Mammalian target of rapamycin (mTOR) plays a
critical role in regulating cellular metabolism by integrating
signals from nutrients, growth factors and the environment. mTOR
exists in two distinct mTOR complexes (mTORCs), mTORC1 and mTORC2,
which are conserved across species from yeast to humans. mTORC1 is
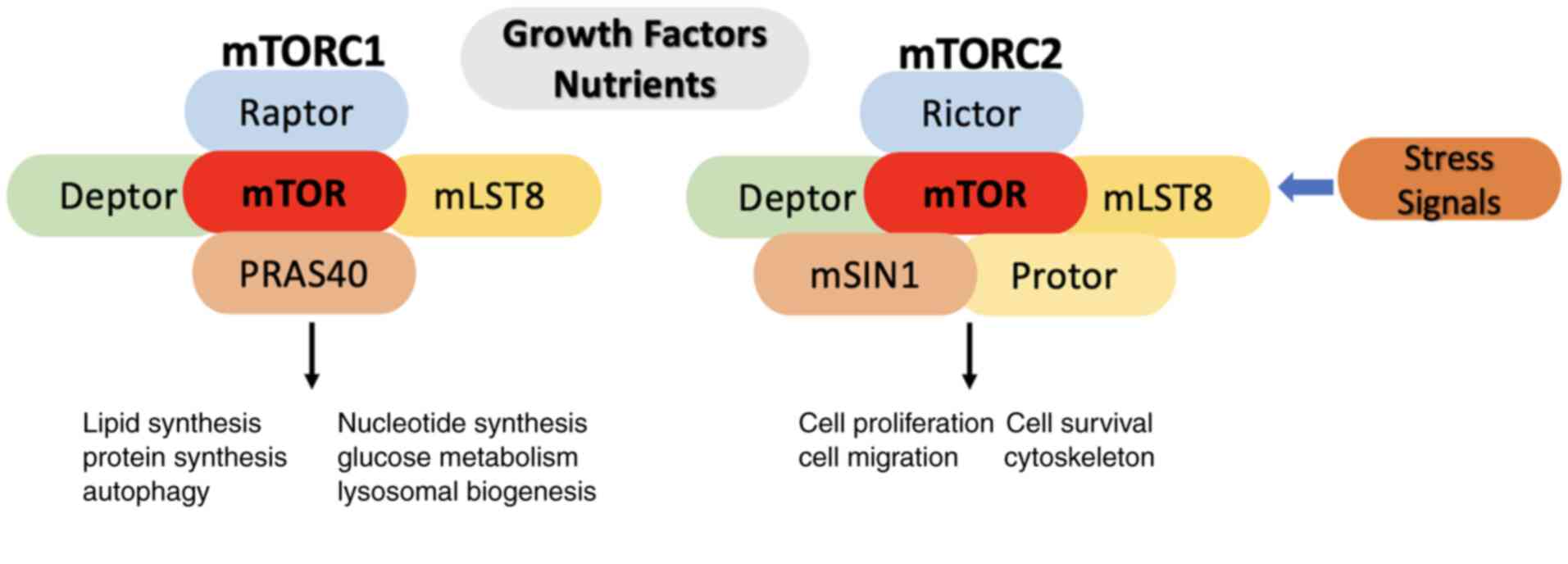
a protein complex that consists of mTOR, Raptor, Deptor, mLST8, and
PRAS40. It is the primary target of the drug rapamycin that
allosterically reduces its activity, while mTORC2 consists of mTOR,
Debtor, Raptor, mLST8, mSIN1 and Protor, and its inhibition occurs
indirectly (1-4)
(Fig. 1A). Rapamycin and its
analogs are currently utilized in clinical settings. They are being
tested for various conditions, including cancer, immune disorders,
metabolic syndromes and neurodegenerative diseases, exhibiting
potential as anti-aging agents that may enhance human and animal
health. Insight into mTOR-mediated metabolic shifting in target
cells, such as tumors and immune cells in their microenvironments,
can significantly advance the development of targeted therapeutic
approaches. The proper functioning of mTORC1 and mTORC2 is
essential for maintaining metabolic equilibrium, preventing
diseases and extending the health area (5). Studies have revealed that
AMP-activated protein kinase (AMPK) affects cancer cell metabolism
through several mechanisms. The activation of AMPK has been
correlated with the inhibition of aerobic glycolysis which is
well-known as the Warburg Effect. The Warburg effect has been known
as one of the cancer hallmarks and is characterized by enhanced
glycolysis even in the presence of oxygen (5-7).
