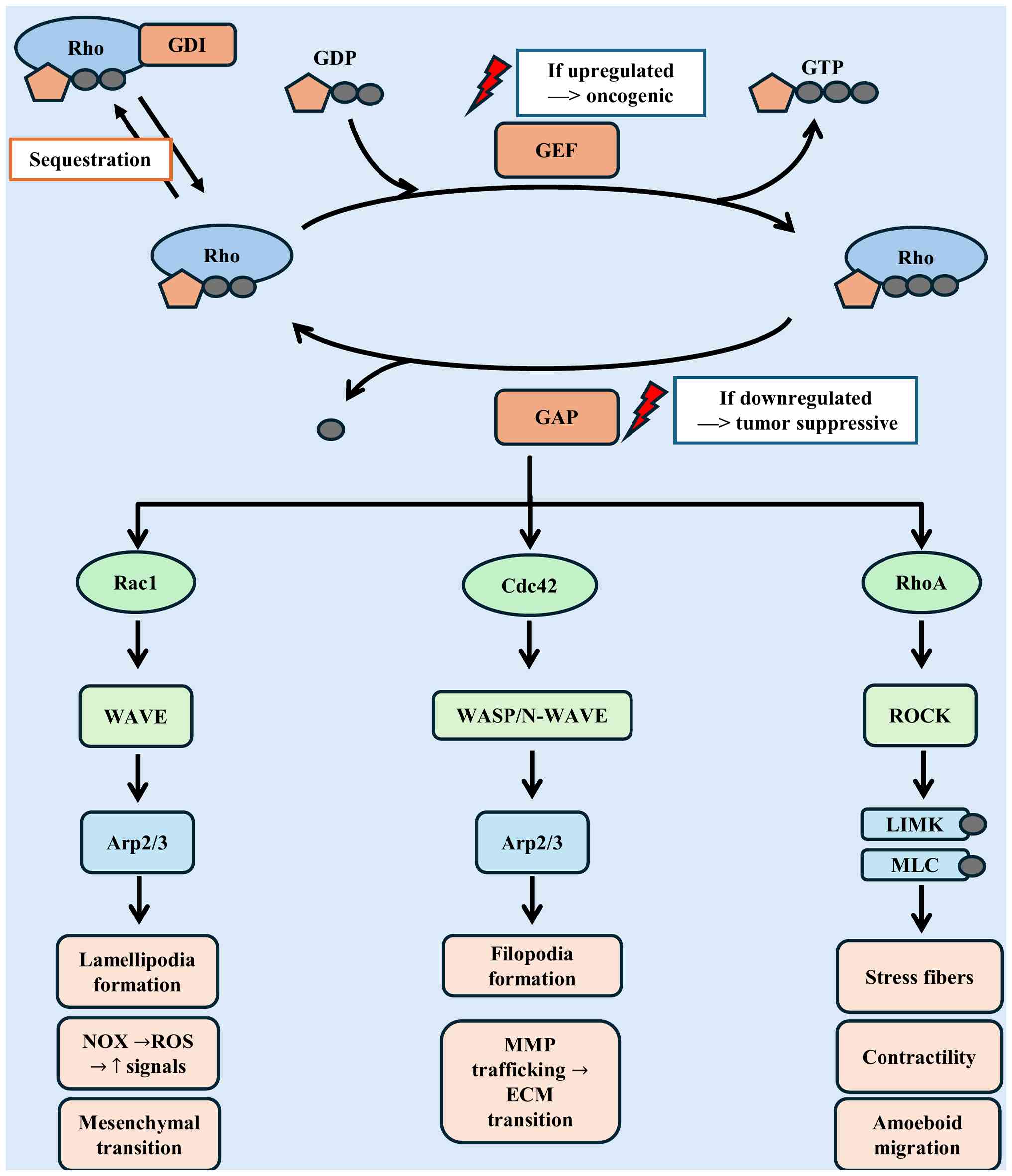 | Figure 3.Regulation and downstream signaling
of Rho GTPases in cancer metastasis. Extracellular stimuli,
including growth factors, integrin-ECM interaction, and chemokines,
activate Rho GTPases (RhoA, Rac1, Cdc42). These proteins function
as molecular switches, cycling between inactive GDP-bound and
active GTP-bound states. Their activity is tightly regulated by
GEFs, GAPs and GDIs, where GEF hyperactivation or GAP loss leads to
sustained Rho GTPase signaling. Activated RhoA drives ROCK-mediated
stress fiber assembly and actomyosin contractility, facilitating
amoeboid motility; Rac1 promotes lamellipodia formation via
WAVE/Arp2/3 and generates localized ROS through NOX complexes,
amplifying pro-migratory signals and mesenchymal motility; Cdc42
orchestrates filopodia formation, establishes polarity, and
regulates MMP trafficking for ECM remodeling. Collectively, this
coordinated signaling network drives cytoskeletal remodeling,
directional migration, ECM degradation, and ultimately facilitates
cancer cell invasion and metastatic dissemination. ECM,
extracellular matrix; GEF, guanine nucleotide exchange factor; GAP,
GTPase-activating protein; GDI, guanine nucleotide dissociation
inhibitor; ROS, reactive oxygen species; MMP, matrix
metalloproteinase; NOX, NADPH oxidase. |
Post-transcriptional regulation by
miRNA
Beyond the immediate protein-level regulation by
GEFs and GAPs, Rho GTPase signaling is finely tuned at the
post-transcriptional level by miRNA. These small non-coding RNAs
typically bind to the 3′ untranslated regions of target mRNAs,
leading to transcript degradation or translational repression. By
directly targeting the mRNAs encoding Rho GTPases themselves, as
well as their activators (GEFs) and inhibitors (GAPs and GDIs),
microRNAs (miRNAs or miRs) constitute a critical and dynamic
regulatory layer that shapes the cytoskeletal dynamics essential
for metastasis (70,76). This endogenous regulatory mechanism
provides a conceptual basis for therapeutic RNA-based approaches,
including siRNA-mediated silencing strategies.
In cancer, global dysregulation of miRNA expression,
through genomic deletion, amplification, or altered processing,
frequently disrupts this precise balance. The loss of
tumor-suppressive miRNAs or the overexpression of oncogenic miRNAs
(oncomiRs) leads to the hyperactivation of pro-invasive Rho GTPase
pathways. Numerous miRNAs have been experimentally validated as key
regulators of this axis. For instance, the miR-200 family is a
well-established suppressor of EMT and invasion. It directly
targets the ZEB1 and ZEB2 transcription factors, which themselves
repress epithelial genes. The restoration of an epithelial
phenotype by miR-200 is accompanied by indirect downregulation of
RhoA activity, promoting a less motile state. Conversely, the loss
of miR-200, common in advanced carcinomas, unleashes ZEB1/2,
driving EMT and RhoA-mediated invasion (76–78).
Other miRNAs act more directly on the Rho GTPase
machinery. For example, miR-34a functions as a tumor suppressor and
regulates key oncogenic pathways, including MET and SIRT1, thereby
modulating Rho GTPase signaling and inhibiting cancer cell
migration and invasion (79). The
metastasis suppressor miR-31 directly targets the mRNAs of several
GEFs (RDX and RhoA), thereby reducing Rac1 and RhoA activation and
impairing invasion in breast and colorectal cancer models (80). On the other hand, oncogenic miRNAs
such as miR-21 and miR-155 are frequently overexpressed in cancers.
miR-21 promotes migration and invasion by targeting tumor
suppressors such as programmed cell death protein 4, which in turn
leads to increased RhoB and Cdc42 activity (81). Similarly, miR-155 enhances
metastatic potential by repressing RhoA, shifting the balance
towards Rac1-driven mesenchymal migration (76,82).
Additionally, certain miRNAs exhibit context-dependent roles. For
example, miR-142 directly targets Rac1 and can either suppress or
promote cancer cell invasion depending on tumor type and cellular
context (83,84).
Therapeutically, this regulatory axis presents a
unique opportunity. The development of synthetic miRNA mimics (to
restore tumor-suppressive miRNAs such as miR-31 or miR-200) or
antagomiRs (to inhibit oncomiRs such as miR-21 and miR-155)
represents a promising strategy to indirectly normalize Rho GTPase
signaling. This approach is particularly relevant for cancers
resistant to conventional therapies, offering a way to rewire the
underlying signaling networks that drive adaptive invasion and
metastasis (85). However, the
development of miRNA therapeutics faces significant challenges,
including delivery efficiency, potential off-target effects, and
the need for careful dosing due to the ability of a single miRNA to
regulate hundreds of genes.
In the end, miRNA-mediated regulation adds a
sophisticated layer of control over the Rho GTPase network. While
therapeutically complex, the ability to manipulate these networks
with RNA-based agents connects the fundamental biology of
cytoskeletal regulation to the emerging field of RNA therapeutics.
This paves the way for the discussion of a more direct and potent
approach: the use of siRNA for the specific silencing of individual
Rho GTPase components (Table
II).
| Table II.Post-transcriptional regulation of
Rho GTPases signaling by miRNAs in cancer metastasis. |
Table II.
