Introduction
What is the pathogenesis of neuroblastoma in
childhood? How can the aggressiveness of high-risk neuroblastomas
be interpreted? What is the contribution of viruses and viral
infections in the pathogenesis of neuroblastoma in childhood? With
what means is research trying to investigate the hypothesis of
viral involvement in the pathogenesis of neuroblastoma in
childhood? Will this hypothesis finally be proven false or true?
What is the value of modelling in virology? What is the role of
modelling in current and future cancer research? Professor Ugo
Rovigatti, Professor of Molecular Biology in the Department of
Experimental and Clinical Medicine at the University of Florence in
Florence, Italy, tried to answer these questions during a webinar
on neuroblastoma in children, which was organised virtually on
December 12, 2020, by the Paediatric Virology Study Group (PVSG) of
the Institute of Paediatric Virology based on the island of Euboea
in Greece. Professor Ugo Rovigatti's CV is available at https://spandidos-publications.com/COVER_LEGENDS/ijo_58_3_cover_legend.pdf.
Questions and answers
Question:
First of all, Professor Ugo Rovigatti, thank you for
your feedback and your wishes regarding the foundation of the
Institute of Paediatric Virology on the island of Euboea in Greece.
What is neuroblastoma, what is its incidence in childhood and how
is it staged?
Answer:
Although neuroblastoma is a rare disease affecting
~10.5 children per million of total children aged 1-15 years, it is
the most common solid tumour of early childhood, i.e., children of
pre-schooling age. Therefore, it is only the sixth among paediatric
tumours in terms of its frequency, but in view of the
aggressiveness of its high-risk form, it causes a
disproportionately high lethality, ~15% among paediatric cancers.
The reason is the fact that high-risk neuroblastoma is in a greater
part untreatable, although major progresses were recently made
therapeutically. I will come back to this, but to give a general
overview, it is as if there are two diseases. One is rather benign,
where treatment gives good prognoses. There is also the early
childhood form IV-S, before 1.5 years of age, which - although
apparently aggressive and metastatic to several sites -
spontaneously regresses, often without clinical intervention, i.e.,
without radio-/chemo-therapy. However, the more severe form in
older children is very difficult to cure despite aggressive
treatments and even autologous hematopoietic stem cell
transplantation, treatments which, however, have been associated
with survival improvements. Some recent success seems to be
associated with so-called immunotherapy (IMT), in particular the
usage of monoclonal antibodies, which target a glycosphingolipid:
GD2. In this time-line and perspective of therapeutic improvements,
the 2009 International Neuroblastoma Risk Group (INRG) Staging
System (INRGSS) article was certainly a milestone (1), in the sense that i) it allowed the
achievement of an internationally standardized diagnostic
stratification of neuroblastoma cases; ii) most of all, the staging
system was performed pre-operatively, differently from previous
staging [International Neuroblastoma Staging System (INSS)], thus
allowing much better interventions; and iii) an important impact in
this classification is provided by the so-called image defined risk
factors, which allow differentiation for example between stages L1
and L2. Imaging specialists and radiologists, therefore, hold key
role in this new classification. Needless to say, INRGSS also needs
and will be improved in the near future as different staging
systems, such as the survival-tree regression (STR), and the least
absolute shrinkage and selection operator (LASSO), are being
compared by the Children's Oncology Group and other international
organizations (2).
Question:
How does the prognosis of high-risk neuroblastoma
cases differ compared to other neuroblastoma cases or children with
other neurogenic origin tumours?
Answer:
I already alluded to the dramatic difference in
terms of prognosis between high- and low-risk neuroblastomas. The
question is very appropriate in the sense that it deals with the
central-core of the neuroblastoma enigma and could also be
summarized as: ‘Why cells, which morphologically appear
undistinguishable, as malignant small blue-round cells, behave so
differently in terms of prognosis?’ And this question intersects
with our basic understanding of neuroblastoma, in the sense that
the first molecular biology discoveries in the field certainly shed
some light on this enigma. In particular, the seminal paper by
Manfred Schwab working in Mike Bishop and Harold Varmus lab
discovering the MYCN amplification (MYCNA) in high-risk
neuroblastoma, but not in its less aggressive forms, set the stage
for this paediatric tumour present and future understanding
(3). Furthermore, being this one
of the first genetic aberrations discovered in cancer cells, it
pioneered all future work in molecular oncology. We recently
published an overview perspective on these issues: The shift from
our cancer understanding in terms of ‘clonal outgrowth’, basis of
radio-/chemo-therapy approaches to that of ‘cancer gene network’,
basis of targeted gene therapies (TGTs), starts from discoveries
such as the MYCNA in neuroblastoma (4). In 1984, Brodeur et al
(5) initially demonstrated MYCNA
as a marker of high-risk neuroblastoma and then several hundred
clinical studies confirmed this as a hallmark of ominous prognosis
(6). To conclude with
neuroblastoma prognosis, this can be excellent in low-stage disease
with a progression-free survival (PFS) and overall survival (OS) of
≥90%. However, in high-risk neuroblastomas, prognosis is only ~40%
and in cases of recurrent or relapsing neuroblastomas, this figure
goes down to 10%.
Question:
The variation of clinical outcomes in neuroblastoma
cases indicates distinct genetic and environmental factors
affecting the development of this malignancy. Could you describe to
us the main factors involved in its pathogenesis?
Answer:
This impinges again on the enigmatic nature of
neuroblastoma. Therefore, we can just be speculative and try to
summarize a plethora of papers and investigations on this issue.
Epidemiological work for decades could not identify among several
potential carcinogens, pollutants and specific behaviours, one or
some that could act as specific factors associated with its onset.
Genetic and pedigree studies have also identified congenital
mutations, which appear to be associated with rarer familial forms
(7). However, these are the
iceberg tip of neuroblastoma cases, which are mostly not-congenital
and the aggressive ones are diagnosed after 18 months of age. Among
genetic factors, MYCNA is certainly the most prominent, since it
occurs in approximately one quarter of cases. I already described
the important breakthrough of Schwab et al (3) of MYCNA in high-risk neuroblastoma.
However, the problem of science - or its beauty depending on
different opinions - is that as soon as you solve one important and
interesting question, another or several ones pop out like
mushrooms. MYCNA origin for example is difficult to be explained in
biological and molecular terms. The genomic aberration appears to
be sometimes ‘huge’. For example, we have documented one high-risk
neuroblastoma case, in which MYCN gene was amplified ~1,000 times.