This pathway stimulates the catabolic metabolic processes, which
promote cancer cell metabolism to switch to energy preservation
under severe stress conditions. AMPK stimulation has been shown to
utilize anti-proliferation and propagation effects in various tumor
types (6).
The importance of cancer cell metabolism
orchestrated through AMPK/mTOR cascades is an attractive avenue for
innovative cancer therapies and highlights the need for the
understanding of these signaling pathways further to advance
wide-ranging combination therapy approaches. AMPK and mTORCs have
critical functions in controlling the cell metabolism in a number
of types of cancer, shaping the anabolic and catabolic pathways
that regulate cell growth, proliferation and survival. AMPK serves
as a key energy sensor, activated in response to low cellular
energy levels, while the action of mTOR becomes evident under high
nutrient conditions (3-7).
AMPK deploys dual roles in controlling the
intracellular environment of tumor immunity, affecting both immune
and tumor cells. AMPK is the key factor between the immune cell
microenvironment and the metabolism energy of tumor cells (8). Recently, it has been shown that AMPK
exhibits anticancer immune activity by cooperating with dominant
components at the cancer microenvironment, affecting the roles of
T-lymphocytes, myeloid suppressor cells and macrophages (9). Furthermore, AMPK can inhibit the
production of chemokines and cytokines (10). Taken together, AMPK plays a master
role in the modulation of tumor microenvironment.
Moreover, the mTOR signaling pathway plays a
critical role in the regulation of the tumor microenvironment
(11). The mTOR cascade plays
regulatory roles in the growth, differentiation, survival and
function of adaptive and innate immune cells (12). It controls the phenotypic, the
functional, and direct reprogram in tumor-immune cells
microenvironment. In addition, mTOR stimulates the inflammatory
process and promotes the replenishment of the immune cell to tumor
environment, resulting in stimulating anticancer effects or
supporting tumor cell development, proliferation and metastasis
(13). Therefore, misregulated
mTOR pathways in tumors can modify the tumor microenvironment,
disrupting the cancer immune microenvironment.
Research has demonstrated critical associations
between the AMPK/mTOR signaling pathway and ferroptosis in
carcinogenesis (14). Ferroptosis
is an iron dependent programmed cell death process that has gained
increasing attention from researchers for targeting cancer growth
(15). Ferroptosis is closely
associated with mTOR, and utilizing mTOR-related therapy has the
potential to stimulate the rate of ferroptosis in the targeting of
several types of cancer (16). It
has also been demonstrated that AMPK confines the stimulation of
ferroptosis and subsequently, the inhibition of AMPK therapy
restores the treatment and restores the responsiveness to
ferroptosis activators (17).
Moreover, it has been demonstrated that amentoflavone stimulates
ferroptosis by stimulating the ROS/AMPK/mTOR signaling axis to
reduce the uterine cell cancer viability and prorogation by
stimulating the ferroptosis and apoptosis (18). Thus, AMPK/mTOR signaling pathway is
the central player in ferroptosis process.
Furthermore, AMPK and mTOR have been recently shown
to be involved in mitochondrial dynamics in cancer progression
(19). AMPK plays a crucial role
in the regulation of mitochondrial activity and cytoskeleton
modification, with an increased AMPK activity in cells with a low
migratory ability; this induces the mitochondrial fission, leading
to reduced oxidative phosphorylation (OXPHOS), low adenosine
triphosphate (ATP) biogenesis and the reduction of ameboid myosin
II-dependent cell migration (20).
Moreover, the abnormal expression of MTERF1 in colorectal cancer
cells modulates the p-AMPK/mTOR pathway of mitochondrial
dysfunction (21). Therefore, the
AMPK/mTOR axis can be a good way to treat different
mitochondrial-related diseases such as cancer and metabolic
syndromes.
The AMPK/mTOR signaling pathway has also recently
been shown to be involved metabolically in condensate formation.
