Introduction
Acute myeloid leukemia (AML) is a hematological
malignancy characterized by the clonal expansion and
differentiation blockade of myeloid progenitor cells (1). The incidence of AML is age-dependent,
and a 5-year relative survival rate among patients in the United
States is only 30.5% (1,2). Although most patients with AML receive
induction and consolidation chemotherapy, 20–50% eventually develop
chemoresistance (3). Furthermore,
the high relapse rate of AML leads to poor prognosis and low
remission rates (4). Targeted
therapies have shown efficacy in these refractory or relapsed
patients (5). Due to the high
heterogeneity of AML, identifying new targeted small-molecule
inhibitors remains critical (6).
Previous studies have shown that the PI3K-Akt
pathway is aberrantly activated in 50–80% of patients with AML and
serves a pivotal role in leukemic cell proliferation (6,7).
PI3Kδ, a member of the Class I PI3K family, is highly expressed in
leukocytes (8). Several PI3Kδ
inhibitors have demonstrated anti-proliferative and pro-apoptotic
effects in AML cells (9,10). YY-20394 (linperlisib) is an oral and
highly selective PI3Kδ inhibitor with less activity against PI3Kγ,
giving a kinase inhibition profile that is nearly two orders of
magnitude more selective for PI3Kδ, which may improve tolerability
compared with other PI3K inhibitors (11). In clinical studies, YY-20394 has
shown promising results in hematologic malignancies, including
peripheral T-cell lymphoma, B-cell lymphoma and follicular
lymphoma, with an overall response rate exceeding 60% and a
manageable safety profile (11–13).
However, the effects of YY-20394 in AML remains unclear.
ABT199 (venetoclax), a Bcl-2 inhibitor, has been
approved in combination with hypomethylating agents or low-dose
cytarabine for newly diagnosed patients with AML who are elderly or
unfit for intensive chemotherapy (14). Nevertheless, resistance to ABT199
has been observed in a subset of patients with AML (15). For example, FMS-like tyrosine kinase
3 (FLT3)-internal tandem duplications (ITDs) mutation could
increase the expression of anti-apoptotic Bcl-2 family proteins and
is often associated with AML resistance to ABT199 (16). Research has suggested that targeting
the PI3K/Akt pathway may enhance the efficacy of ABT199 in AML
(17). In the present study, the
sensitivity of different AML cell lines to YY-20394 and ABT199 was
investigated. Furthermore, the effects of YY-20394 alone and in
combination with ABT199 on AML cells were assessed, providing new
insights for the treatment of AML.
Materials and methods
Cell culture
Three AML cell lines, MV-4-11 (human leukemia cells;
cat. no. CL-0572), U937 (human monocytic leukemia cells; cat. no.
CL-0239) and THP-1 (human histiocytic lymphoma cells; cat. no.
CL-0233), and their corresponding cell-specific media were
purchased from Procell Life Science & Technology Co., Ltd. All
cells were authenticated by short tandem repeat profiling and grown
at 37°C under 5% CO2 and 95% relative humidity. For U937
cells, 5 and 10 µM YY-20394 were used for treatment. For MV-4-11
cells, treatments included 120 nM YY-20394, 30 nM ABT199 and a
combination of 120 nM YY-20394 with 30 nM ABT199. The negative
control (NC) group received an equal volume of drug-free culture
medium.
Half-maximal inhibitory concentration
(IC50) determined by cell counting Kit-8 (CCK-8)
assay
A CCK-8 assay was performed using the Cell Counting
Kit-8 (APeXBIO Technology LLC). A total of 100 µl cell suspension
was spread into a 96-well plate (1.5×104 cells per well)
and incubated at 37°C in a 5% CO2 environment. The cells
were divided into two groups: The experimental group (As), which
received the drug (YY-20394 or ABT199), and the control group (Ac),
which received no drug. Additionally, a blank group (Ab),
consisting only of culture medium, was included. Following previous
studies for reference, the concentration gradients for the drugs
were as follows: For YY-20394 (18), the concentrations used in MV4-11
cells were 10, 50, 100, 500, 1,000 and 5,000 nM; in THP-1 cells,
they were 500, 1,000, 2,000, 5,000, 1×104 and
2×104 nM; and in U937 cells, the concentrations were
1,000, 5,000, 1×104, 2×104, 5×104
and 1×105 nM. For ABT199, the concentrations used in
MV4-11 cells were 5, 25, 100, 200, 300, 400 and 600 nM (19,20);
in THP-1 cells, the concentrations used were 1,000,
1×104, 2×104, 3×104,
4×104 and 5×104 nM (21); and in U937, they were 1,000, 5,000,
8,000, 1×104, 1.5×104, 2×104 and
4×104 nM.
Following cell adherence to the plate, the diluted
drugs were incubated with the cells for either 24 or 48 h.
Subsequently, 10 µl CCK-8 solution was added to each well and
incubated for 4 h. The optical density (OD) value at λ=450 nm was
measured using a plate reader. The percentage of cell growth
inhibition was calculated using the following formula: Cell growth
inhibition (%)=1-[(As-Ab)/(Ac-Ab)] ×100%. The IC50 value
of the drug was determined through linear regression analysis.
Specifically, drug concentrations were transformed to their
logarithmic (log) values, and the IC50 values were
calculated using the ‘log(inhibitor) vs. normalized
response-variable slope’ model in GraphPad software (version 8;
Dotmatics). Growth inhibition curves were also plotted based on
this analysis.
Combination index (CI) value detected
by CCK-8 assay
The experimental groups were divided into the
YY-20394, ABT199 and YY-20394+ABT199 combination groups. The drug
concentration was a multiple of the IC50 value (1, 0.5,
0.25, 0.125, 0.0625 and 0.03125 times). Cell growth inhibition was
measured using a CCK-8 assay as described. Subsequently, CI values
were calculated using CompuSyn software (version 1.0; ComboSyn,
Inc.), and Fa-CI plots of equivalent dose-effect ratios were drawn
based on the obtained data. The strength of drug-drug interactions
in the combination of YY-20394 and ABT199 could be quantitatively
determined by the magnitude of the CI values: CI >1 was
antagonistic, CI=1 was additive, 0.7< CI <1 was weakly
synergistic, 0.3< CI <0.7 was synergistic and CI <0.3 was
strongly synergistic.
Determining apoptosis by the dual
acridine orange/ethidium bromide (AO/EB) staining
According to the instructions, cell apoptosis was
detected using dual AO/EB staining using a
normal/apoptotic/necrotic cell detection kit (Jiangsu KeyGen
Biotech Co., Ltd.). The results were observed under a fluorescence
microscope at 510 nm. According to the cell morphology and staining
results, the following four types of cells were counted (the total
number of cells >200): i) Normal cells were defined as round
cells with uniformly green-stained nucleoplasm and consistent size
and shape; ii) necrotic cells were ellipsoidal with uniformly
orange-yellow-stained nucleoplasm and consistent size and shape;
iii) early apoptotic cells were indicated by green nucleoplasm and
cells exhibiting irregular shapes, such as crescent-like
morphology; and iv) late apoptotic cells where the nucleoplasm was
orange, chromatin was condensed, the nucleus was fragmented into
punctate structures of varying sizes and cytoplasmic blebbing was
observed. Apoptosis rate=(early apoptotic cells + late apoptotic
cells)/total number of cells ×100%. Cell necrosis rate=necrotic
cells/total number of cells ×100%.