Post-transcriptional regulation of
Rho GTPases signaling by miRNAs in cancer metastasis.
| First author/s,
year | miRNA | Role in cancer | Validated molecular
targets | Effect on Rho
GTPase signaling pathways | Functional outcome
in metastasis | (Refs.) |
|---|
| Saliani et
al, 2021; | miR-200 family | Tumor
suppressor | ZEB1, ZEB2 | Indirect
downregulation of RhoA | Suppresses EMT,
inhibits invasion | (76–78) |
| Brabletz et
al, 2011; |
|
|
|
|
|
|
| Hur et al,
2013 |
|
|
|
|
|
|
| Augoff et
al, 2011 | miR-31 | Tumor
suppressor | RDX, RhoA | Reduces Rac1 and
RhoA | Impairs invasion
and metastasis | (80) |
| Slabáková et
al, 2017 | miR-34a | Tumor
suppressor | MET, SIRT1 | Modulates Rac1 and
RhoA signaling | Inhibits cell
migration and invasion | (79) |
| Gan et al,
2024 | miR-21 | OncomiR | Programmed cell
death protein 4, PTEN | Enhances RhoB and
Cdc42 activity | Promotes migration,
invasion, and EMT | (81) |
| Qian et al,
2024 | miR-155 | OncomiR | RhoA, SOCS1 | Suppresses RhoA and
enhances Rac1 signaling | Shifts balance to
mesenchymal migration | (82) |
| Xie et al,
2021; Pahlavan et al, 2020 | miR-142 |
Context-dependent | Rac1, ADCY9 | Directly targets
Rac1 mRNA | Can suppress or
promote invasion depending on context | (83,84) |
Therapeutic targeting: siRNA against
Rho GTPases
Pharmacological inhibition of Rho GTPases is
challenging due to their reliance on protein-protein interactions,
motivating alternative strategies such as RNAi. siRNAs are
incorporated into the RNA-induced silencing complex, which directs
sequence-specific mRNA degradation, resulting in potent and
selective knockdown of target proteins (8).
Preclinical studies demonstrate that silencing RhoA,
Rac1, or Cdc42 disrupts cytoskeletal structures, including
lamellipodia, filopodia and stress fibers, impairs focal adhesion
turnover, and suppresses migration, invasion and metastatic
colonization in various cancer models (7,8,71).
Tumor-type-specific dependencies and intra-tumoral heterogeneity
can influence efficacy, emphasizing the need for biomarker-guided
patient selection.
The clinical potential of siRNA therapeutics is
validated by FDA-approved agents such as patisiran, highlighting
feasibility for systemic administration. However, oncology
applications face significant hurdles. Naked siRNAs are unstable in
circulation, prone to nuclease degradation, and can elicit innate
immune responses. Their negative charge limits passive cellular
uptake, while non-specific gene silencing and unintended immune
activation remain key concerns (85,86).
These challenges are intensified in solid tumors due to dense
extracellular matrix, irregular vasculature, and high interstitial
pressure.
Consequently, advanced delivery platforms are
essential to overcome these limitations. Lipid nanoparticles (LNPs)
can encapsulate siRNA and facilitate cellular uptake via
endocytosis, enabling efficient intracellular delivery and gene
silencing in preclinical cancer models (86–88).
Recent advances in LNP design, including lipoprotein-mimicking
nanocarriers and ligand-targeted formulations, have further
improved tumor specificity and delivery efficiency (89). Polymeric nanocarriers, including
cationic polymers like polyethyleneimine or biodegradable polymers
such as poly(lactic-co-glycolic acid) (PLGA), form stable
polyplexes and allow controlled release, while surface
modifications enhance tumor specificity (87). Exosome-based systems leverage
natural biocompatibility for improved tissue targeting and
endosomal escape (90,91). Stimuli-responsive carriers release
siRNA in response to TME cues, such as low pH or high ROS,
enhancing localized silencing (87,88,92).
Each platform balances delivery efficiency, toxicity
and manufacturability, requiring careful optimization for specific
applications. The ultimate promise of siRNA may lie in
combinatorial strategies. Co-delivering siRNAs against Rho GTPases
with mitochondrial inhibitors, such as doxycycline, could
synergistically disrupt both cytoskeletal dynamics and metabolic
support, potentially overcoming compensatory resistance mechanisms
that limit monotherapies (Table
III).
| Table III.Delivery platforms for siRNA-mediated
targeting Rho GTPases in cancer metastasis. |
Table III.
Delivery platforms for siRNA-mediated
targeting Rho GTPases in cancer metastasis.
| First author/s,
year | Delivery
platform | Composition | Delivery
mechanism | Advantages | Limitations | Preclinical
evidence (model and outcome) | (Refs.) |
|---|
| Hou et al,
2024; Jin et al, 2024; Zeng et al, 2024 | LNPs | Ionizable or
cationic lipids, PEG-lipid | Endocytosis and
membrane fusion | Clinically
validated, high loading capacity, scalable | Immunogenicity and
liver tropism | Rac1 siRNA delivery
in murine models of metastasis model ↓ metastatic burden | (86–88) |
| Jin et al,
2024 | Polymeric
nanoparticle | PEI, PLGA,
chitosan |
Endocytosis,‘proton-sponge’ effect | Tunable, controlled
release | Cytotoxicity (PEI),
aggregation | RhoA siRNA-loaded
PLGA nanoparticles in in vivo cancer models ↓ tumor
growth | (87) |
| Kamerkar et
al, 2017; Ubanako et al, 2024 | Exosomes | Natural lipid
bilayer with surface proteins | Membrane fusion,
receptor-mediated uptake | Biocompatibility,
low immunogenicity, inherent tissue tropism | Low yield, limited
loading efficiency | Rac1 siRNA-loaded
exosomes ↓ melanoma metastasis in vivo | (90,91) |
| Jin et al,
2024; Zeng et al, 2024; Thomas et al, 2020 | Stimuli-Responsive
Systems |
pH/ROS/enzyme-sensitive polymers | TME-triggered
release | Enhanced
specificity, reduced off-target toxicity | Complex design,
stability concerns | RhoA siRNA released
under acidic TME conditions improves localized gene silencing | (87,88,92) |
Limitations and future directions
Despite the compelling rationale for siRNA targeting
of Rho GTPases outlined in the previous section, several
significant limitations must be overcome for clinical success.
Tumor heterogeneity and adaptive metabolic and cytoskeletal
plasticity represent a central challenge. When a specific Rho
GTPase such as RhoA is silenced, metastatic cells can activate
compensatory pathways to maintain invasiveness. This includes
upregulation of related family members (RhoC compensating for RhoA
loss), activating parallel cytoskeletal regulators, or inducing EMT
programs, effectively bypassing the therapeutic blockade (70). It is important to note that
functional redundancy varies by context; for example, while RhoA
and RhoC are often pro-metastatic, RhoB can exhibit
tumor-suppressive functions, highlighting the need for precise
target selection (68,93).
The second major barrier is delivery. The hostile
physiology of solid tumors, characterized by a dense ECM, aberrant
vasculature and hypoxic cores, severely restricts the penetration
and uniform distribution of siRNA carriers. Furthermore, systemic
administration faces obstacles such as rapid renal clearance,
nuclease degradation, and potential unintended immune activation,
all of which reduce bioavailability and raise safety concerns
(94). These barriers are far more
pronounced than in hematological malignancies, creating a uniquely
difficult environment for siRNA-based therapies in solid
cancers.
Future progress therefore hinges on innovative
strategies that simultaneously address biological compensation and
physical delivery barriers. To counteract adaptive resistance,
rational combination therapies are essential. This includes the
co-targeting of multiple Rho GTPases or, more powerfully, the
integration of Rho pathway inhibition with agents targeting
complementary vulnerabilities. As a core thesis of the present
review, a promising strategy approach may be the combination of Rho
GTPase siRNA with mitochondrial inhibitors (doxycycline). This
strategy simultaneously disables the cytoskeletal ‘engine’ and its
metabolic ‘fuel’, limiting the cancer's compensatory escape
mechanisms that may occur when using either drug alone (23,93).
Concurrently, the next generation of delivery
platforms must be engineered to address three critical objectives.
First, enhanced specificity can be achieved through the
incorporation of targeting ligands, such as Arginyl-glycyl-aspartic
acid (RGD) peptides or antibodies, that selectively recognize
receptors overexpressed on metastatic cells. Second, controlled
activation is essential, with carriers designed to release their
siRNA payload only in response to defined TME cues, including
acidic pH or elevated protease activity. Third, improved
penetration strategies are required to overcome the dense
extracellular matrix and abnormal stromal architecture of solid
tumors. Approaches such as incorporating ECM-degrading enzymes or
developing size-tunable nanoparticles are being actively explored
to ensure deeper intratumoral delivery and more effective
silencing.
Platforms incorporating these features, such as the
targeted and stimuli-responsive systems currently in preclinical
development, hold immense promise for improving the therapeutic
index by maximizing on-target effects while minimizing systemic
toxicity (87,88,92).
While the path to clinical application is fraught
with challenges, the ability of siRNA to directly silence
‘undruggable’ primary regulators of metastasis remains a highly
attractive and valid therapeutic goal. Overcoming the intertwined
obstacles of tumor plasticity, delivery efficiency, and adaptive
resistance through intelligent combinatorial and delivery
strategies will be paramount. Success in this endeavor will not
only yield new anti-metastatic agents but could also establish a
novel treatment paradigm centered on disrupting the integrated
metabolic and mechanical dependencies of cancer dissemination.
Crosstalk between mitochondria and Rho
GTPases in cancer metastasis
Rationale for crosstalk
discussion
The preceding sections have established both
mitochondria and Rho GTPases as powerful, independent drivers of
metastasis, governing bioenergetic adaptation and cytoskeletal
remodeling, respectively. However, a more complete, systems-level
understanding reveals that their functions are deeply intertwined,
creating an integrated functional axis that is essential for
metastatic competency (11,16). This extensive bidirectional
crosstalk ensures that the energetic supply is precisely matched to
mechanical demand.
The mechanistic basis for this interplay is
well-documented. Mitochondria provide the ATP required to directly
fuel actin polymerization and focal adhesion turnover, while
mitochondrial-derived ROS act as crucial second messengers that
oxidize cysteine residues in regulatory proteins, thereby
activating key Rho GTPase signaling pathways such as Src and PI3K
(95–98). More specifically, mitochondrial ROS,
particularly hydrogen peroxide (H2O2), oxidize redox-sensitive
cysteine residues in regulatory proteins, leading to activation of
Src-family kinases and modulation of RhoGEFs and RhoGAPs. This
process differentially regulates Rac1-driven protrusion and
RhoA-mediated contractility in a context-dependent manner (17,99–102).
Importantly, ROS signaling is highly dose-dependent: Low to
moderate ROS levels promote pro-migratory signaling, whereas
excessive ROS induces oxidative damage and suppresses metastatic
potential (17,18,38).