Developments (condensates) are a mutual strategy for cells to
complete various functions, such as DNA repair, chromatin
association, transcriptional activity and signal transduction
(22). Recently, abnormal phase
separation and alteration have been stated to be associated with
cancer and other neurodegenerative diseases (23). AMPK activation occurs during
metabolic stress; this activation stimulates the formation of
condensate and stress particles that promote cancer cell survival
and progression under different nutrients deficiency (24). In addition, condensate development
organizes the necessary metabolic enzymes to stimulate the
metabolic switching in the tumor microenvironment (25). Moreover, a recent study
demonstrated that the formation of EMT supports carcinogenesis, and
metastasis is notably initiated and governed by phase separation
(condensate formation) (26).
These data highlight the critical role of the AMPK/mTOR pathway in
regulating condensate particle formation; these data may provide a
novel approach for cancer treatment by targeting these metabolic
vulnerabilities.
Disruptions in these signaling pathways may
inadvertently cause tumor progression, emphasizing the importance
of novel therapeutic interventions targeting this complex network.
Moreover, understanding the complex balance of the action between
AMPK and mTORCs can lead to the development of novel approaches to
combat cancer growth, while diminishing the threat of unintentional
consequences associated with modifying metabolic switches.
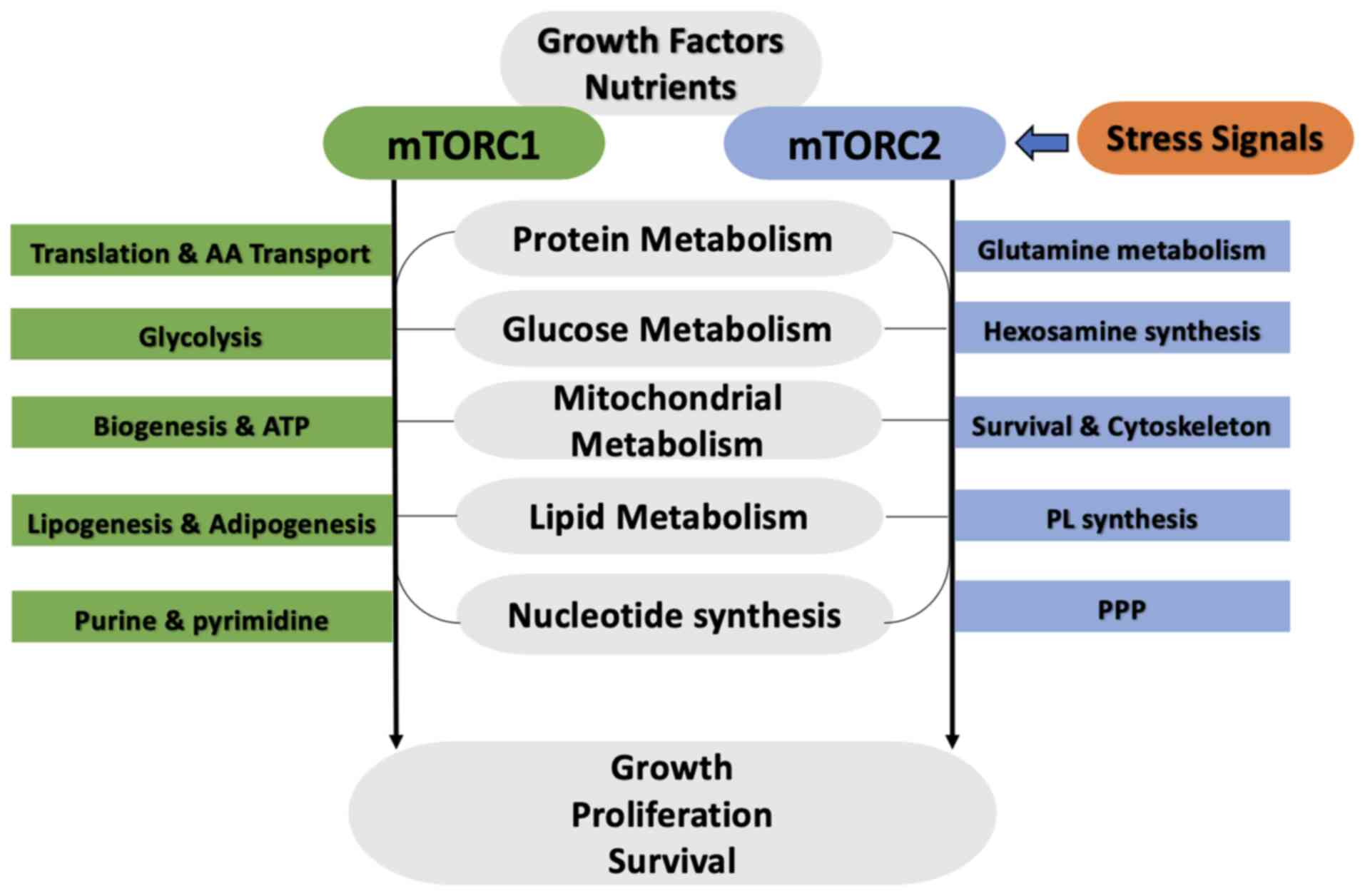
Both mTORCs function as central regulators of the
use of glucose in normal and cancer cells, markedly contributing to
metabolic rewiring, which describes different malignancies. mTOR1
and mTOR2 can regulate the expression of glycolytic enzymes,
glucose transporters and transcription machinery that stimulate the
expression of various genes related to glycolysis pathway effectors
(27).