Cell cycle assay
Drug-exposed cells were washed with PBS and fixed
homogeneously in pre-cooled 95% ethanol at 4°C overnight. After
washing with PBS, the cells were stained with propidium iodide (PI)
solution (Beijing Solarbio Science & Technology Co., Ltd.)
containing RNase A and incubated in the dark at 37°C for 30 min.
The cell cycle distribution was analyzed using a flow cytometer
(NovoCyte; Agilent Biosciences). Flow cytometry data were analyzed
using FlowJo software (BD Biosciences). Briefly, target cell
populations were first gated based on forward scatter (FSC) and
side scatter (SSC) parameters to exclude debris and non-viable
fragments. Doublets and cell aggregates were subsequently excluded
by gating on FSC-A vs. FSC-H. Cell cycle distribution was then
determined based on DNA content in the PE channel. To ensure
analytical accuracy and consistency across groups, a gating
template was established from NC group and subsequently applied to
all other experimental groups.
Reverse transcription-quantitative PCR
(RT-qPCR)
The mRNA levels of Akt, mTOR, myeloid cell
leukemia-1 (Mcl-1), Bcl-2 interacting mediator of cell death (Bim),
Bcl-2, B-cell lymphoma-extra large (Bcl-xL), Bcl-2 antagonist
killer 1 (Bak) and Bcl-2 associated X (Bax) were assessed using
RT-qPCR in drug-exposed cells. GAPDH was used as an internal
reference gene. Specific primers are presented in Table SI. Total RNA was extracted from
cell samples using TRIzol reagent (Tiangen Biotech Co., Ltd.), and
cDNA was synthesized using the FastQuant cDNA First Strand
Synthesis Kit (Tiangen Biotech Co., Ltd.). The PCR reaction mixture
was prepared by combining SuperReal PreMix Plus (SYBR Green;
Tiangen Biotech Co., Ltd.) and specific primers, following the
manufacturer's instructions. The PCR amplification protocol was as
follows: 95°C for 15 min, followed by 40 cycles of 95°C for 10 sec,
55°C for 30 sec and 72°C for 32 sec. A final extension step was
performed at 95°C for 15 sec, 60°C for 60 sec and 95°C for 15 sec.
The amplification results were detected using an ABI 7300
fluorescence quantitative PCR instrument (Applied Biosystems;
Thermo Fisher Scientific, Inc.), and relative gene expression was
analyzed using the 2−ΔΔCq method (22).
Western blot analysis
The protein levels of Akt (Akt Polyclonal Antibody;
1:1,000; ImmunoWay Biotechnology Company; cat. no. YT0185),
phosphorylated (p)-Akt [Akt (phospho Ser473) Polyclonal Antibody;
1:1,000; ImmunoWay Biotechnology Company; cat. no. YP0006), ERK
(Mouse Anti-ERK1/2 antibody; 1:1,000; BIOSS; cat. no. bsm-33337M),
p-ERK [Phospho-ERK1/2 (Thr202/Tyr204) Antibody; 1:1,000; Affinity
Biosciences; cat. no. AF1015), Mcl-1 [MCL1 Rabbit mAb (hp7u);
1:1,000; Nature Biosciences; cat. no. A57858], Bim [Bim Rabbit mAb
(7kqv); 1:1,000; Nature Biosciences; cat. no. A83449], Bcl-2
(Rabbit Anti-Bcl-2 antibody; 1:1,000; BIOSS; cat. no. bs-0032R),
Bcl-xL [Bcl-xL Rabbit mAb (kn97); 1:1,000; Nature Biosciences; cat.
no. A66923], Bak [Bak Rabbit mAb (RX7I); 1:1,000; Nature
Biosciences; cat. no. A53931], Bax (Rabbit Anti-Bax antibody;
1:1,000; BIOSS; cat. no. bs-0127R) and c-Myc [c-Myc Rabbit mAb
(paYu); 1:1,000; Nature Biosciences; cat. no. A23647] were assessed
using western blot in drug-exposed cells. Actin (1:3,000; BIOSS;
cat. no. bs-0061R) was used as an internal reference.
RIPA lysis buffer (Beyotime Biotechnology) was added
to the cell samples, and proteins were extracted by ultrasound
homogenization on ice. Protein concentration was determined using
the BCA protein assay (Beijing Solarbio Science & Technology
Co., Ltd.) and adjusted to 1.5 mg/ml (~21 µg/lane). Proteins were
separated on 5 and 10% polyacrylamide gels and then transferred to
activated PVDF membranes. A color-pre-stained protein marker
(10–180 kDa; Biodragon) was used to indicate the molecular weight
of the target protein. The membrane was blocked with 5% bovine
serum albumin (Wuhan Servicebio Technology Co., Ltd.) at room
temperature for 1 h. The membrane was then incubated overnight at
4°C with the primary antibody specific to the target protein.
Subsequently, the membrane was incubated with the corresponding
secondary antibody (1:5,000; Nature Biosciences; Goat Anti-Mouse,
cat. no. M00001; Goat Anti-Rabbit, cat. no. R00001) at room
temperature for 1 h, and the results were detected using an ECL
reagent kit (Harbin HaiGene Biotech Co., Ltd.) through
chemiluminescence.
Statistical analysis
One-way analysis of variance followed by Tukey's
post hoc test was performed using GraphPad Prism for multiple group
comparisons. All pairwise comparisons were two-tailed. All data are
presented as the mean ± standard deviation. A P-value of <0.05
was considered to indicate a statistically significant
difference.
Results
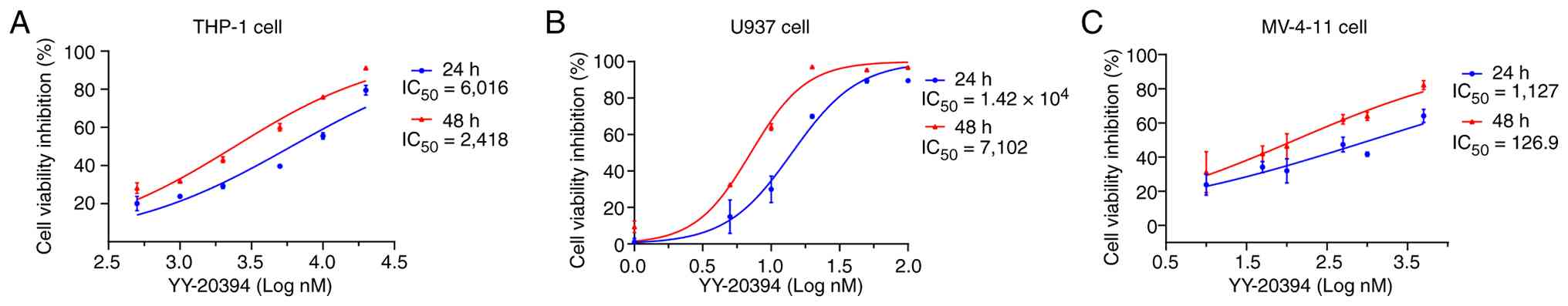
Differential sensitivity of YY-20394
in different AML cell lines
The IC50 values of YY-20394 were
evaluated to assess the sensitivity in different AML cell lines. In
THP-1 cells, the 24 and 48 h IC50 values of YY-20394
were 6,020 and 2,420 nM, respectively (Fig. 1A). In U937 cells, the 24 and 48 h
IC50 values of YY-20394 were 1.42×104 and
7,102 nM, respectively (Fig. 1B).