Conversely, Rho GTPase-driven cytoskeletal dynamics and the
establishment of cell polarity govern the trafficking and
positioning of mitochondria via proteins such as Miro and DRP1,
ensuring that mitochondria are recruited to regions of high energy
demand, such as the leading edge of migratory cells (11,30).
This reciprocal reinforcement creates a feedforward loop that
potently optimizes the cell's migratory and invasive capacity.
Therefore, this section will dissect the molecular
nodes of this metabolic-mechanical interplay, examining both
signaling and bioenergetic integration. The academic exercise of
understanding this crosstalk will provide the fundamental rationale
for designing innovative combination therapies that simultaneously
disrupt the energetic supply and the cytoskeletal machinery of
metastasis. This synergistic strategy of targeting complementary
vulnerabilities, represents a promising approach to overcome the
adaptive resistance that frequently limits the efficacy of
single-target interventions (23,103).
Functional convergence of mitochondria
and Rho GTPases
Building on the rationale for their crosstalk, the
functional integration of mitochondria and Rho GTPases occurs at
several key nodes that directly control cell motility. This
convergence ensures that bioenergetic supply is precisely coupled
to mechanical demand during invasion.
At the leading edge of migratory cells,
mitochondrial ATP is indispensable for fueling the actin
polymerization that drives protrusion formation. This localized
energy supply directly supports the extension of lamellipodia and
filopodia, processes governed by Rac1 and Cdc42, respectively
(11,104,105). Beyond ATP, mitochondria-derived
ROS, particularly H2O2, function as spatially restricted signaling
molecules under conditions of increased metabolic flux, hypoxia and
oncogenic stimulation (17,24,99,106).
At low to moderate concentrations, H2O2 oxidizes redox-sensitive
cysteine residues within protein tyrosine phosphatases (PTPs),
including PTP1B, leading to their transient inactivation (107). This oxidation-dependent inhibition
shifts signaling toward kinase-dominant states, resulting in
sustained phosphorylation and activation of Src-family kinases,
which in turn enhances Rho GTPase signaling pathways, including
phosphorylation-dependent activation of RhoGEFs (17,99,108).
This, in turn, amplifies signaling through downstream effectors
such as ROCK (promoting actomyosin contractility) and the
WAVE/Arp2/3 complex (reinforcing lamellipodia assembly), thereby
consolidating directional motility (104,109). Functionally, Rac1 preferentially
promotes lamellipodia formation and mesenchymal migration via
WAVE/Arp2/3 signaling, whereas RhoA drives stress fiber formation
and ROCK-mediated actomyosin contractility, supporting contractile
or amoeboid movement (68,69). In addition, Rac1-associated oxidant
signaling can further reinforce pro-migratory pathways,
contributing to a positive feedback between redox signaling and
cytoskeletal remodeling (32,99).
Conversely, the cytoskeletal machinery exerts
significant control over mitochondrial function and localization.
Rho GTPase-mediated activation of ROCK generates actomyosin
contractility, which influences mitochondrial dynamics by
modulating the activity of fission factors such as DRP1.
Furthermore, cellular polarity established by Cdc42 signaling
guides the trafficking of mitochondria along microtubules via
adaptor proteins such as Miro1-dependent adaptor complexes, which
link mitochondria to kinesin motors, ensuring their strategic
positioning at the cell's leading edge to sustain localized energy
production (110,111). In addition to general
mitochondrial trafficking mechanisms, mitochondrial Rho GTPases,
particularly MIRO1 and MIRO2, have emerged as critical regulators
of organelle positioning along the cytoskeleton. These proteins
function as adaptor molecules linking mitochondria to
microtubule-based motor complexes, including kinesin, thereby
enabling directed transport toward regions of high energetic demand
such as the leading edge of migrating cancer cells. This spatial
positioning is essential for sustaining localized ATP production
required for actin polymerization and focal adhesion dynamics.
Importantly, previous studies indicate that MIRO-dependent
trafficking actively contributes to metastatic behavior by
modulating cytoskeletal signaling and Rho GTPase activity, further
reinforcing the metabolic-mechanical coupling underlying invasion
(16,110,112). This spatial redistribution enables
localized ATP delivery and ROS signaling at the leading edge,
thereby reinforcing Rac1-dependent protrusive activity and
directional migration (30,113). For instance, in breast cancer
cells, mitochondrial translocation to invadopodia is a Rho
GTPase-dependent process essential for invasive capacity.
Bidirectional interaction reinforces the
coordination between metabolic output and cytoskeletal remodeling,
which then recruits more mitochondria to sustain the invasive
activity. Importantly, this metabolic-mechanical coupling is
regulated in a dose-dependent manner, as low to moderate ROS levels
preferentially sustain Rac1-driven protrusion and controlled RhoA
activity, whereas excessive ROS disrupt cytoskeletal organization
and suppress migration (17,18,38).
The integration of this metabolic output with mechanical activity
is not merely supportive but is a fundamental driver of efficient
and sustained invasion.
Molecular nodes of intersection
The functional convergence of mitochondria and Rho
GTPases is orchestrated by specific molecular nodes that serve as
direct signaling conduits. Understanding the cellular energy
sensors, redox-sensitive pathways, and the machinery controlling
organelle dynamics is crucial for appreciating how metabolic status
is translated into mechanical action.
A primary node of integration involves the
energy-sensing kinases AMPK and mTOR. These sensors directly couple
the cell;s metabolic state to its migratory machinery. Under
conditions of metabolic stress, including hypoxia, nutrient
deprivation, or increased AMP/ATP ratio, AMPK is activated and
directly suppresses RhoA-driven cytoskeletal contractility through
phosphorylation-dependent mechanisms. Specifically, AMPK inhibits
downstream effectors such as ROCK and MLC, thereby reducing
actomyosin contractility and limiting energy-consuming processes
such as cell contraction and migration (114–116). This establishes a metabolic
checkpoint that restrains RhoA-mediated mechanical output under
low-energy conditions.
Conversely, under nutrient-rich conditions and
growth factor stimulation, mTORC1 becomes activated and promotes
Rac1-dependent cytoskeletal remodeling. Mechanistically, mTORC1
enhances actin polymerization and lamellipodia formation by
regulating cytoskeletal effectors such as the WAVE complex and
Arp2/3, thereby facilitating Rac1-driven protrusive migration
(117–119). In addition, mTOR signaling
supports anabolic metabolism and protein synthesis required for
sustained migratory activity.
Together, these pathways establish a
context-dependent regulatory axis in which AMPK suppresses
RhoA-mediated contractility under energy stress, whereas mTORC1
promotes Rac1-driven protrusion under nutrient-replete conditions,
thereby coordinating metabolic status with distinct modes of cell
migration.
A second critical node is the mitochondrial
production of ROS. As described above, mitochondrial ROS act as
redox signaling intermediates that sustain activation of Src and
FAK, thereby amplifying Rac1- and RhoA-dependent pathways (99–101).
Finally, the physical distribution of mitochondria,
controlled by dynamics proteins, constitutes a third key node. The
master fission GTPase DRP1 is activated by various cues, including
phosphorylation at Ser616 (activation) and Ser637 (inhibition), and
creates small, mobile organelles (120). These mitochondria are trafficked
along microtubules to the leading edge by adaptor proteins such as
Miro, ensuring a localized supply of ATP to power Rac1- and
Cdc42-mediated actin polymerization (11,30,121).
Beyond its role as a trafficking adaptor, emerging evidence
indicates that mitochondrial Rho GTPases such as MIRO2 can directly
modulate RhoA signaling pathways. Mechanistically, MIRO2-dependent
mitochondrial positioning has been linked to regulation of
cytoskeletal dynamics through MYO9B-mediated control of RhoA
activity, thereby influencing cell migration and invasion. This
reveals an additional layer of bidirectional crosstalk in which
mitochondrial trafficking not only responds to cytoskeletal demands
but also actively regulates Rho GTPase signaling (16).
Dysregulation of any of these nodes can profoundly
amplify metastatic potential. For instance, oncogenic signaling
that constitutively activates mTORC1 can lead to hyperactive,
Rac1-driven invasion (119,122,123).
The understanding that these intersections are critical regulatory
hubs underscores their high value as targets for multi-faceted
therapeutic intervention (Fig.
4).