This means that the whole region of MYCN (called amplicon,
typically large = one million base pairs - 1 MB - or even more) is
amplified 1,000 times: In our genome of just >3 GB, this means
that one third of this cell genome is totally aberrant (8). This is certainly difficult to explain
in terms of random events, such as ephemeral point mutations,
casually happening during DNA replications (9). It also strongly points towards
natural selection for such a catastrophic event (10). For its origin, we have proposed a
model based upon the micro-foci inducing virus (MFV) infection,
since we can experimentally show that MFV infection of normal cells
initially MYCN diploid modifies them into cells, which are
transformed, tumorigenic and with higher MYCNA (100X). That
epigenetic factors also play a relevant role in neuroblastoma was
initially documented by induction of their differentiation with
retinoic acid in the study by Thiele et al (11). Several groups, including ours, have
also documented the possible phenotypic transition/transformation
of mesenchymal cells with stemness markers, into neuroectodermal
clones sometimes with transformed cell features (12-14)
and the group of Rogier Versteeg tentatively associated some of
these phenotypes with a network of transcription factors (15). Additional alterations in telomerase
or other epigenetics involved genes have also been described in
high-risk neuroblastoma (16).
However, in view of genetic elements variations, including
mutations, deletions, amplifications, segmental chromosomal
alterations etc., as well as alterations of epigenetic elements,
the overall picture still looks as an unsolved puzzle (16). Using the science of logics that was
born in Greece almost 2.5 thousand years ago, we are missing the
understanding of the first mover. Aristotle, in his book
‘Metaphysics’, used to talk about ‘the unmoved mover’ (ὃ οὐ
κινούμενον κινεῖ). The alternative explanation of alterations
occurring randomly and cumulatively is extremely unlikely, also in
view of the patients' very young age (9,10).
Questions:
What are the genomic landscapes and how are they
involved in the pathogenesis of high-risk neuroblastoma?
Answer:
Genomic landscapes in neuroblastoma have been
extensively studied, starting from the mentioned MYCNA present in
20-25% of cases. Some associations have been clearly identified.
For example, MYCNA is typically associated with chromosome 1p
deletions and 17q gains, while in another fraction of tumours the
association is between 11q deletions, the alpha thalassaemia mental
retardation X-linked mutations and associated death-associated
protein 6 loss: The so-called telomere maintenance mechanism or
alternative lengthening of telomeres (16). However, the definition of such
alterations has been often elusive. For example, at the beginning
of the 1990s, a real hunt had started for the identification of the
gene or genes, which are present in the chromosome 1p region, which
is often lost in high-risk neuroblastomas, generally with MYCNA.
Therefore, this could be most likely explained by the loss of a
tumour suppressor gene (TSG). Despite several publications, efforts
and competitions between the major groups working on both sides of
the Atlantic, as well as in Japan, the 1p TSG has not materialized
so far, although some interesting candidates were proposed
(6). The neuroblastoma field is
more realistically now directing its attention towards
understanding how such genetic/genomic landscapes are generated and
most of all why. In fact, although some of the described
alterations are also targetable by TGT, mutations in high-risk
neuroblastomas appear to be so numerous and with relatively low
frequency to impede utilization by so called precision medicine
(16,17). This becomes today a general theme
of cancer genetics and genomics, that we have recently addressed in
two publications (4,10). In other words, the idea of
targeting mutations specifically present in a particular type of
cancer - although good in theory - is invalided by tumour
heterogeneity (TH), a concept which is now becoming more recognized
in cancer biology and molecular biology (4,16).
The other essential concept to explain TH is the
Darwinian selection. Cancer cells are extremely prone to change
their genetic make-up, in order to respond to environmental
selection, such as chemotherapy or TGT (10). In high-risk neuroblastomas, not
only inter-TH, but especially intra-TH plays an essential role, as
mutations in the RAS-MAPK pathway often appear in relapses
(18). In these cases, therefore,
mutations more than an essential role by themselves appear to have
a consequential role. They seem to be a consequence of the
Darwinian selection (10). This
may also be a more general problem of cancer genomic landscapes and
obviously the important question then arises: Which are the
Darwinian selection forces or factors behind (4)? In this respect, it may be instructive
to read again the article by Gatenby et al (19).
Question:
How frequent are cancer clusters of neuroblastoma
cases in specific geographic areas over a period of time and how
could they be related to the pathogenesis of neuroblastoma?
Answer:
Clusters of neuroblastoma cases are rather rare for
at least two reasons. Firstly, neuroblastoma is a rare childhood
malignancy, with a frequency ~50 times lower than paediatric
leukaemia. Secondly, although leukaemia clusters have been
described in several instances, less is known about neuroblastoma
clusters. Recently, neuroblastoma space time clusters have been
reported in the USA, Argentina and Spain (16). Another report from the UK described
several instances of neuroblastoma time-clusters (20). For these researchers, such temporal
clusters, detected by studying 227 cases during a period of 44
years, appear to be ‘mini-epidemics’ that happen often and in
several different locations (20).
The reason of the appearance of cancer-clusters, especially in
children, has been often and heatedly debated. They seem to emerge
in children rather than in adults, in view of the immune system,
which is still underdeveloped earlier in life (21). However, such clusters are
especially evident and have been more extensively studied in
paediatric leukaemia, which is more frequent. In such situations,
childhood leukaemia incidence is, for a limited time, several folds
higher than national and international average (21). Most debated hypotheses for
leukaemia clusters are the ‘population-mixing’ and the ‘delayed
infection’, respectively by Kinlen (22) and Greaves (23). The first one hypothesizes that an
infectious agent is carried by an exposed population - typically
from densely inhabited areas. Isolated and unexposed groups will
lack herd immunity against such an agent. Therefore, the
immigration of exposed populations towards more isolated regions
will generate such mini-epidemics or leukaemia clusters (22). The second hypothesis is more
general and vague. It suggests a higher risk for segregated
childhood populations, for example, groups with higher income, but
it does not clarify whether one specific or several possible
infections may cause the trigger; the second hypothesis is
privileged by Greaves (23).
However, both hypotheses agree on the presence of and causality by
an infectious agent, either one X-virus according to Kinlen
(22) or any possible virus - but
also other agents such as bacteria - according to Greaves (23). Our findings on the MFV associated
to a neuroblastoma cluster is in agreement with the hypothesis of
Kinlen (22), although it could
also be an example of delayed infection. However, the MFV model
more clearly explains how and why genetic/genomic aberrations are
generated in cancer cells.