The 24 and 48 h IC50 values of YY-20394 in MV-4-11 cells
were 1,127 and 126.9 nM, respectively (Fig. 1C). Comparatively, MV-4-11 cells were
more sensitive to YY-20394, while THP-1 and U937 cells were less
sensitive. YY-20394 exhibited a smoother dose-response profile in
U937 cells, which were selected for further experiments.
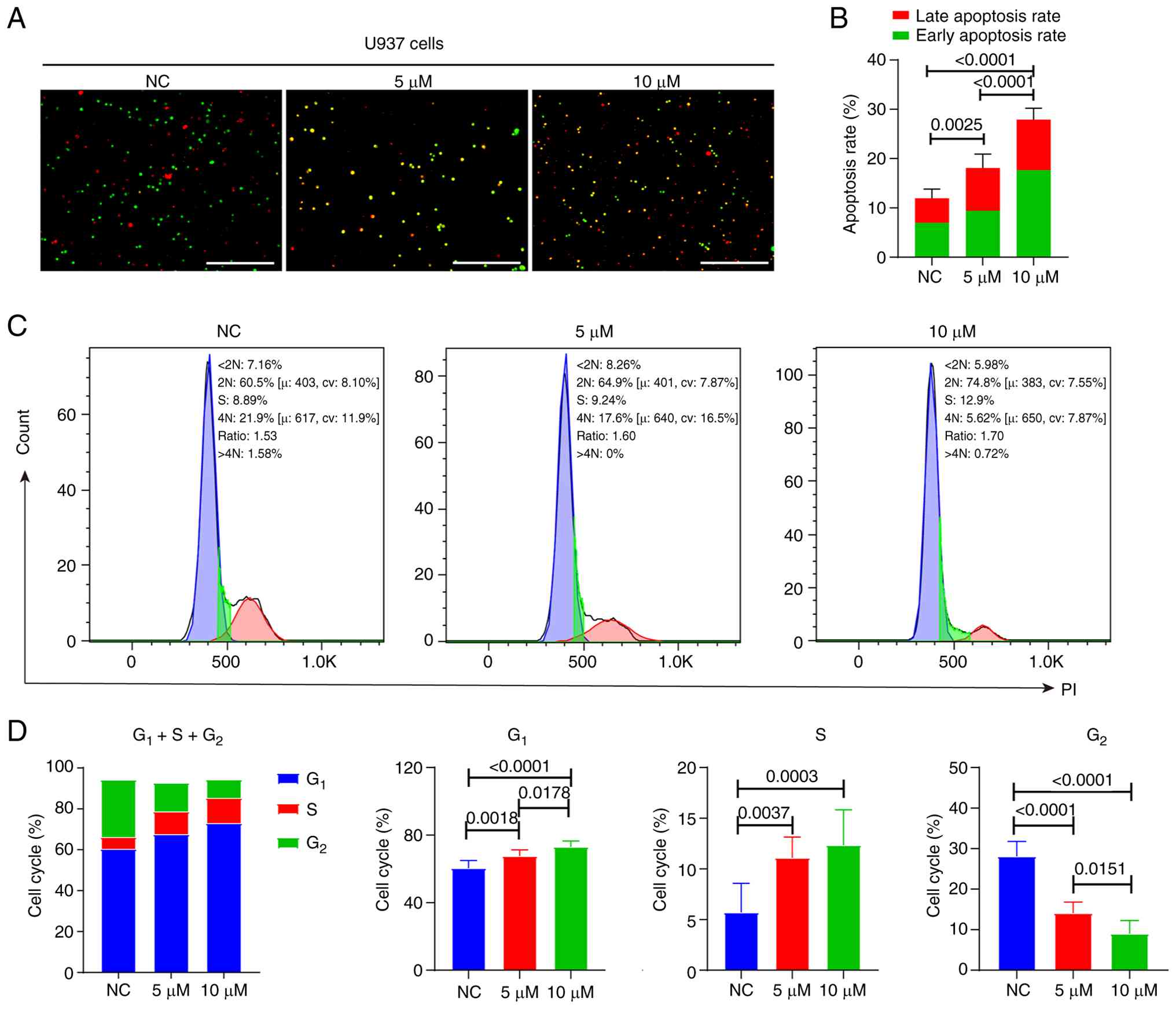
YY-20394 inhibits U937 cell
proliferation, promotes apoptosis and arrests the G1/S
phase
Cell apoptosis and the cell cycle are important
biological processes that collectively determine the proliferation
and survival of malignant cells. The dual AO/EB assay revealed that
YY-20394 significantly induced apoptosis in U937 cells
(P<0.001), with the apoptosis rate being significantly higher in
the 10 µM group compared with the 5 µM group (P<0.0001)
(Fig. 2A and B). Flow cytometry
analysis was used to assess the effects of YY-20394 on the cell
cycle (Fig. 2C and D). Compared
with the NC group, treatment with 5 and 10 µM of YY-20394
significantly increased the proportion of cells in the
G1 and S phases (P<0.01) while significantly reducing
the population in the G2 phase (P<0.0001), indicating
that YY-20394 interfered with cell cycle progression at the
G1/S transition and early S phase. Overall, YY-20394
could inhibit U937 cell proliferation, promote apoptosis and induce
arrest at G1/S transition and in early S phase.