Decades ago, Otto Warburg found that cancer cells
use aerobic glycolysis as fuel source to keep the building blocks
that are required for cell growth and propagation. This specific
characteristic of tumor cells is well-known as the Warburg effect,
whereas OXPHOS is the main energy source for the maintenance of the
development and proliferation of normal cells (27-30).
Warburg's discovery was recognized and confirmed in various human
tumors with radioactive glucose analogs using (fluorodeoxyglucose)
to examine glucose utilization in normal and tumor tissues
(30). Currently, the Warburg
effect is one of the cancer hallmarks, in addition to other
characteristics, such as immortality, resistance to cell death,
avoiding growth defeat, stimulating invasion, angiogenesis and
metastasis (28-30).
Additionally, a high mutation rate with genomic variability,
escaping the immune defense and tumor-increasing inflammation are
up-and-coming hallmarks that are related to carcinogenesis.
Modified glucose uptake over aerobic glycolysis is an ineffective
mode to generate energy, as it provides two ATPs per glucose
molecule. By contrast, OXPHOS provides 36 ATPs from glucose
oxidation via the Krebs cycle (30-33).
Previous studies have confirmed that the majority of tumor cells
shift the glucose metabolism from OXPHOS to aerobic glycolysis even
in the presence of O2. Tumor cells convey glucose
metabolic intermediates to biosynthetic macromolecule new building
blocks pathways. This activity sustains tumor development by
providing metabolic intermediates to refill cancer cells and
producing reducing molecules, such as nicotinamide adenine
dinucleotide phosphate (NADPH) for different metabolic activities
(33-35).
In differentiated cells, glucose oxidation is an
oxygen-dependent process, either through OXPHOS (with
O2) or fermentation (without O2) (35-38).
In expanding cells, mainly tumor cells, pyruvate from the oxidation
of glucose is transformed into lactic acid through aerobic
glycolysis, also known as the Warburg effect, even in the presence
of oxygen. In addition, glucose oxidation provides the intermediate
substrates for the pentose phosphate pathway (PPP) to generate
ribose-5-phosphate (R5P) required for nucleotide synthesis and
NADPH to sustain cellular redox homeostasis (39). Furthermore, glycolytic substrates
are essential for synthesis 3-phosphoglycerate, a precursor for the
serine synthesis cascade, providing the crucial molecules for the
biosynthesis of various (non-essential) amino acids. Moreover, the
transamination reaction converts pyruvate to alanine and citrate
transfers the mitochondria into the cytoplasmic matrix for the
biogenesis of fatty acids (FAs) (35,40-42).
This indicates that tumor cells efficiently use different glucose
metabolism to maintenance progression and survival.
mTORC1 stimulates glycolysis by promoting and
regulating two main transcription factors [hypoxia inducible factor
(HIF)1a and Myc] (Fig. 2). mTORC1
similarly stimulates the upregulation of glycolytic gene through
the transcriptional controller of Myc (43,44).