Question:
The viral aetiology of neuroblastomas has been
proposed very early, almost 50 years ago, but it has not been
proven. Which oncogenic or non-oncogenic viruses have been detected
in cases with neuroblastoma up to now? How do you explain the
presence of these viruses in neuroblastoma cases?
Answer:
The fact that neuroblastic tumours may be associated
with previous viral infection has been discussed throughout the
years, starting from Robert Bolande's definition of
neurochristopathies and their associated tumours 50 years ago
(24). However, it should be
clarified that there are at least three different types of studies
and approaches to this problem. Firstly, certain laboratories, for
example, have just detected infections, which are present in
neuroblastoma patients. This has been even utilized to
neuroblastoma patients' advantage. The new chimeric antigen
receptor T-cell technology, which directs T-cells against an
appropriate tumour target, took advantage of neuroblastoma cases
with concomitant Epstein-Barr virus (EBV) infections. By
co-targeting T-cells against EBV capsid antigens and GD2
glycosphingolipid, a much stronger response could be elicited
(25). A few instances of
concomitant infections have been described: EBV, hepatis C virus
(HCV), varicella-zoster virus (VZV), etc.
Secondly, another approach has been taken by
researchers, which hypothesized that a certain family of viruses
could be associated with high-risk neuroblastoma - Herpes family
viruses in one project, Polyoma type viruses, such as BK virus
(BKV) in another, etc. These approaches typically detect some
positivity in a limited percentage of cases. This could be
exemplified by the detection of BKV presence in neuroblastoma cells
obtained by Flaegstad et al >20 years ago (26). Obviously, such studies must then
convince us, the scientific community, that the presence of that
particular virus is significantly higher or more frequent in
neuroblastoma patients. In the case of BKV, differences were rather
slim and the project was eventually dropped (27).
Thirdly, our approach for detecting, isolating and
studying MFV has been completely different. We started by studying
in the laboratory tumours from a cancer-cluster of neuroblastoma
cases in Morgan City (LA, USA) (13). In view of a strong TH, we then
tested the hypothesis that an infectious agent was present by
adding ultra-filtered supernatants to normal cells. This finally
led to electron microscopy studies, which disclosed the presence of
a cytoplasmic virus (13). We have
then employed several experimental systems both in vitro and
in vivo, which document the transforming capability and
tumorigenicity of MFV. Furthermore, the molecular mechanism was
investigated leading to the qualified conclusion that MFV can
induce molecular aberrations similar to those present in the
original tumours, i.e., MYCNA (8,13).
This is particularly compelling, since origin of such genomic
aberrations is an unsolved enigma not only of the molecular biology
of neuroblastoma, but also of cancer cells in general (8,10).
Question:
Recent studies have proposed Zika viral therapy as
an adjunctive treatment for neuroblastoma by targeting tumour cells
that can lead to recurrent disease and treatment failure. We would
like your comment on this possibility.
Answer:
Zika virus has become a health concern in the past
few years, especially in view of their infections during pregnancy,
which can lead to foetal/neonatal microcephaly. This was
particularly alarming in 2016, when a Zika virus epidemic was
spreading throughout Brazil and South America during the past
Olympic Games. The idea to employ now Zika virus against
neuroblastoma or other neuroectodermal tumours, such as
glioblastomas, is based on the Zika virus targeting of neural cells
(28). However, the general idea
of employing viruses for targeting tumours and tumour cells is a
very old one. We were previously discussing about a viral
hypothesis for neuroblastoma aetiology, but the proposal of
employing so-called oncolytic viruses for cancer therapy dates back
at least 70 or even 80 years. There are many examples of
viro-therapy attempts in these 70-80 years: from hepatitis viruses
to EBV, from West-Nile virus (WNV) to adenovirus and more recently:
paramixo-, herpes, picorna- and pox-viruses as well as
enteroviruses. Unfortunately, no real candidate has finally arisen
from these studies, in view of great toxicities and other problems.
For all these reasons, I am rather sceptical about Zika virus
attempts in neuroblastoma or other neuroectodermal malignancies. We
should not forget that malignancies in general, and especially
neuroblastoma, are based upon transformation of stem cell targets.
Zika virus lytically also infects neural stem cells, thus causing
microcephaly of foetuses and neonates. Therefore, one can always
imagine a scenario, in which defective Zika virus particles could
even be associated with transforming/malignant effects. I honestly
believe that excessive manipulations of dangerous pathogens are not
particular useful unless very specific and well demonstrated
instances indicate so. However, there is not so far - despite
experimental studies for >70 years - an acceptable candidate for
oncolytic therapy, especially for high-risk neuroblastomas. A
similar philosophy may be deduced and should be applied on the
extensive research on pathogenic, lethal and pandemic coronaviruses
(or influenza viruses) performed between 2009 and 2019, since it
has not produced so far important treatments, nor was capable of
preventing - as we are all personally and dramatically experiencing
today - one of the worst pandemic outbreaks in decades. We also
tend to forget that a sizeable portion of us, as scientists, with
Simon Wein-Hobson as one of the leading most outspoken figures,
strongly opposed such gain-of-function experiments [Rey et
al (29)]. In conclusion, any
intervention with biological agents should be carefully monitored
and evaluated with the strictest scientific criteria. If there is
no evidence of scientific or clinical advantage, it should be
abandoned.
Question:
Could you further describe to us your MFV model and
its involvement in aggressive neuroblastoma genetic/genomic
aberrations?
Answer:
I have already partially described the MFV model.
So, I will add some further essential elements, which may be
helpful for your PVSG. Firstly, I will analyse the clear presence
of clusters of neuroblastoma cases; secondly, the peculiar aspects
of the experimental animal models and thirdly, the presence of very
high MYCNAs in initial tumour samples and their study in derived
models.
So, let's start with the first. Although rarer,
neuroblastoma clusters have been described in the past in different
continents and regions. In the UK, there is evidence in favour of
temporal clusters, but most of the other instances are based on
space-time clusters (Argentina, Spain; Florida, Louisiana, USA). I
have already briefly described the theories on cancer-cluster
onset, mostly based on childhood leukaemia studies. In our model as
well as in other instances - for example in the extensively studied
clusters of Sellafield (UK) and of Fallon (NE, USA) - presence of a
specific virus [X-virus according to Kinlen (22)] better explains the epidemiological
data. Was the Morgan City cluster isolated? Probably not, as
paediatricians alerted us that excess cases were diagnosed
throughout Southern Louisiana and in the neighbour State of Texas
in 1987-1988, but we were able to study only this isolated cluster
in Morgan City (LA, USA) (13).