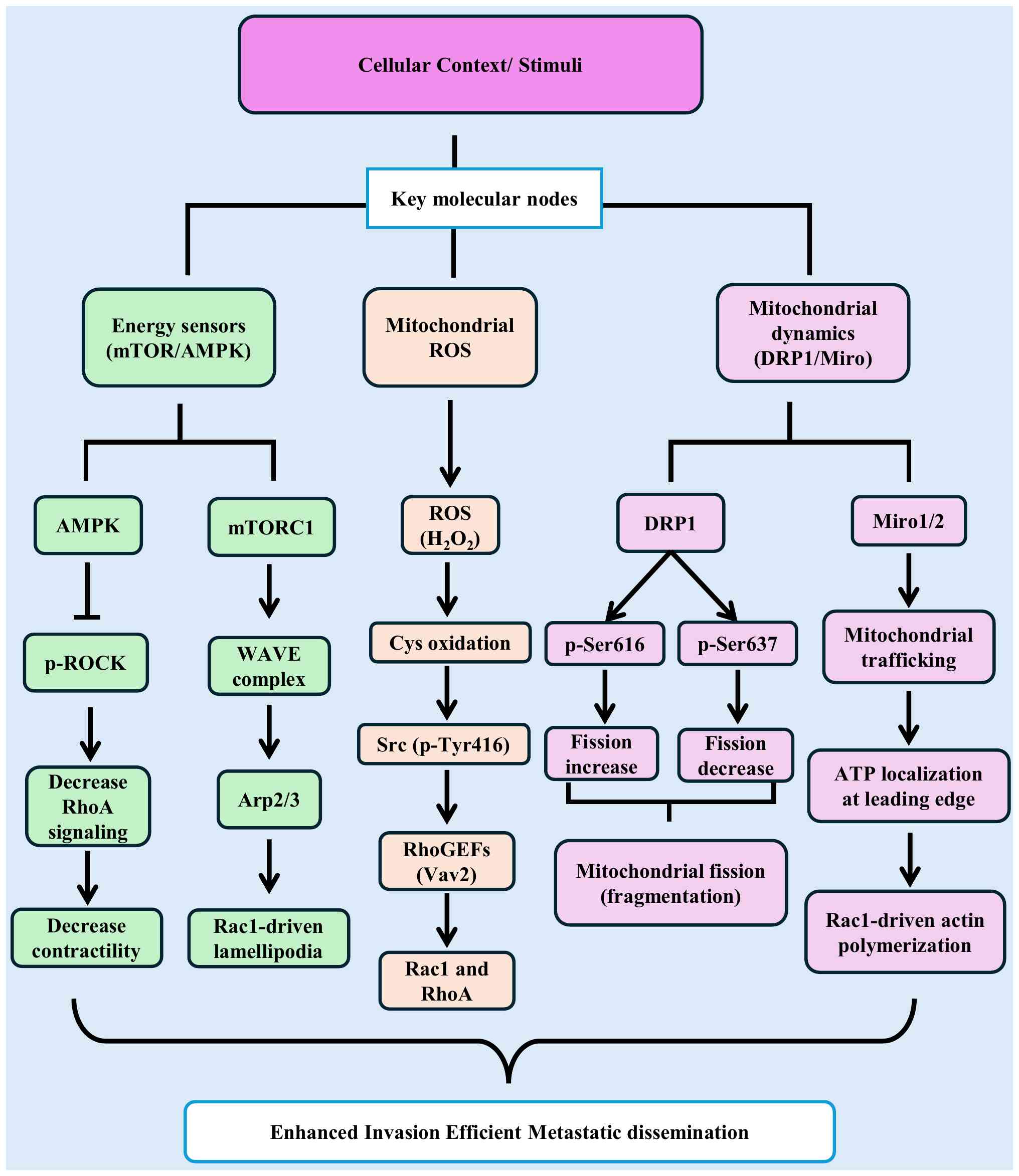 | Figure 4.Molecular nodes integrating
mitochondrial function with Rho GTPase-mediated cytoskeletal
regulation in cancer metastasis. Metastatic signaling converges on
three key molecular nodes linking mitochondrial function to Rho
GTPase-mediated cytoskeletal remodeling. Energy-sensing kinases
AMPK and mTORC1 translate nutrient and energy availability into
distinct cytoskeletal outputs: AMPK inhibits RhoA-ROCK signaling
via phosphorylation-dependent mechanisms, reducing actomyosin
contractility, whereas mTORC1 promotes Rac1-driven protrusion
through activation of the WAVE-Arp2/3 complex, leading to
lamellipodia formation (68,75).
Mitochondrial ROS, particularly H2O2, act as redox signaling
molecules by oxidizing cysteine residues in regulatory proteins,
leading to activation of Src (p-Tyr416) and downstream Rho-GEFs,
thereby promoting Rac1 and RhoA signaling (17,99,100,102,119). In parallel, mitochondrial
dynamics, are regulated by DRP1 phosphorylation (Ser616 promotes
fission; Ser637 inhibits fission) and Miro1/2-mediated trafficking,
which enables redistribution of mitochondria and localized ATP
supply to support Rac1- and Cdc42-driven actin polymerization
(67). These pathways form a
bidirectional feedback loop in which mitochondrial metabolism (ATP
and ROS) drives Rho GTPase activation, while Rho-dependent
cytoskeletal remodeling regulates mitochondrial positioning.
Together, these nodes converge to drive cytoskeletal remodeling,
contractility, extracellular matrix degradation, and directional
migration, enhancing invasive and metastatic potential.
Therapeutically, this network highlights actionable targets,
including AMPK activators (+), mTORC1 inhibitors (−), DRP1 or
mitochondrial trafficking inhibitors (−), and context-dependent ROS
modulators (±). ROS, reactive oxygen species; p-, phosphorylated;
DRP1, dynamin-related protein 1. |
Complementarity in therapeutic
targeting
The detailed molecular crosstalk between
mitochondria and Rho GTPases, outlined in the preceding sections,
reveals a fundamental vulnerability: Their interdependence. This
relationship provides a powerful rationale for combinatorial
therapeutic strategies designed to simultaneously disrupt both the
metabolic and mechanical engines of metastasis, thereby overcoming
the adaptive resistance that limits single-target therapies.
The logic of this approach is rooted in direct
mechanistic complementarity. Mitochondria-targeting agents, such as
the antibiotics tigecycline and doxycycline, inhibit OXPHOS. This
action has a dual effect; first, it depletes the local ATP supply
required for energy-intensive processes such as actin
polymerization, and second, it reduces the generation of
mitochondrial ROS, which function as essential second messengers
for activating pro-migratory signaling pathways through Src kinase
and Rho GTPases (23).
Specifically, inhibition of mitochondrial ROS disrupts
redox-dependent activation of Src and downstream RhoGEF signaling,
while ATP depletion impairs Rac1-driven actin polymerization and
RhoA-mediated contractility, effectively collapsing both protrusive
and contractile components of migration.
When administered alone, cancer cells may
compensate for mitochondrial inhibition by upregulating glycolysis.
However, when combined with siRNA that directly silences key Rho
GTPases (RhoA, Rac1, or Cdc42), the cytoskeletal machinery itself
is dismantled. The cell is left with neither the efficient energy
source (OXPHOS) nor the functional apparatus to utilize alternative
energy for migration and invasion (8).
The potential efficacy of this combinatorial
approach arises from the mechanistic interdependence between
mitochondrial metabolism and Rho GTPase signaling. Mitochondrial
inhibition limits ATP production and disrupts ROS-dependent
activation of Src and RhoGEF-mediated signaling, thereby impairing
both the energetic and signaling inputs required for cytoskeletal
remodeling. Concurrently, direct silencing of Rho GTPases disables
the downstream execution of migration by preventing Rac1-driven
protrusion and RhoA-mediated contractility. Together, this dual
targeting strategy restricts both the supply of metabolic resources
and the cellular machinery required for invasion, thereby limiting
the ability of cancer cells to engage compensatory adaptive
responses (70,103).
Although the translational implementation of such
combinations presents challenges, particularly the co-delivery of
two therapeutic agents and managing potential toxicities, advances
in nanomedicine, including multi-agent lipid nanoparticle systems,
are actively addressing these hurdles. In conclusion, targeting the
integrated mitochondria-Rho GTPase axis represents a
mechanistically grounded strategy to exploit a central
vulnerability of metastatic cells. By simultaneously disabling the
‘fuel’ and the ‘engine’, this combination therapy offers a
promising path toward more durable and effective control of cancer
dissemination (Fig. 5).
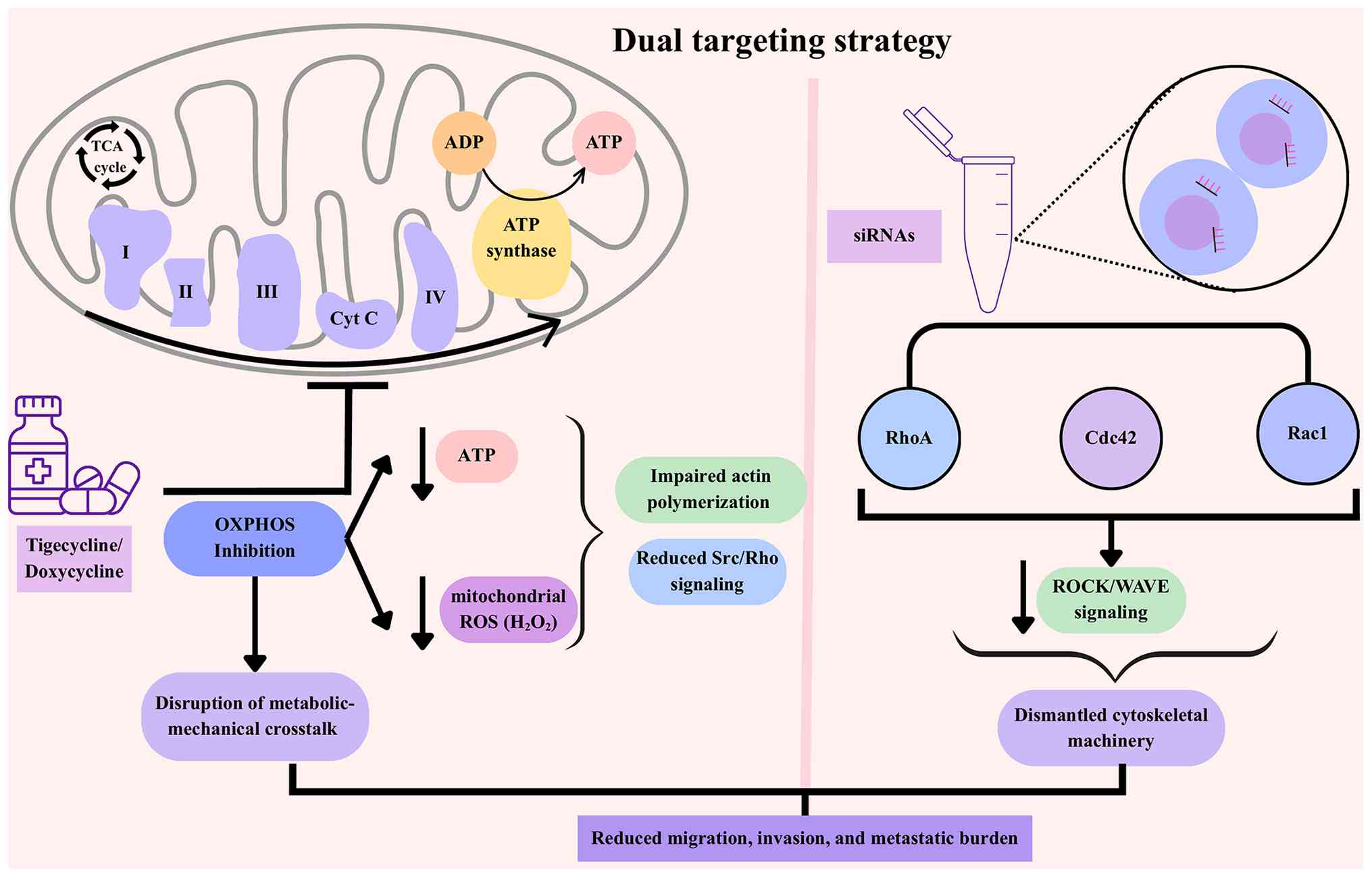 | Figure 5.Mechanistically complementary dual
targeting of mitochondria and Rho GTPases to inhibit cancer
metastasis. Mitochondria-targeting agents, such as tigecycline or
doxycycline, inhibit OXPHOS, leading to depletion of ATP and
reduction of mitochondrial ROS. This limits the energy supply
necessary for actin polymerization and dampens pro-migratory
signaling via Src kinase and Rho GTPases. Simultaneously,
siRNA-mediated silencing of Rho GTPases (RhoA, Rac1 and Cdc42)
directly dismantles the cytoskeletal machinery required for
migration and invasion. Individually, cells may compensate, by
upregulating glycolysis or switching migration modes, but combined
targeting creates a synergistic effect, blocking both the “fuel”
and the “engine” of metastasis. Preclinical studies demonstrate
that this dual approach is expected to reduce invasive capacity
more effectively than monotherapies. OXPHOS, oxidative
phosphorylation; ROS, reactive oxygen species; siRNAs, small
interfering RNAs. |
Overcoming limitations of single
approaches
The rationale for dual targeting must be balanced
against the intrinsic drawbacks of each monotherapy. Unless these
challenges are addressed, promising strategies are unlikely to
translate into clinical application.