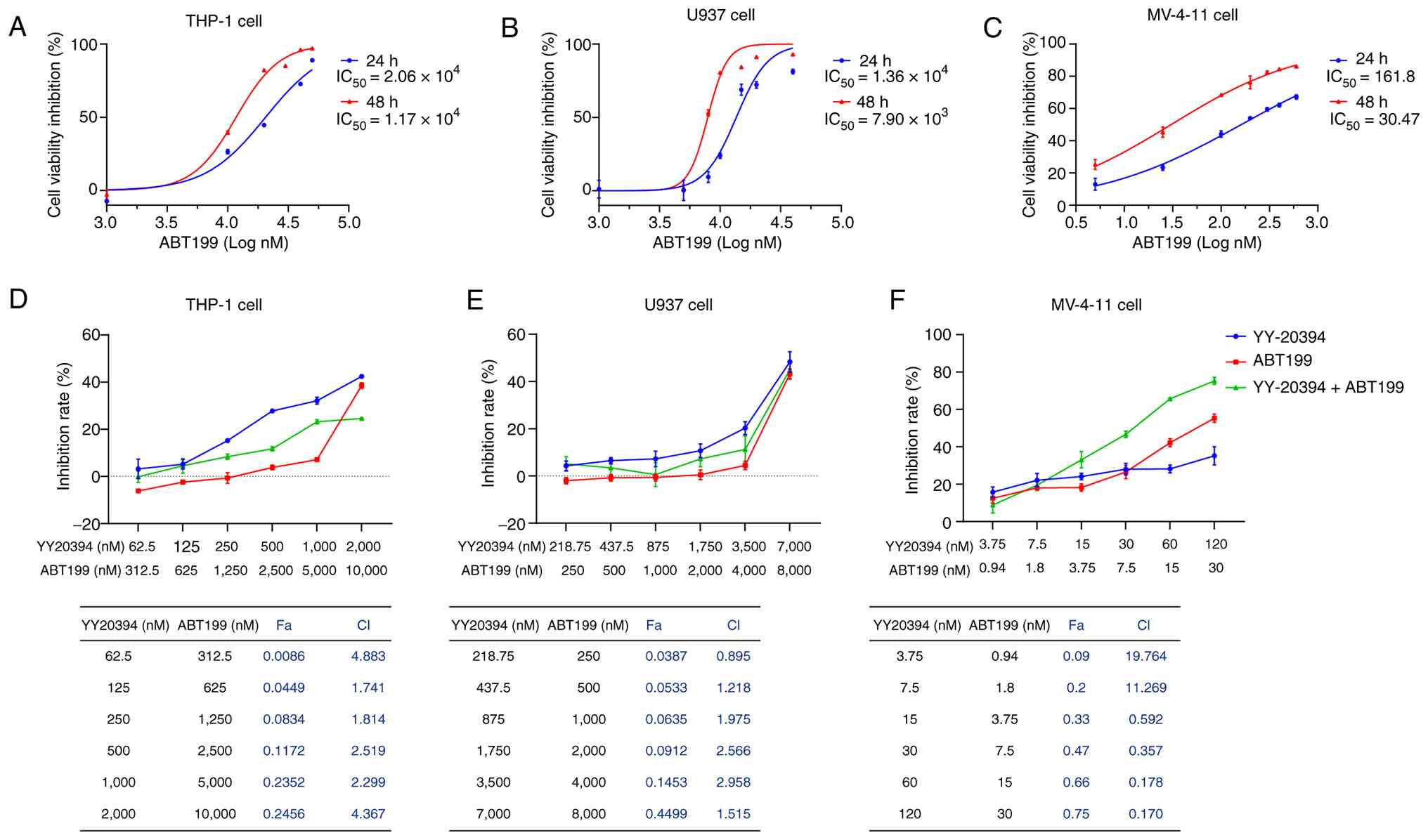
YY-20394 and ABT199 have a synergistic
effect in MV-4-11 cells
The percentage of cell proliferation inhibition of
ABT199 was assessed to determine its sensitivity in different AML
cell lines (Fig. 3A-C). The results
revealed that the IC50 values of ABT199 were
2.06×104, 1.36×104 and 161.80 nM for 24 h in
THP-1, U937 and MV-4-11 cells, respectively, and
1.17×104, 7.90×103 and 30.47 nM for 48 h,
respectively. To assess the potential synergistic effect of
YY-20394 and ABT-199, three AML cell lines were treated with a
range of doses based on the respective IC50 values of
each drug. No significant synergistic effect was observed in THP-1
cells (CI>1, Fig. 3D), and only
a negligible synergistic effect was detected in U937 cells
(CI=0.895, Fig. 3E). Notably,
synergistic interactions were observed in MV-4-11 cells, with the
CI value of the optimal dose combination being 0.17 (Fig. 3F). Based on these results, the
concentration with the strongest synergistic effect was selected
for further experiments with MV-4-11 cells.
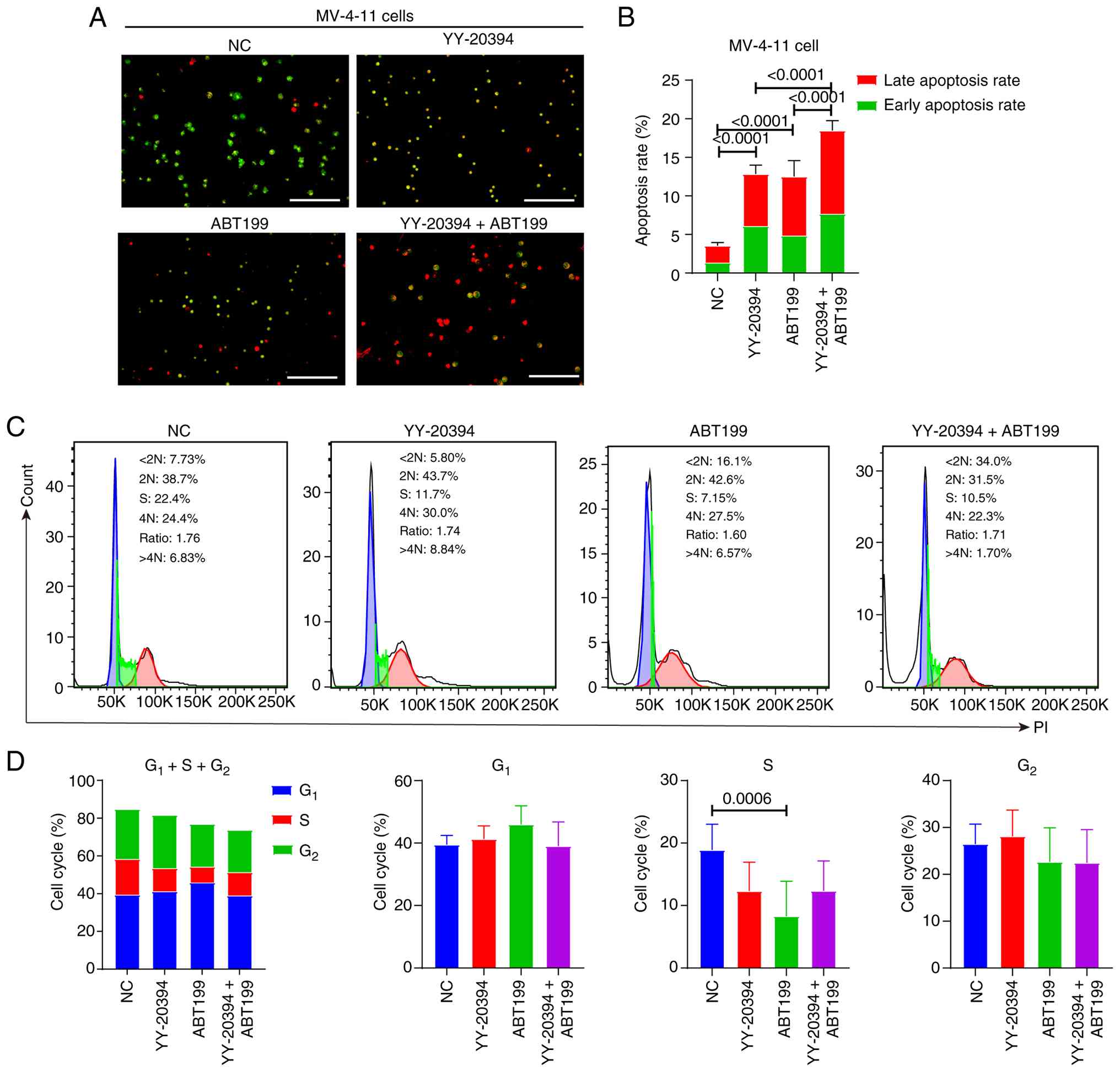
YY-20394 and ABT199 synergistically
promote apoptosis in MV-4-11 cells without inducing cell cycle
arrest
Dual AO/EB staining indicated that YY-20394 and
ABT199 alone significantly promoted apoptosis in MV-4-11 cells
compared with the NC group (P<0.001) (Fig. 4A and B). Moreover, the combination
of the two drugs exhibited a higher apoptosis rate than either drug
alone. Flow cytometry analysis further revealed that ABT199
significantly reduced the S phase in MV-4-11 cells (P=0.0006), with
YY-20394 showing a similar trend (P=0.0510) (Fig. 4C and D). By contrast, there were no
significant differences in G1 and G2 phases
between the two drugs alone, in combination or compared with the NC
group (all P>0.05). These findings suggest that YY-20394 and
ABT199 may influence DNA replication in MV-4-11 cells.