Secondly, since the beginning of this isolation and
discoveries, it has been of paramount importance for us to develop
animal as well as cell culture models. The first animal models were
based on rats, while later we evaluated carcinogenesis in nu/nu
mice (13,30). The great interest of rat models was
based on their capability to recapitulate several facets of
paediatric neuroblastomas. Not only neuroblastoma tumours appeared
in all the litters from experimentally infected mothers, but other
aspects, such as the opsoclonus myoclonus syndrome (OMS), ataxia,
the raccoon eyes typical of children with neuroblastoma at
diagnosis, watery diarrhoea due to the vasoactive intestinal
peptide, etc. (13,30).
And thirdly, the dramatic presence of MYCNA at a
level (1,000X amplification) only rarely described was another red
flag for these tumours. Amplification, however, seemed to disappear
when growing tumour cells in tissue culture and rapidly so (after
3-4 passages) (13). In trying to
rationalize what was happening in vitro, it was realized
that two components were present in vitro. Proliferating
cells had a flat, mesenchymal and Schwann-like appearance (S cells)
and grew attached to the bottom of the flasks, while on top of them
micro-foci of small-round-blue cells with neural markers (N cells)
could form. Only N cells of micro-foci displayed high/very high
MYCNA, while S cells remained MYCN diploid (13). This induction of oncogene
amplification has been studied in subsequent years as a model for
the genesis of cancer-specific aberrations, possibly not just for
neuroblastoma (8,30).
Question:
How could this model be used in the understanding of
neuroblastoma pathogenesis? How could this model be involved in the
novel therapies against aggressive neuroblastoma?
Answer:
I will consider these two questions separately and
consequentially. Neuroblastoma is a puzzle made with plenty of tale
stones for which we still lack a solution. Neuroblastoma is also
associated with a number of uncommon if not paradoxical facets,
such as the OMS or racoon eyes syndrome mentioned before, the MYCNA
which is rather frequent being present in 20-25% of cases and the
peculiar behaviour of the intriguing subset of neuroblastoma named
IV-S disease. Our MFV model has the potential to accommodate in a
logical framework most if not all of these aspects. Take the IV-S
disease as an example. Since the seminal study by D'Angio et
al (31), we know that
neuroblastoma in neonates or very young children can also appear as
a widespread and metastatic disease - throughout the body, in
blood, in bone marrow but not inside bones - and yet suddenly and
spontaneously regresses before 18 months of age (the threshold was
considered before at 1 year). This phenomenon is so dramatic and
reproducible that clinicians typically wait to treat IV-S patients
with radio-/chemo-therapy, since they know that nature will
spontaneously find the therapy. Our model with MFV offers a simpler
explanation. If this is caused by an infecting virus that Homo
sapiens is used to live with, it is quite possible that the
slowly developing immune system eventually gets rid of the problem.
I am particularly thinking about the cellular immunity (natural
killer cells, specialized T-cells, etc.), which ontogenically
appears at around 1 year of age. But it is particularly in the area
of molecular genetics that our model has great potential, since we
have demonstrated in several experiments and publications that MFV
infections cause MYCNA, also at dramatic levels (100X) with certain
cell lines, such as SK-N-AS, VA-N-BR, etc. (8,13,30).
Genetic aberrations are still another enigma of high-risk
neuroblastoma and cancer cells in general for which the MFV model
provides a potential explanation, as we have discussed in papers
about cancer modelling (4,10,16).
Cancer modelling takes into account all aspects of a
particular theory and even its consequences, for example its
therapeutic implications. This can even become a method for
invaliding or falsifying a particular hypothesis, as we have
recently critically discussed TGTs and their therapeutic
implications (4). In high-risk
neuroblastomas, there has been a slow progress, since a sizeable
portion of cases still has recurrences and dies - up to 60%).
Unfortunately, this figure is even higher in relapsing/progressing
cases since survival becomes only 10%. A few decades ago, one of
the first form of cancer IMT was initiated for high-risk
neuroblastomas at the Memorial Sloan Kettering Cancer Center
(MSKCC) by Nai-Kong Cheung and Brian Kushner with a monoclonal
antibody against the glycosphingolipid GD2. Several clinical trials
showed that passive anti-GD2 IMT provides a 20% PFS improvement in
these patients (32). However, the
reasons for this excellent result are not very clear, particularly
since extensive knowledge has been accumulated on genomic
landscapes and genetic markers for high-risk neuroblastomas, but no
specific marker has been associated with GD2 presence/persistence
and with its therapeutic targeting successes (16). In a recent publication in Cancer
Letters, I proposed an alternative explanation (16). The MFV model shows that genomic
alterations, such as MYCNA, are induced by MFV infections, so that
this could the cause or one of the triggers of the disease. A clear
link between GD2 and MFV could be envisaged by carefully analysing
the sialic acid receptors for this family of viruses, i.e., the
Reoviridae. Since another similar glycosphingolipid, GM2, is
the receptor for the type 1 Lang strain, it is hypothesized that
GD2 is the receptor for MFV and similar Reoviridae (16). This proposal and interpretation are
strengthened by recent clinical data from the study by Kushner
et al (33) at MSKCC. The
IMT treatment of MYCNA-positive cases provides excellent survival
results with PFS of 82% and OS of 94% (33). These results are rather similar to
what can be achieved in low stage disease, not in cases which are
usually considered as high-risk neuroblastomas (33).
Question:
How is your model evaluated in the following
years?
Answer:
There are several ways to evaluate the possible
relevance and especially heuristic consequences and therapeutic
applications of the MFV model. Before indicating at least some of
the steps that I would consider essential for evaluating the MFV
model, I will also briefly comment on how to practically perform
such an evaluation. Since I will retire from active teaching duties
from the University of Florence at the end of this academic year, I
am planning to invest then all of my working time into research,
especially on the MFV model. I am also presently considering moving
to different countries, in view of the fact that research in Italy
is very poorly funded and appears to be dominated by a patronizing
system - at least at the university level where I worked for three
decades. This means that research projects are often funded more on
the basis of personal liaisons and friendships rather than for real
scientific merits. I am presently considering five different
locations: Two in the USA and three in Europe, where I could
possibly relocate. There are at least four major themes, which will
be investigated in order to evaluate the MFV model.
Firstly, one will be the extension of my most recent
article and presentations on anti-GD2 passive IMT (16). As described in my previous answer,
the striking effects of anti-GD2 IMT do not find an obvious
explanation with current studies on genomic landscapes, but could
be explained by the qualified hypothesis of GD2 glycosphingolipid
being associated with MFV receptor recognition and cell entrance.