Understanding these limitations highlights the
distinct constraints associated with each therapeutic class.
Mitochondrial inhibitors, such as OXPHOS disruptors, are effective
at impairing tumor bioenergetics, but their clinical use is
hindered by dose-limiting toxicities in energy-demanding tissues
such as myocardium and neurons. In addition, tumor metabolic
plasticity, including rapid shifts to glycolysis, further
undermines durability of therapeutic response (55,124,125). By contrast, siRNA-based therapies
face pharmacological barriers, including rapid renal clearance,
nuclease-mediated degradation, inefficient cellular uptake and
endosomal sequestration. These factors collectively compromise
their therapeutic index, particularly in solid tumors characterized
by dense stroma and abnormal vasculature (86,94).
These limitations directly impact the
mitochondria-Rho GTPase axis, as insufficient delivery prevents
effective disruption of both metabolic signaling (ROS and ATP) and
cytoskeletal remodeling pathways (RhoA/ROCK and Rac1/WAVE), thereby
allowing persistence of the metastatic feedback loop. Importantly,
this bidirectional metabolic-mechanical loop provides a mechanistic
basis for resistance to single-target therapies. Disruption of
either mitochondrial function or Rho GTPase signaling alone is
often insufficient, as cancer cells can compensate through the
reciprocal pathway, maintaining invasive and metastatic
potential.
Despite its strong mechanistic rationale, the
dual-targeting strategy itself faces several translational
challenges. A primary obstacle is the efficient co-delivery of two
distinct therapeutic modalities, as siRNAs and small-molecule
mitochondrial inhibitors differ in stability, pharmacokinetics, and
cellular uptake pathways. Achieving synchronized delivery and
intracellular release remains technically challenging. In addition,
simultaneous targeting of metabolic and cytoskeletal pathways may
increase the risk of systemic toxicity, particularly in highly
energy-dependent normal tissues. Furthermore, tumor heterogeneity
and pharmacodynamic variability may limit uniform therapeutic
responses across patients.
To overcome these pharmacological and biological
barriers, nanocarriers-based systems have emerged as promising
platforms for co-delivery. LNPs, clinically validated for siRNA
delivery, can be co-loaded with small-molecule inhibitors and
further engineered with tumor-specific ligands to enhance
selectivity and reduce systemic exposure (89,126).
Biodegradable polymers such as PLGA enable co-encapsulation of
hydrophilic siRNA and hydrophobic drugs, with tunable release
kinetics and responsiveness to the TME (127). Hybrid lipid-polymer constructs
further optimize stability, drug loading, and release dynamics.
Preclinical studies support the translational
potential of such co-delivery systems, demonstrating enhanced
therapeutic efficacy when metabolic and signaling pathways are
simultaneously targeted (103).
For instance, lipid nanoparticle-mediated delivery of siRNA
targeting oncogenic drivers has been shown to suppress tumor
progression and improve therapeutic outcomes in vivo
(128). Mechanistically, dual
targeting disrupts both bioenergetic adaptation and cytoskeletal
reprogramming, thereby limiting the ability of tumor cells to
engage compensatory escape mechanisms.
Taken together, these results underscore the
necessity of rationally designed co-delivery systems to fully
exploit the therapeutic potential of simultaneously targeting
metabolic and cytoskeletal pathways.
Future perspectives
The synthesis of evidence presented herein
establishes the mitochondria-Rho GTPase crosstalk as a critical,
actionable axis in metastasis. Moving forward, the therapeutic
potential of dual-targeting strategies must be systematically
evaluated and advanced through a focused research agenda.
Future preclinical investigations must extend
beyond conventional cell line models to systems that capture the
complexity of metastatic disease. These include patient-derived
xenografts, genetically engineered mouse models and 3D organoid
cultures that recapitulate tumor heterogeneity, stromal
interactions, and the stresses of the metastatic cascade. Within
these systems, studies should precisely delineate the mechanistic
consequences of dual inhibition, including its impact on
mitochondrial ROS signaling, localized ATP availability,
cytoskeletal dynamics, and the activation of compensatory pathways
such as alternative GTPase expression or enhanced glycolytic flux.
Particular emphasis should be placed on quantifying how dual
targeting alters ROS-dependent Src activation, RhoGEF/RhoGAP
balance, and the spatial coordination of Rac1-driven protrusion
versus RhoA-mediated contractility, as well as mitochondrial
positioning at the leading edge.
Translational innovation will be essential. The
development of advanced co-delivery platforms, such as
tumor-targeted lipid nanoparticles or engineered exosomes capable
of delivering both siRNA and mitochondrial inhibitors, is a
critical prerequisite for clinical application. Exosome-based
delivery systems, in particular, offer enhanced biocompatibility,
reduced immunogenicity, and improved targeting efficiency, making
them promising candidates for next-generation RNAi therapeutics
(91).
Furthermore, the potential of integrating this
dual-targeting approach with existing standard-of-care therapies,
such as immune checkpoint inhibitors or targeted agents, should be
explored to create multi-pronged treatment strategies. In parallel,
the identification of predictive biomarkers, such as gene
expression signatures reflecting mitochondrial dependency and Rho
GTPase signaling activity, will be crucial for patient
stratification and therapeutic optimization (14,68,70).
Beyond intracellular metabolic-mechanical
crosstalk, emerging evidence highlights intercellular mitochondrial
transfer as an additional layer contributing to tumor progression
and metastasis. Cancer cells can exchange mitochondria with stromal
and immune cells through mechanisms such as tunneling nanotubes and
extracellular vesicles, enabling recipient cells to acquire
functional mitochondria and enhance metabolic flexibility,
resistance to oxidative stress, and survival under adverse
microenvironmental conditions (129). This process is closely linked to
cytoskeletal dynamics and depends on mitochondrial trafficking
machinery, including proteins such as MIRO1 and MIRO2, further
extending metabolic-mechanical crosstalk beyond single-cell systems
(20,95). From a therapeutic perspective,
targeting mitochondrial transfer and trafficking mechanisms may
represent an additional strategy to disrupt metastatic progression.
Combining mitochondrial inhibition with approaches that interfere
with cytoskeletal regulation or mitochondrial transport, such as
Rho GTPase silencing, could further enhance therapeutic efficacy by
disrupting both intracellular energy distribution and intercellular
metabolic support.
By addressing these challenges, future research can
translate the compelling biology of the metabolic-mechanical
interface into a new therapeutic paradigm. The goal is to develop
next-generation interventions that simultaneously dismantle the
energetic and mechanical underpinnings of cancer dissemination,
offering a potent strategy to control the process responsible for
most of the cancer-related mortality.
Conclusion
Metastasis remains the principal cause of
cancer-related mortality, driven by the remarkable adaptability and
plasticity of tumor cells (4,130).
The present review has synthesized evidence establishing that
mitochondrial metabolism and Rho GTPase signaling are not
independent drivers of metastasis but rather functionally
integrated into a coordinated metabolic-mechanical axis (11,16).
This bidirectional crosstalk establishes a dynamic feedback loop in
which mitochondria supply ATP and ROS to fuel cytoskeletal
remodeling, while Rho GTPase-mediated cytoskeletal organization
governs mitochondrial positioning and function (30,102).
This reciprocal interaction optimizes the spatial and energetic
requirements of cell migration and represents a fundamental
mechanism supporting metastatic efficiency (30,131).
Importantly, this interdependence provides a
mechanistic explanation for the limited efficacy of single-target
therapies (35). Disruption of
either mitochondrial metabolism or Rho GTPase signaling alone is
frequently insufficient, as tumor cells can compensate through the
reciprocal pathway, maintaining invasive and metastatic potential.
This highlights the need for therapeutic strategies that
simultaneously target both metabolic and cytoskeletal components of
this axis to overcome adaptive resistance.
To overcome this adaptive resistance,
dual-targeting approaches combining siRNA-mediated silencing of Rho
GTPases with mitochondrial inhibitors, such as doxycycline,
represent a rational strategy to overcome compensatory resistance.