YY-20394 and ABT199 synergistically
induce MV-4-11 cell apoptosis, which may be associated with
c-Myc
ABT199 specifically targets Bcl-2. Mcl-1 and Bcl-2
could inhibit apoptosis by binding to the BH3-specific protein Bim,
thereby preventing Bim from activating Bax and Bak (23). Whether YY-20394 and ABT199
synergistically promote apoptosis via the Bcl-2 pathway was
investigated (Fig. 5A-E). The
results revealed changes in the levels of anti-apoptotic proteins
Mcl-1, Bcl-2 and Bcl-xL that were inconsistent with the observed
apoptotic phenotype in MV-4-11 cells (Fig. 5A, C and D). Specifically, Mcl-1
transcription (P=0.001) and protein (P=0.0783) levels were
increased in YY-20394 alone compared with the NC group. Mcl-1
transcription levels decreased while protein levels increased in
the combination group compared with either agent alone.
Furthermore, no inhibitory effects on Bcl-2 or Bcl-xL were observed
at transcription level with ABT199 alone or in combination (all
P>0.05) (Fig. 5B), while
significant upregulations were observed in protein levels
(P<0.05) (Fig. 5E). Bim and Bak
transcription levels increased while protein levels decreased in
the YY-20394 group compared with the NC group. Additionally, levels
of pro-apoptotic factors Bim, Bak and Bax decreased in the
combination group compared with NC or ABT199 alone, which contrasts
with the observed pro-apoptotic phenotype (Fig. 5C-E).
 | Figure 5.Effect of YY-20394 and ABT199 on
apoptotic pathways in MV-4-11 cells. MV-4-11 cells were treated
with 120 nM of ABT199, 30 nM of YY-20394 or a combination of both
for 48 h. (A) The mRNA levels of anti-apoptotic factors Mcl-1,
Bcl-2 and Bcl-xL were assessed using RT-qPCR. (B) The mRNA levels
of pro-apoptotic factors Bim, Bak and Bax were assessed using
RT-qPCR. (C) The protein results of Mcl-1, Bcl-2, Bcl-xL, Bim, Bak
and Bax were assessed using western blotting. (D) The protein
levels of Mcl-1, Bcl-2, and Bcl-xL were semi-quantified using
densitometric analysis. (E) The protein levels of Bim, Bak and Bax
were semi-quantified using densitometric analysis. (F) The protein
results of c-Myc were assessed using western blotting and (G) the
statistical result of densitometric analysis. Data are presented as
mean ± SD from three independent experiments. RT-qPCR, reverse
transcription-quantitative PCR; Mcl-1, myeloid cell leukemia-1;
Bim, Bcl-2 interacting mediator of cell death; Bcl-xL, B-cell
lymphoma-extra large; Bak, Bcl-2 antagonist killer 1; Bax, Bcl-2
associated X. |
Due to the critical role of c-Myc in cancer cell
survival and apoptosis regulation (24), the protein levels of c-Myc were
evaluated. The results demonstrated that the combination treatment
significantly reduced c-Myc protein levels compared with the NC
(P=0.001) and ABT199 alone (P=0.0007) (Fig. 5F and G). Therefore, the synergistic
pro-apoptotic effects of the combined treatment may be associated
with c-Myc suppression.
Synergistic effect of YY-20394 and
ABT199 in MV-4-11 cells is associated with inhibition of p-Akt and
increase of p-ERK
Previous studies have suggested that the effects of
PI3Kδ inhibitors on tumor cell proliferation or apoptotic signaling
may be mediated through the regulation of PI3K/Akt and ERK pathways
(25,26). Whether the synergistic effects of
YY-20394 and ABT199 on MV-4-11 cells are associated with these
pathways was investigated (Fig.
6A). Compared with the NC, p-Akt levels were reduced in both
the YY-20394 alone (P=0.0074) and in combination (P=0.0149)
(Fig. 6B-D). By contrast, p-ERK
levels increased in the ABT199 alone and in combination compared to
the NC (P=0.0253). Additionally, the p-ERK/ERK ratio was higher in
the combination group than in the ABT199 alone (P=0.0291) (Fig. 6E-G).
Discussion
PI3K inhibitors have demonstrated limited clinical
success due to the adverse effects of inhibiting other isoforms,
highlighting the need to develop subtype-specific PI3K inhibitors
(27). PI3Kδ is commonly expressed
in most AML cells (21), making it
a therapeutically relevant target. YY-20394, a highly selective
PI3Kδ inhibitor, has demonstrated efficacy in various hematologic
malignancies (28). However, its
role in AML remains unclear. The present study demonstrated that
YY-20394 could inhibit cell viability in MV-4-11, THP-1 and U937
cells, consistent with the effects of other PI3Kδ inhibitors
(29,30). Although U937 cells exhibited the
lowest sensitivity, YY-20394 suppressed their proliferation,
promoted apoptosis, and induced G1/S and early S phase
arrest in a concentration-dependent manner. These effects
contribute to its antitumor activity in AML cells.
Due to the issue of acquired resistance to ABT199,
it is often not recommended as a monotherapy for AML (20). The present findings demonstrate that
the combination of YY-20394 and ABT199 exerts synergistic effects
in MV-4-11 cells, reducing cell survival and enhancing apoptosis
more effectively than either agent alone. MV-4-11 cells are an AML
cell line positive for FLT3-ITDs, a common driver mutation
associated with poor prognosis and present in ~25% of AML cases
(31,32). While ABT199 monotherapy has limited
efficacy in FLT3-ITD-mutated AML (33), the combination of ABT199 and PI3Kδ
inhibitors has shown promise in preclinical studies (21,34).
The present study supports previous research, suggesting that
FLT3-ITD status may be associated with increased sensitivity to
this combination strategy. Due to clonal variability among
different FLT3-ITD-positive AML cell lines, the broader
applicability of these findings should be further validated in
additional models, such as MOLM-13 and MOLM-14.
Changes in apoptosis-related molecules were explored
following treatment with YY-20394 and ABT199. The results showed
inconsistent changes between the transcriptional and protein levels
of several apoptosis-related molecules in the combination group,
suggesting the involvement of post-transcriptional regulation
(35). For example, A-1210477 has
been reported to disrupt the Mcl-1-Bim complex while stabilizing
Mcl-1 protein, thereby promoting Mcl-1 accumulation independently
of transcription (36). Consistent
with the findings of Yao et al (21), inhibition of Bcl-2 or Bcl-xL by
either ABT199 alone or in combination was not observed. Instead,
significantly increased protein levels of Mcl-1, Bcl-2 and Bcl-xL
were observed. ABT199 primarily functions as a BH3 mimetic that
binds to Bcl-2 and inhibits its functional activity, rather than
reducing its transcription or protein abundance. One possible
explanation for this increase is activation of compensatory
survival signaling pathways in parallel cascades (37,38),
which may be associated with the increased p-ERK protein levels
observed in the present study. A previous study has shown that
ABT199 can induce compensatory activation of ERK1/2 and
subsequently promote downstream Mcl-1 expression, a phenomenon
frequently observed in FLT3-ITD AML cells (39). Meanwhile, PI3K inhibition has also
been reported to induce compensatory ERK activation through the
RAS/RAF/MEK/ERK pathway (38). ERK
activation has been shown to maintain cellular homeostasis by
regulating cell cycle-associated pathways (such as cyclin D1) and
promoting anti-apoptotic signaling (such as Mcl-1) (40). Previous research showed that such
adaptive signaling may enable cancer cells to tolerate therapeutic
stress and potentially contribute to treatment resistance (41). Therefore, future studies could
explore whether MEK or ERK inhibition may further enhance the
synergistic pro-apoptotic effects of YY-20394 and ABT199.