Dinutuximab and other monoclonals utilized for therapy will be
employed initially in in vitro systems to assess effects on
MFV infection. Subsequently, the previously described animal
systems will be utilized and when indicated we will also
investigate patient specimens from current anti-GD2 trials or
treatments.
Secondly, the core-part of this project will be the
molecular and cellular evaluation of this model. As previously
described, we know that MFV infections can elicit experimental
MYCNA up to 100-fold in SK-N-AS, VA-N-BR, etc. Besides testing
additional cell lines, also from different paediatric and adult
tumours, we want to investigate and better understand the mechanism
of such catastrophic events. One explanation, which is being
evaluated today is whether extensive genomic aberrations may be
associated with chromothripsis or similar events of chromosome
shattering and repasting (16).
This was discovered in approximately one out of five neuroblastoma
cases in the study by Molenaar et al (34), and we have evidence that MFV
infection could cause it (10).
Stem cells will be also investigated as targets of MFV induced
aberrations and malignant transformation (16).
Thirdly, I previously alluded to the very exciting
results of Kushner et al (33) at MSKCC and similar results were
obtained at St. Jude and by the International Society of Paediatric
Oncology European Neuroblastoma Group (SIOPEN), a cooperative group
that is committed to paediatric neuroblastoma research (16). These data suggest that the
excellent survivals are indeed linked to peculiar cases with MYCNA,
which raises the possibility of MFV infections, associated with
anti-GD2 IMT. Therefore, IMT could be particularly efficacious for
a target involving MFV. We will investigate how many of such
targets could be envisaged and tested for efficient therapies in
either high-risk neuroblastoma or additional paediatric and adult
tumours.
And lastly, in testing this MFV model, we will
expand our analysis to additional paediatric tumours, such as
medulloblastomas, lymphomas - especially of Burkitt's lymphoma (BL)
type-, and leukaemias, as well as adult ones, where MFV-like
viruses have been either isolated or hypothesized, such as
small-cell lung cancer - similar to neuroblastoma-, prostate
cancer, lymphomas, breast cancer, etc.
Question:
What is the value of modelling in virology? Could
you give us examples?
Answer:
Modelling in virology has an exceptional value. In
fact, virology could not exist as it is today without virology
modelling. There are myriads of examples, but let's concentrate
only on outstanding virological discoveries, which were awarded a
Nobel Prize in Medicine. Max Theiler was awarded the prize in 1951
for yellow-fever first vaccine; John Enders, Frederick Robbins and
Thomas Weller for isolating and growing poliovirus in tissue
cultures - this led to polio vaccines; Peyton Rous was awarded the
prize in 1966 for his discovery of a cancer-causing virus, now
known as the Rous sarcoma virus; Renato Dulbecco, David Baltimore
and Howard Temin were awarded the prize in 1975 for their work on
oncogenic viruses (Polyoma/SV40 and retroviruses); Baruch Blumberg
in 1976 for his work on hepatitis B; Mike Bishop and Harold Varmus
in 1989 for their discovery of viral and cellular retroviral
oncogenes. In 2008, Harald zur Hausen was awarded the prize for his
discovery of human papillomaviruses (HPVs) cervical carcinogenesis
(35) together with Françoise
Barré-Sinoussì and Luc Montagnier for discovering human
immunodeficiency virus (HIV). Similarly, very appropriate was last
year, 2020, the prize awarded to Michael Houghton, Harvey Alter and
Charles Rice for their identification, characterization and growth
of HCV, allowing screening and vaccination. In all these instances,
virological modelling was extensively utilized to reproduce under
experimental conditions both in vitro and in vivo
essential facets of the infections, thus leading to major
life-savers, such as specific vaccines. If you think about it, the
discoveries of Harald zur Hausen, Baruch Blumberg, and the polio
and HCV trios - just to mention some groups - have saved or cured
the lives of hundreds of thousands of people. Our virological
discovery of a novel virus in paediatric cancers and especially in
cancer-clusters of childhood is more modest in terms of frequency,
but scientifically certainly not unimportant and I want to defend
it. I think there are three aspects of our viral model, which still
deserve attention, because they were ignored or not sufficiently
emphasized.
We were probably not the first to identify in
animal, in vitro or in vivo models the presence of
viruses like MFV. From the end of the 1960's, Elisabeth Gateff,
then at the University of Freiburg and also collaborating with the
Deutsches Krebs Forschung Zentrum in Heidelberg in Germany,
described with other colleagues, similar viral particles in tumours
and stem cells of Drosophila melanogaster (DM). Her findings
are quite interesting, because the DM particles, whose size is
quite different from the ones isolated by us in neuroblastoma or
paediatric lymphoma, were finally identified as Reovirus particles
in 1980(36). Furthermore, in all
the instances in which DM particles were isolated, the tissue of
origin was either malignant - for example a DM brain tumour - or
were cultures of stem cells. Therefore, this is another
characteristic feature of MFV and similar viruses. Differently from
HPVs, which actively replicate only in differentiated epithelium,
MFV-related viruses (MFRVs) need to infect stem cells or cancer
stem cells.
There was another instance in which these types of
viruses were identified and then the notion was somehow ‘dropped’,
during the chase for the ‘equatorial belt virus’. The presence of a
virus was hypothesized by the great physician, Denis Burkitt, who
noticed the appearance of paediatric lymphomas, typically in the
head-neck region, which seemed to follow a geographic and altitude
pattern around the equatorial Africa. The ensued race for
discovering if/which virus was responsible led not only to the
discovery of EBV by Anthony Epstein and his assistant Ivonne Barr,
who received BL samples in London. At the same time, several
isolations of novel Reoviridae family viruses were obtained by
Thomas Bell and colleagues at the Imperial Cancer Research Fund
(ICRF) in Uganda. These discoveries were documented in prestigious
journals, such as the British Journal of Medicine (37), but finally dropped in favour of EBV
by the scientific community. Our re-discovery of MFRVs in cases of
BL in Switzerland - where it is difficult to object that Reoviruses
would be isolated in view of poor hygienic conditions - strongly
argues in favour of an important role for this family of viruses in
BL pathogenesis (30).
Furthermore, as it was indicated in the original papers in the
1960s, we also discovered a few cases with presence of both EBV and
MFV. As previously indicated for the replicative difference between
HPVs and MFV, the issue of co-infection should be investigated
again, since different families of viruses could have complementary
roles in malignant transformation. EBV for example appears to be an
immortalizing virus while MFV behaves as a clastogenic and
transforming virus.