By concurrently disrupting the cytoskeletal ‘engine’ and its
metabolic ‘fuel’, this approach aims to achieve more effective
suppression of metastatic progression (8,23,103).
However, despite this strong mechanistic rationale, several
translational challenges must be addressed. These include limited
delivery to metastatic niches, efficient co-delivery of siRNA and
small-molecule agents, potential systemic toxicity in
energy-dependent tissues, and variability in therapeutic response
due to tumor heterogeneity.
Emerging nanocarrier-based systems, including lipid
nanoparticles and biodegradable polymer platforms, offer promising
solutions by enabling co-encapsulation, targeted delivery, and
controlled release of therapeutic agents (126,127). Furthermore, validating this
approach requires advanced preclinical models that capture
metastatic complexity and the identification of biomarkers to guide
patient selection in future clinical trials (35).
Looking forward, integrating this dual-targeting
paradigm into precision oncology frameworks offers a transformative
opportunity. Targeting this integrated metabolic–mechanical
network, rather than its individual components, provides a
conceptual and therapeutic framework for overcoming resistance and
achieving durable control of metastatic disease. Collectively, this
work provides a mechanistic roadmap for the development of
next-generation therapies aimed at addressing one of the most
urgent unmet needs in oncology.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AD drafted the initial manuscript. ZA restructured
the manuscript's organization and ideas, created the figures,
helped in creating the table, and performed proofreading. AK and
AAK helped in the revision and the thorough editing of the
manuscript. MES wrote the final draft and edited the manuscript.
Data authentication is not applicable. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript or to generate images, and subsequently,
the authors revised and edited the content produced by the
artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Sig Transduct Target Ther. 5:282020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
4
|
Lambert AW, Pattabiraman DR and Weinberg
RA: Emerging biological principles of metastasis. Cell.
168:670–691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clayton NS and Ridley AJ: Targeting rho
GTPase signaling networks in cancer. Front Cell Dev Biol.
8:2222020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jung H, Yoon SR, Lim J, Cho HJ and Lee HG:
Dysregulation of rho GTPases in human cancers. Cancers.
12:11792020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma N, Xu E, Luo Q and Song G: Rac1: A
regulator of cell migration and a potential target for cancer
therapy. Molecules. 28:29762023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crosas-Molist E, Samain R, Kohlhammer L,
Orgaz JL, George SL, Maiques O, Barcelo J and Sanz-Moreno V: Rho
GTPase signaling in cancer progression and dissemination. Physiol
Rev. 102:455–510. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Q, Zhuo Z, Qiu X, Luo R, Guo K, Wu H,
Jiang R, Li J, Lian Q, Chen P, et al: Adverse clinical outcomes and
immunosuppressive microenvironment of RHO-GTPase activation pattern
in hepatocellular carcinoma. J Transl Med. 22:1222024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caino MC and Altieri DC: Molecular
pathways: Mitochondrial reprogramming in tumor progression and
therapy. Clin Cancer Res. 22:540–545. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Gregorio J, Petricca S, Iorio R,
Toniato E and Flati V: Mitochondrial and metabolic alterations in
cancer cells. Eur J Cell Biol. 101:1512252022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Markov N, Sabirova S, Sharapova G,
Gomzikova M, Brichkina A, Barlev NA, Egger M, Rizvanov A and Simon
HU: Mitochondrial, metabolic and bioenergetic adaptations drive
plasticity of colorectal cancer cells and shape their
chemosensitivity. Cell Death Dis. 16:2532025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
LeBleu VS, O'Connell JT, Gonzalez Herrera
KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A,
Domingos Chinen LT, Rocha RM, et al: PGC-1α mediates mitochondrial
biogenesis and oxidative phosphorylation in cancer cells to promote
metastasis. Nat Cell Biol. 16:992–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
García-Heredia JM and Carnero A: Role of
mitochondria in cancer stem cell resistance. Cells. 9:16932020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boulton DP, Hughes CJ, Vaira V, Del Gobbo
A, Palleschi A, Locatelli M, Danis E, Raza M, Neumann AJ, Purdy SC,
et al: MIRO2 promotes cancer invasion and metastasis via MYO9B
suppression of RhoA activity. Cell Rep. 44:1151202025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Junco M, Ventura C, Santiago Valtierra FX
and Maldonado EN: Facts, dogmas, and unknowns about mitochondrial
reactive oxygen species in cancer. Antioxidants. 13:15632024.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
An X, Yu W, Liu J, Tang D, Yang L and Chen
X: Oxidative cell death in cancer: Mechanisms and therapeutic
opportunities. Cell Death Dis. 15:5562024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Palma FR, Gantner BN, Sakiyama MJ, Kayzuka
C, Shukla S, Lacchini R, Cunniff B and Bonini MG: ROS production by
mitochondria: Function or dysfunction? Oncogene. 43:295–303. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Yang Z, Zhang S and Li J:
Miro-mediated mitochondrial transport: A new dimension for
disease-related abnormal cell metabolism? Biochem Biophys Res
Commun. 705:1497372024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boulton DP and Caino MC: Emerging roles
for Mitochondrial Rho GTPases in tumor biology. J Biol Chem.
300:1076702024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, An Y, Ren M, Wang H, Bai J, Du W
and Kong D: The mechanisms of action of mitochondrial targeting
agents in cancer: Inhibiting oxidative phosphorylation and inducing
apoptosis. Front Pharmacol. 14:12436132023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karp I and Lyakhovich A: Targeting cancer
stem cells with antibiotics inducing mitochondrial dysfunction as
an alternative anticancer therapy. Biochem Pharmacol.
198:1149662022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mathur S, Srivastava P, Srivastava A, Rai
NK, Abbas S, Kumar A, Tiwari M and Sharma LK: Regulation of
metastatic potential by drug repurposing and mitochondrial
targeting in colorectal cancer cells. BMC Cancer. 24:3232024.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Isazadeh H, Oruji F, Shabani S, Behroozi
J, Nasiri H, Isazadeh A and Akbari M: Advances in siRNA delivery
approaches in cancer therapy: Challenges and opportunities. Mol
Biol Rep. 50:9529–9543. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johansson JM, Du Rietz H, Hedlund H,
Eriksson HC, Oude Blenke E, Pote A, Harun S, Nordenfelt P, Lindfors
L and Wittrup A: Cellular and biophysical barriers to lipid
nanoparticle mediated delivery of RNA to the cytosol. Nat Commun.
16:53542025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Corazao-Rozas P, Guerreschi P, André F,
Gabert PE, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A
and Quesnel B: Mitochondrial oxidative phosphorylation controls
cancer cell's life and death decisions upon exposure to MAPK
inhibitors. Oncotarget. 7:39473–39485. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Porporato PE, Filigheddu N, Pedro JMBS,
Kroemer G and Galluzzi L: Mitochondrial metabolism and cancer. Cell
Res. 28:265–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Faubert B, Solmonson A and DeBerardinis
RJ: Metabolic reprogramming and cancer progression. Science.
368:eaaw54732020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alshaabi H, Shannon N, Gravelle R,
Milczarek S, Messier T and Cunniff B: Miro1-mediated mitochondrial
positioning supports subcellular redox status. Redox Biology.
38:1018182021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brillo V, Chieregato L, Leanza L, Muccioli
S and Costa R: Mitochondrial dynamics, ROS, and cell signaling: A
blended overview. Life (Basel). 11:3322021.PubMed/NCBI
|
|
32
|
Capeloa T, Van De Velde JA, Pranzini E,
Ippolito L, Zampieri LX, Tardy M, Vazeille T, Provito A, Carrà G,
Scalera A, et al: Mitochondrial ROS inhibition prevents
doxorubicin-induced breast cancer cell migration and invasion.
iScience. 28:1130312025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim H, Sung JY, Park EK, Kho S, Koo KH,
Park SY, Goh SH, Jeon YK, Oh S, Park BK, et al: Regulation of
anoikis resistance by NADPH oxidase 4 and epidermal growth factor
receptor. Br J Cancer. 116:370–381. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Szanto I: NADPH Oxidase 4 (NOX4) in
cancer: Linking redox signals to oncogenic metabolic adaptation.
Int J Mol Sci. 23:27022022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao J, Zhang J, Yu M, Xie Y, Huang Y,
Wolff DW, Abel PW and Tu Y: Mitochondrial dynamics regulates
migration and invasion of breast cancer cells. Oncogene.
32:4814–4824. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Passaniti A, Kim MS, Polster BM and
Shapiro P: Targeting mitochondrial metabolism for metastatic cancer
therapy. Mol Carcinog. 61:827–838. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumar A, Vaish M, Karuppagounder SS,
Gazaryan I, Cave JW, Starkov AA, Anderson ET, Zhang S, Pinto JT,
Rountree AM, et al: HIF1α stabilization in hypoxia is not
oxidant-initiated. Elife. 10:e728732021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Piskounova E, Agathocleous M, Murphy MM,
Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ
and Morrison SJ: Oxidative stress inhibits distant metastasis by
human melanoma cells. Nature. 527:186–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mollinedo F and Gajate C: Lipid rafts as
signaling hubs in cancer cell survival/death and invasion:
Implications in tumor progression and therapy: Thematic Review
Series: Biology of Lipid Rafts. J Lipid Res. 61:611–635. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pascual G, Avgustinova A, Mejetta S,
Martín M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A,
Hueto JA, et al: Targeting metastasis-initiating cells through the
fatty acid receptor CD36. Nature. 541:41–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ghiglione N, Abbo D, Bushunova A,
Costamagna A, Porporato PE and Martini M: Metabolic plasticity in
pancreatic cancer: The mitochondrial connection. Mol Metab.