In addition, although the transcriptional levels of
the pro-apoptotic proteins Bim, Bak and Bax did not change
significantly in the combination group, their protein levels were
markedly decreased. We hypothesize that there may be two possible
explanations. First, Bax and Bak are terminal effectors of
mitochondrial apoptosis, and the reduction in total protein levels
may reflect extensive apoptosis (42). Further studies are needed to
investigate the expression of other BH3-only proteins (such as p53
upregulated modulator of apoptosis and
phorbol-12-myristate-13-acetate-induced protein 1), as well as to
assess Bax/Bak conformational changes or mitochondrial
translocation to determine whether this decrease is secondary to
apoptosis activation. Second, alternative cell death-related
mechanisms may also contribute to this effect, including
stress-induced apoptosis such as endoplasmic reticulum stress,
caspase-independent apoptosis mediated by AIF and Endo G, death
receptor-mediated extrinsic apoptosis, or even non-apoptotic
programmed cell death pathways such as necroptosis (43).
c-Myc is known to be highly expressed in AML and is
associated with poor prognosis and therapeutic resistance (44,45).
Downregulation of c-Myc has been shown to enhance the activity of
ABT199 more effectively than Mcl-1 inhibition (46). Compared with treatment with the
PI3K/HDAC inhibitor CUDC-907 alone, its combination with ABT-199
did not result in further downregulation of c-Myc (47,48).
Nevertheless, c-Myc served a critical role in the synergistic
effect of CUDC-907 and ABT-199, as the combination caused marked
dysregulation of MYC target genes, which may contribute to AML cell
apoptosis through regulation of mitochondrial function and
induction of DNA damage. Consistent with the present results, the
PI3K/HDAC inhibitor CUDC-907 synergistically induces apoptosis in
AML cells in combination with ABT199, partially through c-Myc
inhibition (47). Furthermore,
preclinical research has reported that FLT3-ITD can specifically
activate c-Myc through the PI3K/Akt signaling pathway (49). Based on these findings, the
combinatory effects of YY-20394 and ABT199 may be linked to c-Myc
downregulation.
Studies have suggested that the in vitro
potency of drugs for hematological malignancies is more comparable
to the average clinical exposure concentration (C-unbound,
average), and such direct comparisons may better reflect clinical
translational potential than other cancers (50). Pharmacokinetic studies in B cell
malignancies have shown that both single-dose (20–140 mg) and
multiple-dose (20–200 mg) administration of YY-20394 resulted in
dose-dependent increases in drug exposure parameters [such as
Cmax, area under the curve (AUC)0-t and
AUC0-∞] (11). For
ABT199, the reported Cmax under the standard 400 mg QD
regimen is ~2.1 µg/ml (51).
However, although higher drug exposure is generally associated with
improved clinical response, it may also increase the risk of
adverse events (52). Consistent
with previous reports, the present results demonstrated substantial
heterogeneity in ABT199 sensitivity among different AML cell lines
(17,53). However, due to the clinically
achievable unbound drug exposure, the relatively high
IC50 values observed in U937 and THP-1 cells may suggest
limited efficacy of YY-20394 or ABT199 monotherapy in these
clinical subtypes (54). Notably,
the 48 h IC50 values were 2-10-fold lower than those at
24 h, indicating that prolonged drug exposure may enhance
antileukemic activity. Therefore, optimization of dosing schedules
to maintain effective exposure may help broaden the therapeutic
window and improve translational potential.
The present study has several limitations. First,
all experiments were conducted in vitro using a limited
panel of AML cell lines, which may not fully reflect the biological
heterogeneity of AML in patients. Second, the mechanistic findings
regarding the involvement of the c-Myc/Akt and ERK pathways are
primarily correlative and require further functional validation.
The precise contribution of Bcl-2 family proteins to the
synergistic pro-apoptotic effects remains incompletely understood.
Further studies are needed to investigate the functional status of
these proteins, including their conformational activation,
mitochondrial translocation and post-translational modifications.
Fourth, no in vivo studies or primary patient-derived AML
samples were included to evaluate the therapeutic efficacy and
translational relevance of YY-20394 alone or in combination with
ABT199. Future studies incorporating animal models, primary AML
samples and more comprehensive mechanistic investigations will be
necessary to further validate and extend the present findings.
An emerging strategy to enhance the therapeutic
index of targeted agents in AML is nanocarrier based delivery.
Nanoparticle formulations can improve pharmacokinetics, enable
co-delivery of synergistic drug pairs, enhance tumor/bone marrow
targeting and reduce off-target hematologic toxicity. Recent
advances in nanoparticle design, including zein-based carriers for
in vivo antitumor delivery (55), antibody functionalized lipid
nanocarriers for targeted RNA/drug delivery (56) and next-generation lipid nanocarriers
enabling novel administration routes (57), illustrate the translational
potential of these platforms. In the context of YY-20394 and
venetoclax, a nanocarrier approach could allow lower systemic doses
while maintaining effective intratumoral concentrations, facilitate
synchronized drug exposure and potentially overcome
microenvironment mediated resistance. Preclinical evaluation of
such co-delivery formulations in FLT3 ITD AML models would
therefore be a promising translational step.
In conclusion, YY-20394 exhibits potential
growth-inhibitory effects across different AML cell lines, making
it a promising therapeutic candidate for AML. Moreover, the
combination of YY-20394 and ABT199 demonstrates synergistic
antitumor activity in MV-4-11 cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by the Health Research Program of
Anhui (grant no. AHWJ2023BAc10019 to YG).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YG, WW and YY conceived of and designed the study,
contributed to manuscript drafting and revised the manuscript. JC
and YL collected and curated data. LZ, QL and JS provided materials
and samples. YG, LZ, QL and JS performed analysis and
interpretation of data and conducted statistical analysis. WW and
YY confirm the authenticity of all raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DiNardo CD, Erba HP, Freeman SD and Wei
AH: Acute myeloid leukaemia. Lancet. 401:2073–2086. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Juliusson G, Hagberg O, Lazarevic VL,
Ölander E, Antunovic P, Cammenga J, Wennström L, Möllgård L, Brune
M, Jädersten M, et al: Improved survival of men 50 to 75 years old
with acute myeloid leukemia over a 20-year period. Blood.