The general and take-home message of this
virological modelling for this family of viruses is that MFV/MFRVs
are ‘normal-passengers’ or viruses persistently infecting Homo
sapiens - also according to the epidemiological data presented
in the study by Kinlen (22),
probably harbouring inside stem cells (36). It should be clarified why certain
isolates are associated with grave malignancies such as
neuroblastomas and lymphomas. However, something similar is clearly
happening with high-risk HPVs, such as HPV 16, 18 and 31 clearly
linked to cervical cancers (35).
Question:
What is the value of cancer modelling in the
understanding of cancer pathogenesis and therapeutic
interventions?
Answer:
Cancer modelling refers to a general re-analysis, or
sometimes meta-analysis, of all the data we have in favour or
against a particular hypothesis to explain cancers and especially
human cancers. We have used this term for the first time in a
longer review article finally published in 2015(4), also in view of my criticism of the
modelling present at that time. I have argued in that paper, and I
am still convinced today, that the falsification of a model in
cancer research does not come or originate only from experiments
performed in the laboratories, but also by the clinical practice
that applies such model for treating cancer patients. In other
words, if the logical consequences of a model lead to a specific
type of therapy and this therapy is not cancer-curative, this
should logically lead to the falsification of the model itself. We
should therefore distinguish three phases in this reassessment of
cancer modelling, which however follow the current trends of cancer
research.
My criticism in 2015 was qualified by an extensive
analysis of the pitfalls of TGTs, at that time the most accepted
cancer therapies. I argued that, as chemotherapy before was based
upon interpretation of cancer as ‘clonal outgrowth’, TGT stands
upon a vision of malignancies as derangements of ‘gene networks’.
We know today that clonal outgrowth is incorrect, since it has been
shown that cancer is dominated by TH, of which intra-TH drives
continuous expansions of new clones, essentially created by new
mutations accrual and by selection. This obviously leads to
selection of chemo-therapy resistant clones. However, also the
‘gene network’ hypothesis is invalided: i) Not only by intra-TH
which obviously affects genes and their expression as first
targets; but also ii) by what appears to be a general misconception
of the model that has emphasized only final or landscape events of
cancer genomes. This concept becomes rather obvious, if one
analyses instead functions, which are considered upstream from such
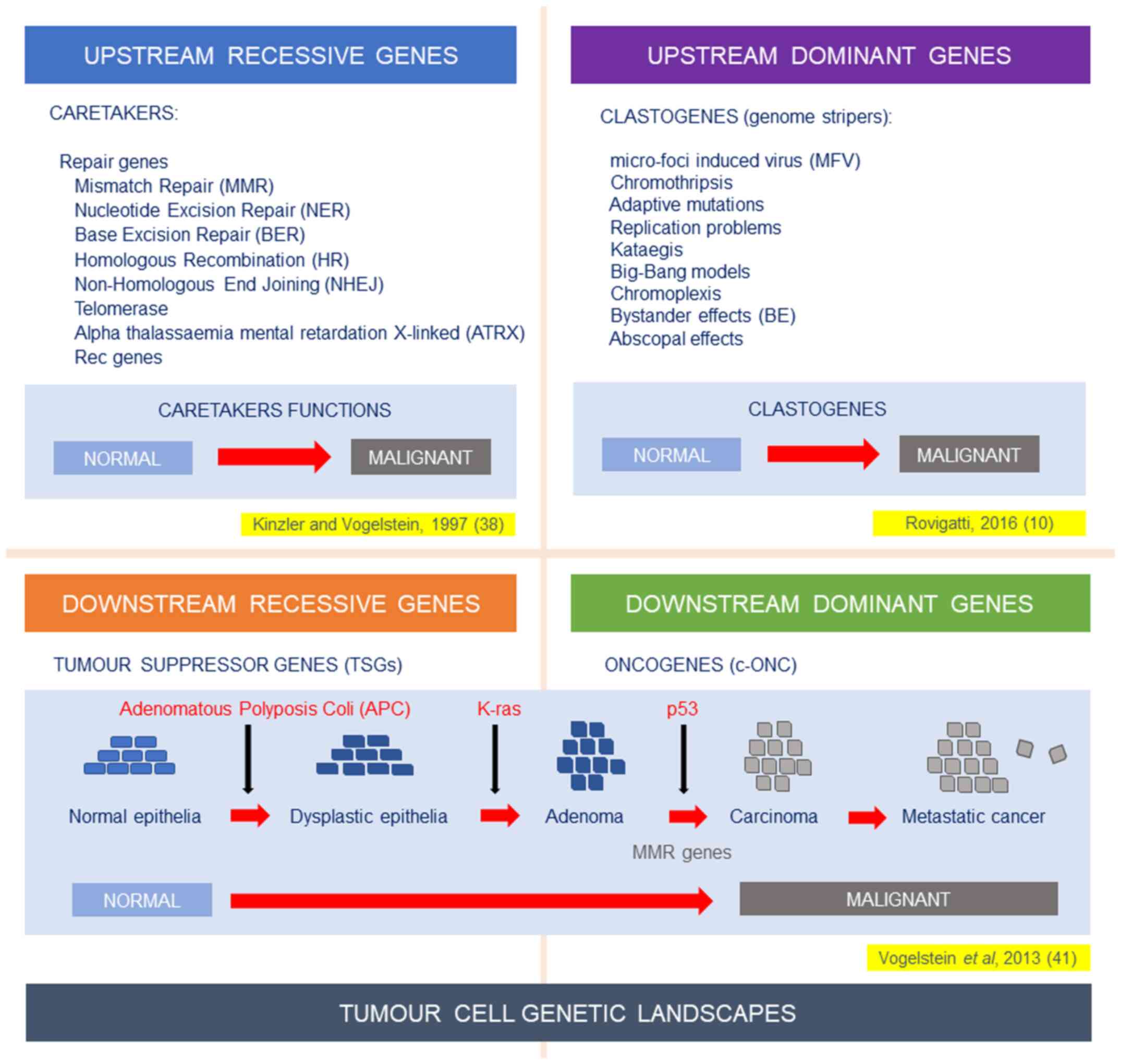
genomic landscapes. In Fig. 1
(upper left panel), I represent what was described till 2016, i.e.,
the recessive function of caretakers described by Kinzler and
Vogelstein (38) several years
before. However, it was becoming clear that novel functions should
be hypothesized as present upstream of the genomic landscape. They
ought to be dominant and clastogenic (Fig. 1, upper right panel).