Myc is mutated in >50% of human cancers, and its increased
expression is frequently associated with enhanced glycolysis and
glutamine metabolism (45,46). mTORC1 stimulation of S6K and 4EBP1
is essential for Myc-stimulated carcinogenesis (47). Researchers have demonstrated that
genes are sensitive to rapamycin and are controlled by Myc
(48). The metabolic shift of
glucose metabolism is mainly orchestrated across the mTOR1
activation, which reacts to external growth signals such as
insulin, amino acids and glucose (49-52).
mTORC1 boosts the glycolysis pathway and enhances the transcription
and translation activities for crucial enzymes that participate in
glycolytic and lactic acid production, such as hexokinase II and
pyruvate kinase M2, that is produced in dividing and cancer cells.
In addition, the mTOR2 pathway stimulates the expression of glucose
transporter (GLUT)1 and phosphofructokinase (PFK) under the
transcription machinery of these factors, thereby accelerating the
glucose uptake in various types of cancer, such as colon,
pancreatic, hepatic, lung and breast cancers (53-56).
This step is essential for maintaining the excessive ATP levels
necessary for energy-intensive activities, such as cell development
and division. Furthermore, mTOR1 stimulates the expression level of
several glycolytic enzymes by enhancing the translation of HIF1a
(57). Evidence indicates that the
role of mTOR is to regulate the gene expression of the enzymes for
the glycolysis pathway and modulate the enzymatic activity through
phosphorylation, promoting the appropriate microenvironment for
cancer cell growth and development (58). Furthermore, the upregulation of
this enzyme by mTOR1 is regulated through the aforementioned
mechanism. In addition, the upregulated mTOR1 pathway due to
tuberous sclerosis complex (TSC)2 mutation specifically stimulates
the one isoform of PFK in acute myeloid leukemia (AML) (59). Moreover, the connection between the
mTOR cascade and the PI3K/AKT signaling cascades has been
demonstrated as the central axis for the glycolysis signaling
pathway. Activated AKT induces the activity of the glycolytic
signaling pathway under hypoxic environments and downregulates the
expression of large non-coding RNA, further enriching tumor cell
adaptation and development (60-62).
mTORC2 increases glycolysis that stimulates class
IIa histone deacetylase phosphorylation, thus inhibiting them,
promoting Fox O1 and O3 acetylation, resulting in the
downregulation of c-Myc in glioblastoma (63). A better understanding of the
function of the mTOR pathway in glycolysis and cancer cell
metabolism will open a new avenue for targeting this pathway
therapeutically, allowing the modification of glycolysis to impede
cancer growth. Targeting the mTOR pathway using an mTOR inhibitor
has shown promising results in clinical breast cancer models by
decreasing glycolytic activity and enhancing metabolic deprivation
in tumor cells (64).
mTOR plays a substantial role in the dependence of
cancer cell on glutamine breakdown, which is increasingly
recognized as a crucial factor in cancer biology and therapeutic
approaches. mTOR is an evolutionarily conserved serine/threonine
kinase that is in two different complexes, mTORC1 and mTORC2; mTOR
plays regulatory roles in numerous biological processes, such as
cell glucose metabolism, growth, lysosome biogenesis, survival,
autophagy and migration, that are required for cell development
(Fig. 2). Therefore, its
equilibriums between the anabolic and catabolic activities
consistent to different bioenvironmental elements (65,66).
One of the crucial roles of mTOR is its role in the metabolism of
glutamine, which has been raised as a critical source of nitrogen
and energy for rapidly proliferating cancer cells. Tumor cells use
glutamine also as an energy source apart from the utilization of
glucose; glutamine is conditionally an essential amino acid, and it
is free in human blood. Therefore, its production may be decreased
under specific different pathophysiological situations and
metabolic conditions (67).
Glutamine synthetase (GS) accelerates the linking reaction between
glutamate and ammonia to procedure glutamine (68). Glutamine has functions in lipid and
protein synthesis, precisely in tumor cells and energy redox
homeostasis such s glucose. It is a precursor for glutamate citric
acid and carbon synthesis (69).
Moreover, glutamine is a nitrogen atom donor for purines,
pyrimidines, and asparagine. Notably, it is also a precursor of
(NAD) and is a source of glucosamine synthesis (67). In addition to the function of
glucose in stimulating tumor cell proliferation, tumor cells are
dependent and addicted to glutamine use to support different
anabolic signaling pathways for constant cell persistence (70).