134:1558–1561. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song Y, Jia Y, Zhang G, Wei S, Li Y, Hu Y,
Hao Q, Wang Z, Fang Q, Tian Z, et al: Machine leaning algorithm on
chemotherapeutic drug resistance related gene classifier in acute
myeloid leukemia. Blood. 136:28–29. 2020. View Article : Google Scholar
|
|
4
|
Ma YR, Zhao T, Ma L, Hu LJ, Duan WB, Jiang
H, Huang XJ and Jiang Q: Variables associated with hematological
remission and survival in patients with acute myeloid leukemia
after induction failure and relapse. Zhonghua Xue Ye Xue Za Zhi.
43:644–650. 2022.(In Chinese). PubMed/NCBI
|
|
5
|
Wysota M, Konopleva M and Mitchell S:
Novel therapeutic targets in acute myeloid leukemia (AML). Curr
Oncol Rep. 26:409–420. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bazinet A and Kantarjian HM: Moving toward
individualized target-based therapies in acute myeloid leukemia.
Ann Oncol. 34:141–151. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nepstad I, Hatfield KJ, Grønningsæter IS
and Reikvam H: The PI3K-Akt-mTOR signaling pathway in human acute
myeloid leukemia (AML) cells. Int J Mol Sci. 21:29072020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng Y, Cu X and Xin M: PI3Kδ inhibitors
for the treatment of cancer: A patent review (2015-present). Expert
Opin Ther Pat. 29:925–941. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang Y, Zheng Y, Hu X, Zhao H and Cui S:
Discovery of potent and selective PI3Kδ inhibitors for the
treatment of acute myeloid leukemia. J Med Chem. 67:6638–6657.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang X, Li F, Chen C, Jiang Z, Wang A,
Liu X, Ge J, Hu Z, Yu K, Wang W, et al: Discovery of
(S)-2-amino-N-(5-(6-chloro-
5-(3-methylphenylsulfonamido)pyridin-3-yl)-4-methylthiazol-2-
yl)-3-methylbutanamide (CHMFL-PI3KD-317) as a potent and selective
phosphoinositide 3-kinase delta (PI3Kδ) inhibitor. Eur J Med Chem.
156:831–846. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang B, Qi J, Song Y, Li Z, Tu M, Ping L,
Liu Z, Bao H, Xu Z and Qiu L: Phase 1 clinical trial of the PI3Kδ
inhibitor YY-20394 in patients with B-cell hematological
malignancies. J Hematol Oncol. 14:1302021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin J, Cen H, Zhou K, Xu X, Li F, Wu T,
Yang H, Wang Z, Li Z, Huang W, et al: A Phase Ib study of
linperlisib in the treatment of patients with relapsed and/or
refractory peripheral T-cell lymphoma. Clin Cancer Res.
30:4593–4600. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang T, Sun X, Qiu L, Su H, Cao J, Li Z,
Song Y, Zhang L, Li D, Wu H, et al: The oral PI3Kδ inhibitor
linperlisib for the treatment of relapsed and/or refractory
follicular lymphoma: A phase II, Single-Arm, Open-label clinical
trial. Clin Cancer Res. 29:1440–1449. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu J, Lu AD, Zhang LP, Zuo YX and Jia YP:
Study of clinical outcome and prognosis in pediatric core binding
factor-acute myeloid leukemia. Zhonghua Xue Ye Xue Za Zhi.
40:52–57. 2019.(In Chinese). PubMed/NCBI
|
|
15
|
Wu D, Li M, Hong Y, Jin L, Liu Q, Sun C,
Li L, Han X, Deng S, Feng Y, et al: Integrated stress response
activation induced by usnic acid alleviates BCL-2 inhibitor ABT-199
resistance in acute myeloid leukemia. J Adv Res. 74:621–635. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Chen Y, Yu L and Yang L: Mechanisms
of venetoclax resistance and solutions. Front Oncol.
12:10056592022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu H, Hussain Z, Xie Q, Yan X, Zeng C,
Zhou G and Cao S: Targeting PI3K/AKT/mTOR pathway to enhance the
anti-leukemia efficacy of venetoclax. Exp Cell Res. 417:1131922022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang K, Huang Y, Duan Y, Guo B, Ke Q,
Liao C and Cen H: Anti-tumor effect of PI3K inhibitor Linperlisib
on diffuse large B-cell lymphoma in vitro. Chin J Oncol Prev Treat.
16:56–61. 2024.(In Chinese).
|
|
19
|
Alkhatabi HA, Zohny SF, Shait Mohammed MR,
Choudhry H, Rehan M, Ahmad A, Ahmed F and Khan MI:
Venetoclax-resistant MV4-11 leukemic cells activate PI3K/AKT
pathway for metabolic reprogramming and redox adaptation for
survival. Antioxidants (Basel). 11:4612022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ren WX, Guo H, Lin SY, Chen SY, Long YY,
Xu LY, Wu D, Cao YL, Qu J, Yang BL, et al: Targeting cytohesin-1
suppresses acute myeloid leukemia progression and overcomes
resistance to ABT-199. Acta Pharmacol Sin. 45:180–192. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao MY, Wang YF, Zhao Y, Ling LJ, He Y,
Wen J, Zheng MY, Jiang HL and Xie CY: BCL-2 inhibitor synergizes
with PI3Kδ inhibitor and overcomes FLT3 inhibitor resistance in
acute myeloid leukaemia. Am J Cancer Res. 12:3829–3842.
2022.PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vela L, Gonzalo O, Naval J and Marzo I:
Direct interaction of Bax and Bak proteins with Bcl-2 homology
domain 3 (BH3)-only proteins in living cells revealed by
fluorescence complementation. J Biol Chem. 288:4935–4946. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao J, Wu S, Wang D, Edwards H, Thibodeau
J, Kim S, Stemmer P, Wang G, Jin J, Savasan S, et al: Panobinostat
sensitizes AraC-resistant AML cells to the combination of
azacitidine and venetoclax. Biochem Pharmacol. 228:1160652024.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Zhang X, Li BS, Zhai X, Yang Z,
Ding LX, Wang H, Liang C, Zhu W, Ding J and Meng LH: Simultaneous
targeting of PI3Kδ and a PI3Kδ-dependent MEK1/2-Erk1/2 pathway for
therapy in pediatric B-cell acute lymphoblastic leukemia.
Oncotarget. 5:10732–10744. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie C, He Y, Zhen M, Wang Y, Xu Y and Lou
L: Puquitinib, a novel orally available PI3Kδ inhibitor, exhibits
potent antitumor efficacy against acute myeloid leukemia. Cancer
Sci. 108:1476–1484. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiang Q, Dong S and Li XH: A Review of
phosphocreatine 3 kinase δ subtype (PI3Kδ) and its inhibitors in
malignancy. Med Sci Monit. 27:e9327722021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zou L, Qi Y, Tang L, Du Y, Xiang M, Chen
X, Ma J and Yang Z: Clinical review considerations of class I PI3K
inhibitors in hematolymphatic malignancies by Center for Drug
Evaluation. Chin J Cancer Res. 34:415–421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Y, Wu T, Yang C, Lu M, Chen Z, Deng
M, Jia Y, Yang Y, Liu X, Wang H, et al: A pyridinesulfonamide
derivative FD268 suppresses cell proliferation and induces
apoptosis via inhibiting PI3K pathway in acute myeloid leukemia.
PLoS One. 17:e02778932022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang C, Gong Y, Gao Y, Deng M, Liu X, Yang
Y, Ling Y, Jia Y and Zhou Y: Design, synthesis and in vitro
biological evaluation of 2-aminopyridine derivatives as novel PI3Kδ
inhibitors for hematological cancer. Bioorg Med Chem Lett.