Alternative modelling needs to be proposed, to
overcome the difficulties of hallmarks of cancer (HoC) and TGT
models. What appears to be their common denominator is the
incapability of solving the TH conundrum. In 2015, we proposed to
utilize additional models instead of the only one which has
dominated for 20 years (HoC) (4).
This strategy follows also the example of our colleagues,
physicists and astrophysicists, who have proposed several different
models. It was therefore proposed that by following different
alternative or complementary modelling, we may reach more rapidly
the solution of the cancer enigma (4). In 2016, following the described
diatribe over origin and mechanisms of TH, the original scheme was
modified by adding dominant functions, which actively cause
clastogenic activities on the genomic landscapes downstream: Both
oncogenes and TSGs (Fig. 1, bottom
panels). Clear examples were already present with chromothripsis,
kataegis and chromoplexis and examples of Big-Bang models from
human colorectal cancers (10,39).
Therefore, the MFV model appears to be probably the first described
example of such upstream-dominant activities (10,13,16).
Its natural history of infections - occurring very early in
ontogenesis, even during the first months of life, its persistence
with life-long immunity and associated clastogenic and mutagenic
events, often through chromothripsis, render the MFV model a
particularly interesting one, which should be further elaborated
and studied in the future (10,16).
Question:
So, where do we go from here? What should be the
future steps?
Answer:
In 2018, cancer modelling history was somehow
changed when the Nobel Prize for Medicine was assigned to James
Allison and Tasuku Honjo for their fundamental discoveries on
inhibitors of immune checkpoints or negative regulators. Heralded
by lay press and editorial commentaries, these discoveries
announced a progressive shift today toward IMT approaches. It is,
however, difficult to believe that IMT will be - as it is now - the
panacea for all human cancers. In fact, many of the expressions of
excessive enthusiasm for IMT today are reminiscent of the hype
surrounding TGTs and targetable or ‘actionable’ genes (40) almost 20 years ago. Since I am
wearing white hair now, I could suggest some caveats and essential
experiments in order to clarify today's picture. Although the
therapeutic benefits of anti-PD1, PDL1, CTLA4, etc., are in some
cases undeniable and obviously very welcomed, we should clarify
what such activities mean in cellular-molecular terms. In other
words, we should clarify their targets and their meanings. My
recent article in Cancer Letters on the IMT passive
targeting of GD2 in paediatric high-risk neuroblastoma could be an
example (16). Since
next-generation sequencing studies and genetic/genomic data do not
show any clear correlation with GD2 and since Kushner et al
(33) demonstrated excellent
results by IMT in children with MYCNA, the qualified hypothesis was
made that GD2 acts as recognition-receptor for MFV (16). Similarly, in most of the
therapeutic successes of today's ‘inhibitors of negative immune
regulators’, we just know that there is an increased action of the
immune system, but we ignore the target(s).
We should be also aware of our previous experiences.
TGTs probably relied on an ‘incomplete model’ (4), where the analysis was frozen at the
lower level of genetic mutations/aberrations (Fig. 1); these are called tumour cell
genetic landscapes. These landscapes are extremely mobile and
variable and they also depend on upstream elements, which are
actively and dominantly hitting the genome. These ‘Aristotle's
first mover(s)’ should be better identified, studied and
therapeutically approached, as it also depends on them the whole
well-being of our genome.
Finally, we should not forget that in all previous
instances, we have caused and we are still causing our oncologic
patients to become cancer-therapies addicts. It is clear now that
most of the mutations evidenced in radio-/chemotherapy treated
patients are due to the specific treatments (radio-/chemotherapy)
following Darwinian mechanisms. Similar effects are elicited and
evident in TGTs. Think twice before starting creating again an army
of cancer patients addicted to immunotherapeutic drugs! This is
also why it is so essential to unveil TH and cancer ‘first movers’.
The antibiotics example is paradigmatic and should lead us in this
search.
Question:
Thank you very much for participating in our
webinar.
Acknowledgements
This article is published in the context of the ‘7th
Workshop on Paediatric Virology’, which was organised virtually by
the Institute of Paediatric Virology (IPV; https://paediatricvirology.org) on December 20th,
2021.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All authors (UR, GP, INM, CK, AP, MT and DAS)
contributed equally to the conception and design of this
manuscript. The original draft of the answers to the article
questions was entirely written by UR. All authors then edited and
critically revised the manuscript, read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
INM, MT and DAS are co-founders of the Institute of
Paediatric Virology (IPV). UR, GP, CK and AP declare that they have
no competing interests. DAS is the Managing Editor of the journal,
but had no personal involvement in the reviewing process, or any
influence in terms of adjudicating on the final decision, for this
article. The World Academy of Sciences Journal is affiliated with
the World Academy of Sciences and the Institute of Paediatric
Virology.
References
|
1
|
Monclair T, Brodeur GM, Ambros PF, Brisse
HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK,
Nuchtern JG, et al: INRG Task Force: The International
Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force
report. J Clin Oncol. 27:298–303. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Naranjo A, Irwin MS, Hogarty MD, Cohn SL,
Park JR and London WB: Statistical Framework in Support of a
Revised Children's Oncology Group Neuroblastoma Risk Classification
System. JCO Clin Cancer Inform. 2:1–15. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Schwab M, Alitalo K, Klempnauer KH, Varmus
HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M and Trent J:
Amplified DNA with limited homology to myc cellular oncogene is
shared by human neuroblastoma cell lines and a neuroblastoma
tumour. Nature. 305:245–248. 1983.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Rovigatti U: Cancer modelling in the NGS
era - Part I: Emerging technology and initial modelling. Crit Rev
Oncol Hematol. 96:274–307. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-myc in untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1124. 1984.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Matthay KK, Maris JM, Schleiermacher G,
Nakagawara A, Mackall CL, Diller L and Weiss WA: Neuroblastoma. Nat
Rev Dis Primers. 2(16078)2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mossé YP, Laudenslager M, Longo L, Cole
KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P,
et al: Identification of ALK as a major familial neuroblastoma
predisposition gene. Nature. 455:930–935. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rovigatti U: Chronic Fatigue Syndrome
(CFS) and Cancer Related Fatigue (CRF): Two ‘fatigue’ syndromes
with overlapping symptoms and possibly related aetiologies.
Neuromuscul Disord. 22 (Suppl 3):S235–S241. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tomasetti C and Vogelstein B: Cancer
etiology. Variation in cancer risk among tissues can be explained
by the number of stem cell divisions. Science. 347:78–81.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rovigatti U: The Long Journey of cancer
Modeling: Ubi Sumus? Quo Vadimus? Mathew J Cancer Sci. 1:1–5.