92:1020892025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Viale A, Pettazzoni P, Lyssiotis CA, Ying
H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V,
et al: Oncogene ablation-resistant pancreatic cancer cells depend
on mitochondrial function. Nature. 514:628–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Senft D and Ronai ZA: Regulators of
mitochondrial dynamics in cancer. Curr Opin Cell Biol. 39:43–52.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Simoes ICM, Morciano G,
Lebiedzinska-Arciszewska M, Aguiari G, Pinton P, Potes Y and
Wieckowski MR: The mystery of mitochondria-ER contact sites in
physiology and pathology: A cancer perspective. Biochim Biophys
Acta Mol Basis Dis. 1866:1658342020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
DeBerardinis RJ and Chandel NS: We need to
talk about the Warburg effect. Nat Metab. 2:127–129. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lamb R, Ozsvari B, Lisanti CL, Tanowitz
HB, Howell A, Martinez-Outschoorn UE, Sotgia F and Lisanti MP:
Antibiotics that target mitochondria effectively eradicate cancer
stem cells, across multiple tumor types: Treating cancer like an
infectious disease. Oncotarget. 6:4569–4584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sciacovelli M and Frezza C:
Oncometabolites: Unconventional triggers of oncogenic signalling
cascades. Free Radic Biol Med. 100:175–181. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nieman KM, Kenny HA, Penicka CV, Ladanyi
A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB,
Hotamisligil GS, et al: Adipocytes promote ovarian cancer
metastasis and provide energy for rapid tumor growth. Nat Med.
17:1498–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Andrzejewski S, Klimcakova E, Johnson RM,
Tabariès S, Annis MG, McGuirk S, Northey JJ, Chénard V, Sriram U,
Papadopoli DJ, et al: PGC-1α promotes breast cancer metastasis and
confers bioenergetic flexibility against metabolic drugs. Cell
Metab. 26:778–787.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
González A and Hall MN: Nutrient sensing
and TOR signaling in yeast and mammals. EMBO J. 36:397–408. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
DeNicola GM, Chen PH, Mullarky E, Sudderth
JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al: NRF2
regulates serine biosynthesis in non-small cell lung cancer. Nat
Genet. 47:1475–1481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Molina JR, Sun Y, Protopopova M, Gera S,
Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et
al: An inhibitor of oxidative phosphorylation exploits cancer
vulnerability. Nat Med. 24:1036–1046. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yap TA, Daver N, Mahendra M, Zhang J,
Kamiya-Matsuoka C, Meric-Bernstam F, Kantarjian HM, Ravandi F,
Collins ME, Francesco MED, et al: Complex I inhibitor of oxidative
phosphorylation in advanced solid tumors and acute myeloid
leukemia: Phase I trials. Nat Med. 29:115–126. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Boreel DF, Beerkens APM, Heskamp S,
Boswinkel M, Peters JPW, Adema GJ, Span PN and Bussink J:
Inhibition of OXPHOS induces metabolic rewiring and reduces hypoxia
in murine tumor models. Clin Transl Radiat Oncol.
49:1008752024.PubMed/NCBI
|
|
56
|
Mukherjee S, Bhatti GK, Chhabra R, Reddy
PH and Bhatti JS: Targeting mitochondria as a potential therapeutic
strategy against chemoresistance in cancer. Biomed Pharmacother.
160:1143982023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Škrtić M, Sriskanthadevan S, Jhas B,
Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N,
et al: Inhibition of mitochondrial translation as a therapeutic
strategy for human acute myeloid leukemia. Cancer Cell. 20:674–688.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Duivenvoorden WC, Popović SV, Lhoták S,
Seidlitz E, Hirte HW, Tozer RG and Singh G: Doxycycline decreases
tumor burden in a bone metastasis model of human breast cancer.
Cancer Res. 62:1588–1591. 2002.PubMed/NCBI
|
|
59
|
Nardo G, Pantziarka P and Conti M:
Synergistic potential of antibiotics with cancer treatments.
Cancers (Basel). 17:592024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kashatus JA, Nascimento A, Myers LJ, Sher
A, Byrne FL, Hoehn KL, Counter CM and Kashatus DF: Erk2
Phosphorylation of Drp1 promotes mitochondrial fission and
MAPK-driven tumor growth. Mol Cell. 57:537–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ježek J, Cooper KF and Strich R: The
impact of mitochondrial Fission-stimulated ROS production on
Pro-apoptotic chemotherapy. Biology (Basel). 10:332021.PubMed/NCBI
|
|
62
|
Lei X, Lin H, Wang J, Ou Z, Ruan Y,
Sadagopan A, Chen W, Xie S, Chen B, Li Q, et al: Mitochondrial
fission induces immunoescape in solid tumors through decreasing
MHC-I surface expression. Nat Commun. 13:38822022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sharma A, Virmani T, Kumar G, Sharma A,
Virmani R, Gugulothu D, Singh K, Misra SK, Pathak K, Chitranshi N,
et al: Mitochondrial signaling pathways and their role in cancer
drug resistance. Cell Signal. 122:1113292024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kuntz EM, Baquero P, Michie AM, Dunn K,
Tardito S, Holyoake TL, Helgason GV and Gottlieb E: Targeting
mitochondrial oxidative phosphorylation eradicates
Therapy-resistant chronic myeloid leukemia stem cells. Nat Med.
23:1234–1240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Milella M, Rutigliano M, Pandolfo SD,
Aveta A, Crocetto F, Ferro M, d'Amati A, Ditonno P, Lucarelli G and
Lasorsa F: The metabolic landscape of cancer stem cells: Insights
and implications for therapy. Cells. 14:7172025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Suárez-Rivero JM, Pastor-Maldonado CJ,
Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I,
Talaverón-Rey M, Suárez-Carrillo A, Munuera-Cabeza M and
Sánchez-Alcázar JA: Mitochondria and antibiotics: For good or for
evil? Biomolecules. 11:10502021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Rodrigues T and Ferraz LS: Therapeutic
potential of targeting mitochondrial dynamics in cancer. Biochem
Pharmacol. 182:1142822020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Haga RB and Ridley AJ: Rho GTPases:
Regulation and roles in cancer cell biology. Small GTPases.
7:207–221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lawson CD and Ridley AJ: Rho GTPase
signaling complexes in cell migration and invasion. J Cell Biol.
217:447–457. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Humphries BA, Wang Z and Yang C: MicroRNA
regulation of the small rho GTPase Regulators-complexities and
opportunities in targeting cancer metastasis. Cancers (Basel).
12:10922020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mardilovich K, Olson MF and Baugh M:
Targeting Rho GTPase signaling for cancer therapy. Future Oncol.
8:165–177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cantelli G, Orgaz JL, Rodriguez-Hernandez
I, Karagiannis P, Maiques O, Matias-Guiu X, Nestle FO, Marti RM,
Karagiannis SN and Sanz-Moreno V: TGF-β-Induced transcription
sustains amoeboid melanoma migration and dissemination. Cur Biol.
25:2899–2914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
George S, Martin JAJ, Graziani V and
Sanz-Moreno V: Amoeboid migration in health and disease: Immune
responses versus cancer dissemination. Front Cell Dev Biol.
10:10918012023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chen YQ, Huang CM, Liu CY, Chiou A, Wang
YK, Tang MJ and Kuo JC: β-PIX controls intracellular
viscoelasticity to regulate lung cancer cell migration. J Cell Mol
Med. 19:934–947. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Pittar A, Buckley EJ, Boyle ST, Ibbetson
SJ and Samuel MS: Enhanced RHO-ROCK signaling is associated with
CRELD2 production and fibroblast recruitment in cutaneous squamous
cell carcinoma. Cytoskeleton (Hoboken). 81:864–871. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Saliani M, Mirzaiebadizi A, Mosaddeghzadeh
N and Ahmadian MR: RHO GTPase-Related long noncoding RNAs in human
cancers. Cancers (Basel). 13:53862021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Brabletz S, Bajdak K, Meidhof S, Burk U,
Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J
and Brabletz T: The ZEB1/miR-200 feedback loop controls Notch
signalling in cancer cells. EMBO J. 30:770–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hur K, Toiyama Y, Takahashi M, Balaguer F,
Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR and Goel A:
MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT)
in human colorectal cancer metastasis. Gut. 62:1315–1326. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Slabáková E, Culig Z, Remšík J and Souček
K: Alternative mechanisms of miR-34a regulation in cancer. Cell
Death Dis. 8:e31002017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Augoff K, Das M, Bialkowska K, McCue B,
Plow EF and Sossey-Alaoui K: miR-31 is a broad regulator of
β1-Integrin expression and function in cancer cells. Mol Cancer
Res. 9:1500–1508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gan L, Zheng L, Zou J, Luo P, Chen T, Zou
J, Li W, Chen Q, Cheng L, Zhang F and Qian B: MicroRNA-21 in
urologic cancers: From molecular mechanisms to clinical
implications. Front Cell Dev Biol. 12:14379512024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Qian H, Maghsoudloo M, Kaboli PJ,
Babaeizad A, Cui Y, Fu J, Wang Q and Imani S: Decoding the promise
and challenges of miRNA-Based cancer therapies: An essential update
on miR-21, miR-34, and miR-155. Int J Med Sci. 21:2781–2798. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xie N, Meng Q, Zhang Y, Luo Z, Xue F, Liu
S, Li Y and Huang Y: MicroRNA-142-3p suppresses cell proliferation,
invasion and epithelial-to-mesenchymal transition via RAC1-ERK1/2
signaling in colorectal cancer. Mol Med Rep. 24:5682021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pahlavan Y, Mohammadi Nasr M, Dalir
Abdolahinia E, Pirdel Z, Razi Soofiyani S, Siahpoush S and Nejati
K: Prominent roles of microRNA-142 in cancer. Pathol Res Pract.
216:1532202020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Rupaimoole R and Slack FJ: MicroRNA
therapeutics: Towards a new era for the management of cancer and
other diseases. Nat Rev Drug Discov. 16:203–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hou X, Zaks T, Langer R and Dong Y: Lipid
nanoparticles for mRNA delivery. Nat Rev Mater. 6:1078–1094. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jin Z, Al Amili M and Guo S: Tumor
Microenvironment-responsive drug delivery based on polymeric
micelles for precision cancer therapy: Strategies and prospects.