82:1291522023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song G, Valdez BC, Li Y, Liu Y, Champlin
RE and Andersson BS: Synergistic cytotoxicity of sorafenib with
busulfan and nucleoside analogs in human FMS-like tyrosine kinase 3
internal tandem duplications-positive acute myeloid leukemia cells.
Biol Blood Marrow Transplant. 20:1687–1695. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Daver N, Schlenk RF, Russell NH and Levis
MJ: Targeting FLT3 mutations in AML: Review of current knowledge
and evidence. Leukemia. 33:299–312. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Konopleva M, Pollyea DA, Potluri J, Chyla
B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, et
al: Efficacy and biological correlates of response in a phase II
study of venetoclax monotherapy in patients with acute myelogenous
leukemia. Cancer Discov. 6:1106–1117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Darici S, Alkhaldi H, Horne G, Jørgensen
HG, Marmiroli S and Huang X: Targeting PI3K/Akt/mTOR in AML:
Rationale and clinical evidence. J Clin Med. 9:29342020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cui J and Placzek WJ: Post-Transcriptional
regulation of Anti-apoptotic BCL2 family members. Int J Mol Sci.
19:3082018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leverson JD, Zhang H, Chen J, Tahir SK,
Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, et al: Potent
and selective small-molecule MCL-1 inhibitors demonstrate on-target
cancer cell killing activity as single agents and in combination
with ABT-263 (navitoclax). Cell Death Dis. 6:e15902015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Piddock RE, Bowles KM and Rushworth SA:
The role of PI3K isoforms in regulating bone marrow
microenvironment signaling focusing on acute myeloid leukemia and
multiple myeloma. Cancers (Basel). 9:292017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Serra V, Scaltriti M, Prudkin L, Eichhorn
PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M,
Rodriguez S, et al: PI3K inhibition results in enhanced HER
signaling and acquired ERK dependency in HER2-overexpressing breast
cancer. Oncogene. 30:2547–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kiyoi H, Kawashima N and Ishikawa Y: FLT3
mutations in acute myeloid leukemia: Therapeutic paradigm beyond
inhibitor development. Cancer Sci. 111:312–322. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lu Y, Liu B, Liu Y, Yu X and Cheng G: Dual
effects of active ERK in cancer: A potential target for enhancing
radiosensitivity (Review). Oncol Lett. 20:993–1000. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ondrisova L and Mraz M: Genetic and
Non-Genetic mechanisms of resistance to BCR signaling inhibitors in
B cell malignancies. Front Oncol. 10:5915772020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wolf P, Schoeniger A and Edlich F:
Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial
membrane. Biochim Biophys Acta Mol Cell Res. 1869:1193172022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mustafa M, Ahmad R, Tantry IQ, Ahmad W,
Siddiqui S, Alam M, Abbas K, Moinuddi n, Hassan MI, Habib S and
Islam S: Apoptosis: A comprehensive overview of signaling pathways,
morphological changes, and physiological significance and
therapeutic implications. Cells. 13:18382024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ohanian M, Rozovski U, Kanagal-Shamanna R,
Abruzzo LV, Loghavi S, Kadia T, Futreal A, Bhalla K, Zuo Z, Huh YO,
et al: MYC protein expression is an important prognostic factor in
acute myeloid leukemia. Leuk Lymphoma. 60:37–48. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gu K, May HA and Kang MH: Targeting
molecular signaling pathways and cytokine responses to modulate
c-MYC in acute myeloid leukemia. Front Biosci (Schol Ed).
16:152024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Carter JL, Hege K, Yang J, Kalpage HA, Su
Y, Edwards H, Hüttemann M, Taub JW and Ge Y: Targeting multiple
signaling pathways: The new approach to acute myeloid leukemia
therapy. Signal Transduct Target Ther. 5:2882020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hege Hurrish K, Qiao X, Li X, Su Y, Carter
J, Ma J, Kalpage HA, Hüttemann M, Edwards H, Wang G, et al:
Co-targeting of HDAC, PI3K, and Bcl-2 results in metabolic and
transcriptional reprogramming and decreased mitochondrial function
in acute myeloid leukemia. Biochem Pharmacol. 205:1152832022.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li X, Su Y, Hege K, Madlambayan G, Edwards
H, Knight T, Polin L, Kushner J, Dzinic SH, White K, et al: The
HDAC and PI3K dual inhibitor CUDC-907 synergistically enhances the
antileukemic activity of venetoclax in preclinical models of acute
myeloid leukemia. Haematologica. 106:1262–1277. 2021.PubMed/NCBI
|
|
49
|
Basit F, Andersson M and Hultquist A: The
Myc/Max/Mxd network is a target of mutated Flt3 signaling in
hematopoietic stem cells in Flt3-ITD-Induced myeloproliferative
disease. Stem Cells Int. 2018:32869492018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kotani N and Ito K: Translatability of in
vitro potency to clinical efficacious exposure: A retrospective
analysis of FDA-approved targeted small molecule oncology drugs.
Clin Transl Sci. 16:1359–1368. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Salem AH and Menon RM: Clinical
pharmacokinetics and pharmacodynamics of venetoclax, a selective
B-cell lymphoma-2 inhibitor. Clin Transl Sci. 17:e138072024.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kobayashi T, Sato H, Miura M, Fukushi Y,
Kuroki W, Ito F, Teshima K, Watanabe A, Fujishima N, Kobayashi I,
et al: Overexposure to venetoclax is associated with
prolonged-duration of neutropenia during venetoclax and azacitidine
therapy in Japanese patients with acute myeloid leukemia. Cancer
Chemother Pharmacol. 94:285–296. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuusanmäki H, Kytölä S, Vänttinen I,
Ruokoranta T, Ranta A, Huuhtanen J, Suvela M, Parsons A, Holopainen
A, Partanen A, et al: Ex vivo venetoclax sensitivity testing
predicts treatment response in acute myeloid leukemia.
Haematologica. 108:1768–1781. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Graham GG and Scott KF: Limitations of
drug concentrations used in cell culture studies for understanding
clinical responses of NSAIDs. Inflammopharmacology. 29:1261–1278.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Abosalim HM, El-Moselhy TF, Sharafeldin N,
Giovannuzzi S, Begines P, Nafie MS, Fahmy SA, Diab MK, Babker A,
Supuran CT, et al: Innovative design and synthesis of dual-acting
hCA IX/CDK-2 inhibitors through hetero ring fused pyrimidine
utilization for cutting-edge anticancer therapy: Zein nanoparticles
for in vivo lung cancer treatment. Bioorg Chem. 166:1090572025.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Nabih NW, Hassan H, Preis E, Schaefer J,
Babker A, Abbas AM, Amin MU, Bakowsky U and Fahmy SA:
Antibody-functionalized lipid nanocarriers for RNA-based cancer
gene therapy: Advances and challenges in targeted delivery.
Nanoscale Adv. 7:5905–5931. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wafik Nabih N, Nafie MS, Babker A, Alameen
AAM and Fahmy SA: Next-generation lipid nanocarriers for
Parkinson's therapy: Nose-to-brain innovations and clinical
prospects. Nanoscale. 17:27826–27848. 2025. View Article : Google Scholar : PubMed/NCBI
|