2016.
|
|
11
|
Thiele CJ, Reynolds CP and Israel MA:
Decreased expression of N-myc precedes retinoic acid-induced
morphological differentiation of human neuroblastoma. Nature.
313:404–406. 1985.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Ross RA, Biedler JL and Spengler BA: A
role for distinct cell types in determining malignancy in human
neuroblastoma cell lines and tumors. Cancer Lett. 197:35–39.
2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rovigatti U: Isolation and initial
characterization of a new virus: Micro-Foci inducing virus or MFV.
C R Acad Sci III. 315:195–202. 1992.PubMed/NCBI
|
|
14
|
Furlan A, Dyachuk V, Kastriti ME,
Calvo-Enrique L, Abdo H, Hadjab S, Chontorotzea T, Akkuratova N,
Usoskin D, Kamenev D, et al: Multipotent peripheral glial cells
generate neuroendocrine cells of the adrenal medulla. Science.
357(eaal3753)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
van Groningen T, Koster J, Valentijn LJ,
Zwijnenburg DA, Akogul N, Hasselt NE, Broekmans M, Haneveld F,
Nowakowska NE, Bras J, et al: Neuroblastoma is composed of two
super-enhancer-associated differentiation states. Nat Genet.
49:1261–1266. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rovigatti U: The glycosphingolipid GD2 as
an effective but enigmatic target of passive immunotherapy in
children with aggressive neuroblastoma (HR-NBL). Cancer Lett.
503:220–230. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fletcher JI, Ziegler DS, Trahair TN,
Marshall GM, Haber M and Norris MD: Too many targets, not enough
patients: Rethinking neuroblastoma clinical trials. Nat Rev Cancer.
18:389–400. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Eleveld TF, Oldridge DA, Bernard V, Koster
J, Colmet Daage L, Diskin SJ, Schild L, Bentahar NB, Bellini A,
Chicard M, et al: Relapsed neuroblastomas show frequent RAS-MAPK
pathway mutations. Nat Genet. 47:864–871. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gatenby RA, Gillies RJ and Brown JS: Of
cancer and cave fish. Nat Rev Cancer. 11:237–238. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Muirhead CR, Tweddle DA, Basta NO and
McNally RJ: Temporal clustering of neuroblastic tumours in children
and young adults from Northern England. Environ Health.
14(72)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Heath CW Jr: Community clusters of
childhood leukemia and lymphoma: Evidence of infection? Am J
Epidemiol. 162:817–822. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kinlen L: Childhood leukaemia, nuclear
sites, and population mixing. Br J Cancer. 104:12–18.
2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Greaves M: Infection, immune responses and
the aetiology of childhood leukaemia. Nat Rev Cancer. 6:193–203.
2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vega-Lopez GA, Cerrizuela S, Tribulo C and
Aybar MJ: Neurocristopathies: New insights 150 years after the
neural crest discovery. Dev Biol. 444 (Suppl 1):S110–S143.
2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pule MA, Savoldo B, Myers GD, Rossig C,
Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al:
Virus-specific T cells engineered to coexpress tumor-specific
receptors: Persistence and antitumor activity in individuals with
neuroblastoma. Nat Med. 14:1264–1270. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Flaegstad T, Andresen PA, Johnsen JI,
Asomani SK, Jørgensen GE, Vignarajan S, Kjuul A, Kogner P and
Traavik T: A possible contributory role of BK virus infection in
neuroblastoma development. Cancer Res. 59:1160–1163.
1999.PubMed/NCBI
|
|
27
|
Giraud G, Ramqvist T, Pastrana DV, Pavot
V, Lindau C, Kogner P, Orrego A, Buck CB, Allander T, Holm S, et
al: DNA from KI, WU and Merkel cell polyomaviruses is not detected
in childhood central nervous system tumours or neuroblastomas. PLoS
One. 4(e8239)2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mazar J, Li Y, Rosado A, Phelan P,
Kedarinath K, Parks GD, Alexander KA and Westmoreland TJ: Zika
virus as an oncolytic treatment of human neuroblastoma cells
requires CD24. PLoS One. 13(e0200358)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Rey F, Schwartz O and Wain-Hobson S:
Gain-of-function research: Unknown risks. Science. 342:311.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rovigatti U, Tam A, Piccin A, Colognato R
and Sordat B: Preliminary Characterization of a New Type of Viruses
Isolated from Paediatric Neuroblastoma and Non-Hodgkin's Lymphoma:
potential Implications for Aetiology. In: Proceedings of the
International Conference on Childhood Leukaemia, London, section
P1-18, ppI-IV, 2004.
|
|
31
|
D'Angio GJ, Evans AE and Koop CE: Special
pattern of widespread neuroblastoma with a favourable prognosis.
Lancet. 1:1046–1049. 1971.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yu AL, Gilman AL, Ozkaynak MF, London WB,
Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay
KK, et al: Children's Oncology Group: Anti-GD2 antibody with
GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J
Med. 363:1324–1334. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kushner BH, LaQuaglia MP, Modak S, Wolden
SL, Basu EM, Roberts SS, Kramer K, Yataghene K, Cheung IY and
Cheung NV: MYCN-amplified stage 2/3 neuroblastoma: Excellent
survival in the era of anti-GD2 immunotherapy. Oncotarget.
8:95293–95302. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Molenaar JJ, Koster J, Zwijnenburg DA, van
Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J,
Westerman BA, van Arkel J, et al: Sequencing of neuroblastoma
identifies chromothripsis and defects in neuritogenesis genes.
Nature. 483:589–593. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
zur Hausen H: Papillomaviruses in the
causation of human cancers - a brief historical account. Virology.
384:260–265. 2009.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Haars R, Zentgraf H, Gateff E and Bautz
FA: Evidence for endogenous reovirus-like particles in a tissue
culture cell line from Drosophila melanogaster. Virology.
101:124–130. 1980.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Bell TM, Massie A, Ross MG, Simpson DI and
Griffin E: Further isolations of reovirus type 3 from cases of
Burkitt's lymphoma. BMJ. 1:1514–1517. 1966.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kinzler KW and Vogelstein B:
Cancer-susceptibility genes. Gatekeepers and caretakers. Nature.
386:761–763, 763. 1997.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sottoriva A, Kang H, Ma Z, Graham TA,
Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, et
al: A Big Bang model of human colorectal tumor growth. Nat Genet.
47:209–216. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fojo T: Novel_target.com. Oncologist.
6:313–314. 2001.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer Genome Landscapes.
Science. 339:1546–1558. 2013.PubMed/NCBI View Article : Google Scholar
|