Biomedicines. 12:4172024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zeng H, Zhang Y, Liu N, Wei Q, Yang F and
Li J: Stimulus-responsive nanodelivery and release systems for
cancer gene therapy: Efficacy improvement strategies. Int J
Nanomedicine. 19:7099–70121. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yaghmur A, Østergaard J and Mu H: Lipid
nanoparticles for targeted delivery of anticancer therapeutics:
Recent advances in development of siRNA and lipoprotein-mimicking
nanocarriers. Adv Drug Deliv Rev. 203:1151362023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kamerkar S, LeBleu VS, Sugimoto H, Yang S,
Ruivo CF, Melo SA, Lee JJ and Kalluri R: Exosomes facilitate
therapeutic targeting of oncogenic KRAS in pancreatic cancer.
Nature. 546:498–503. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ubanako P, Mirza S, Ruff P and Penny C:
Exosome-mediated delivery of siRNA molecules in cancer therapy:
Triumphs and challenges. Front Mol Biosci. 11:14479532024.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Thomas RG, Surendran SP and Jeong YY:
Tumor Microenvironment-stimuli responsive nanoparticles for
anticancer therapy. Front Mol Biosci. 7:6105332020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kazerounian S, Gerald D, Huang M, Chin YR,
Udayakumar D, Zheng N, O'Donnell RK, Perruzzi C, Mangiante L,
Pourat J, et al: RhoB differentially controls akt function in tumor
cells and stromal endothelial cells during breast tumorigenesis.
Cancer Res. 73:50–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Cummings JC, Zhang H and Jakymiw A:
Peptide carriers to the rescue: Overcoming the barriers to siRNA
delivery for cancer treatment. Transl Res. 214:92–104. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Altieri DC: Mitochondria on the move:
Emerging paradigms of organelle trafficking in tumour plasticity
and metastasis. Br J Cancer. 117:301–305. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Denisenko TV, Gorbunova AS and Zhivotovsky
B: Mitochondrial involvement in migration, invasion and metastasis.
Front Cell Dev Biol. 7:3552019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Koundouros N and Poulogiannis G:
Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in
cancer. Front Oncol. 8:1602018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yadav T, Gau D and Roy P:
Mitochondria-actin cytoskeleton crosstalk in cell migration. J Cell
Physiol. 237:2387–2403. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Nakao LS, Olson MF, Vázquez-Medina JP and
Valdivia A: Editorial: Reactive oxygen species (ROS) signaling
during cytoskeleton dynamics. Front Cell Dev Biol. 11:12952632023.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Rudzka DA, Cameron JM and Olson MF:
Reactive oxygen species and hydrogen peroxide generation in cell
migration. Commun Integr Biol. 8:e10743602015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xu Q, Huff LP, Fujii M and Griendling KK:
Redox regulation of the actin cytoskeleton and its role in the
vascular system. Free Radic Biol Med. 109:84–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Muliyil S and Narasimha M: Mitochondrial
ROS regulates cytoskeletal and mitochondrial remodeling to tune
cell and tissue dynamics in a model for wound healing. Dev Cell.
28:239–252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zheng XX, Chen JJ, Sun YB, Chen TQ, Wang J
and Yu SC: Mitochondria in cancer stem cells: Achilles heel or hard
armor. Trends Cell Biol. 33:708–727. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Fung TS, Chakrabarti R, Kollasser J,
Rottner K, Stradal TEB, Kage F and Higgs HN: Parallel kinase
pathways stimulate actin polymerization at depolarized
mitochondria. Curr Biol. 32:1577–1592.e8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Fung TS, Chakrabarti R and Higgs HN: The
multiple links between actin and mitochondria. Nat Rev Mol Cell
Biol. 24:651–667. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Akter S, Madhuvilakku R, Kar AK, Nila IS,
Liu P, Inuzuka H, Wei W and Hong Y: Reactive oxygen species (ROS)
in cancer: From mechanism to therapeutic implications. Sig
Transduct Target Ther. 11:1112026. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Paulsen CE and Carroll KS:
Cysteine-mediated redox signaling: Chemistry, biology, and tools
for discovery. Chem Rev. 113:4633–4679. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kelley LC, Chi Q, Cáceres R, Hastie E,
Schindler AJ, Jiang Y, Matus DQ, Plastino J and Sherwood DR:
Adaptive F-Actin polymerization and localized ATP production drive
basement membrane invasion in the absence of MMPs. Dev Cell.
48:313–328.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
López-Doménech G, Covill-Cooke C,
Ivankovic D, Halff EF, Sheehan DF, Norkett R, Birsa N and Kittler
JT: Miro proteins coordinate microtubule- and actin-dependent
mitochondrial transport and distribution. EMBO J. 37:321–336. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Qu C, Yang W, Kan Y, Zuo H, Wu M, Zhang Q,
Wang H, Wang D and Chen J: RhoA/ROCK Signaling regulates
Drp1-mediated mitochondrial fission during collective cell
migration. Front Cell Dev Biol. 10:8825812022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Cangkrama M, Liu H, Wu X, Yates J, Whipman
J, Gäbelein CG, Matsushita M, Ferrarese L, Sander S, Castro-Giner
F, et al: MIRO2-mediated mitochondrial transfer from cancer cells
induces cancer-associated fibroblast differentiation. Nat Cancer.
6:1714–1733. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Caino MC and Altieri DC: Cancer cells
exploit adaptive mitochondrial dynamics to increase tumor cell
invasion. Cell Cycle. 14:3242–3247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Liu H, Yin H, Wang Z, Yuan Q, Xu F, Chen Y
and Li C: Rho A/ROCK1 signaling-mediated metabolic reprogramming of
valvular interstitial cells toward Warburg effect accelerates
aortic valve calcification via AMPK/RUNX2 axis. Cell Death Dis.
14:1082023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Alasadi A, Fadhil N and Chen S:
Deciphering the critical roles of the AMPK/mTOR signaling pathway
in cancer cell metabolism (review). World Acad Sci J. 7:1–18. 2025.
View Article : Google Scholar
|
|
117
|
Collins SE, Wiegand ME, Werner AN, Brown
IN, Mundo MI, Swango DJ, Mouneimne G and Charest PG: Ras-mediated
activation of mTORC2 promotes breast epithelial cell migration and
invasion. Mol Biol Cell. 34:ar92023. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Soriano O, Alcón-Pérez M,
Vicente-Manzanares M and Castellano E: The crossroads between RAS
and RHO signaling pathways in cellular transformation, motility and
contraction. Genes. 12:8192021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Xu J, Hao S, Han K, Yang W and Deng H: How
is the AKT/mTOR pathway involved in cell migration and invasion?
Biocell. 47:773–788. 2023. View Article : Google Scholar
|
|
120
|
Cho HM, Ryu JR, Jo Y, Seo TW, Choi YN, Kim
JH, Chung JM, Cho B, Kang HC, Yu SW, et al: Drp1-Zip1 interaction
regulates mitochondrial quality surveillance system. Mol Cell.
73:364–376.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Modi S, López-Doménech G, Halff EF,
Covill-Cooke C, Ivankovic D, Melandri D, Arancibia-Cárcamo IL,
Burden JJ, Lowe AR and Kittler JT: Miro clusters regulate
ER-mitochondria contact sites and link cristae organization to the
mitochondrial transport machinery. Nat Commun. 10:43992019.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Ghosh S, Dash P, Das P and Nayak B: Ligand
Targeted Polymeric Nanoparticles for Cancer Chemotherapy. Padhi S,
Behera A and Lichtfouse E: Polymeric nanoparticles for the
treatment of solid tumors [Internet] Cham: Springer International
Publishing; 2022 pp. 251–272. Sep 27–2025, (Environmental Chemistry
for a Sustainable World). Available from:. https://link.springer.com/10.1007/978-3-031-14848-4_9doi:10.1007/978-3-031-14848-4_9.
View Article : Google Scholar
|
|
123
|
Indrajit R and Goutam G: Aberrant
signaling pathways in cancer cells: Application of nanomaterials. J
Cell Signal. 2:281–299. 2021.
|
|
124
|
Blazanin N, Liang X, Mahmud I, Kim E,
Martinez S, Tan L, Chan W, Anvar NE, Ha MJ, Qudratullah M, et al:
Therapeutic modulation of ROCK overcomes metabolic adaptation of
cancer cells to OXPHOS inhibition and drives synergistic anti-tumor
activity. bioRxiv. Sep 20–2024.doi: 10.1101/2024.09.16.613317.
|
|
125
|
Keoh LQ, Chiu CF and Ramasamy TS:
Metabolic plasticity and cancer stem cell metabolism: Exploring the
Glycolysis-OXPHOS switch as a mechanism for resistance and
tumorigenesis. Stem Cell Rev Rep. 21:2446–2468. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Witzigmann D, Kulkarni JA, Leung J, Chen
S, Cullis PR and Van Der Meel R: Lipid nanoparticle technology for
therapeutic gene regulation in the liver. Adv Drug Deliv Rev.
159:344–363. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Pan J, Rostamizadeh K, Filipczak N and
Torchilin VP: Polymeric Co-Delivery systems in cancer treatment: An
overview on component drugs' Dosage ratio effect. Molecules.
24:10352019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Tazawa H, Ishida Y, Suzuki T, Shimizu Y
and Tashiro H: Delivery of lipid nanoparticles containing small
interfering RNA targeting transmembrane serine protease 4 in a
human gastric cancer model using nude mice. Sci Rep. 15:450532025.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang X and Gerdes HH: Transfer of
mitochondria via tunneling nanotubes rescues apoptotic PC12 cells.
Cell Death Differ. 22:1181–1191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Steeg PS: Targeting metastasis. Nat Rev
Cancer. 16:201–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Panchal K and Tiwari AK: Miro
(Mitochondrial Rho GTPase), a key player of mitochondrial axonal
transport and mitochondrial dynamics in neurodegenerative diseases.
Mitochondrion. 56:118–135. 2021. View Article : Google Scholar : PubMed/NCBI
|