The activity of mTOR is essential in this metabolic
flexibility, as it functions as a nutrient sensor, sensing the
activity of a number of amino acids, including glutamine, and
controlling subsequent signaling pathways accordingly. Research
reveals that metabolic glutamine triggers mTORC1 activity through
intermediates produced during its breakdown. For example, glutamate
is subsequently converted to α-ketoglutarate (α-KG), a key
metabolite that stimulates mTORC1 via its interaction with prolyl
hydroxylases, which increases the GTP loading of Rag GTPases
responsible for mTORC1 activation; this controlling mechanism
elucidates how glutamine not only stores energy, but also performs
as an agonist for mTOR metabolic pathway, developing a pro-growth
microenvironment that boosts cancer cell growth and survival
(71).
The mTORC1 pathway stimulates glutamine metabolism
through the c-Myc transcription factor, which controls the
expression of genes required for glutaminolysis, such as
glutaminase 2 and other enzymes in c-Myc-stimulated T-cells
(72). A feedback cycle exists
between mTORC1 activation and glutamine metabolism through Myc
(73,74). In addition, mTORC1 stimulates
glutaminolysis by controlling the activity of cAMP-responsive
element binding 2 (CREB2) by enhancing its degradation (75). These results indicate that mTORC1
stimulates glutamine uptake via negative feedback mechanism of
CREB2, finally leading to GDH stimulation.
Additionally, the association between the mTOR
pathway and glutamine metabolism extends to controlling autophagy
and cellular stress reactions. Under a rich nutrient situation,
mTORC1 stimulates protein production, while hindering autophagy, a
safe process for degrading excessive or malfunctional proteins and
damaged cellular components. When cellular glutamine levels are
low, mTORC1 action is reduced, stimulating autophagy to reutilize
cellular components and maintain metabolic cellular homeostasis
(76).
Notably, the role of mTOR in glutamine use has a
number of clinical implications, as various studies have
investigated the targeting of this signaling pathway with mTOR
inhibitors as a novel therapeutic approach. For example, it has
been demonstrated that some cancers can adapt to mTOR inhibitors by
enhancing glutamine metabolism to promote cancer growth and resist
treatment. In addition, it has also been demonstrated that
glutamine deprivation plus mTOR inhibition can be more effective in
eradicating tumor growth and survival (72-76).
Cancers with highly upregulated mTOR activity are frequently
challenging to standard therapies, and reducing mTOR activity
significantly impairs cancer growth by breaking their metabolic
flexibility. Moreover, mTORC2 is rising as a critical player in
glutamine cellular metabolism as it controls the expression of Myc
(77). It has been shown that the
knockdown of Rictor reduces α-KG levels, which are expected to have
originated from glutaminolysis, indicating that mTORC2 can control
this process (78). Earlier
findings have demonstrated that the mTORC2/AKT axis may not affect
glutamine metabolism or consumption (79). A recent study suggested a
connection between stimulated mTORC2-facilitated AKT
phosphorylation and glutaminolysis through different mechanisms. In
addition, low levels of glutamine or its substrate activate mTORC2,
as revealed by the phosphorylation of AKT. Moreover, low levels of
glutamine reduce the activity of mTORC1, whereas mTORC2 activity
increases (80). Inhibiting the
activity of mTORCs stimulates glutaminolysis, signifying an
additional multifaceted feedback loop, possibly to confirm TCA
replenishing as mTOR signaling declines.
mTORCs play a crucial role in controlling glutamine
metabolism in cancer cells. By serving as a sensor of metabolic
nutrient accessibility, mTORCs not only affect cellular development
and propagation, but are also tortuously associated with the
metabolic needs of tumor cells to promote a favorable
microenvironment for tumor progression. Further studies are
required to elucidate the precise mechanisms behind the regulatory
role of mTORCs for glutamine use. It is crucial to develop
effective therapies that disturb the metabolic routes that tumor
cells manipulate to accomplish their rapacious energy
requirements.
mTOR has a central function in controlling protein
metabolism in cancer cells, functioning as a central hub that
combines numerous signaling cascades, the availability of
nutrients, and energy situations to regulate cellular growth,
propagation and metabolic variation. Understanding the importance
of mTOR in protein biosynthesis and metabolism is necessary for
illustrating how malignant cells achieve their elevated need for
proteins, crucial constituents for cell structure, role, and
signaling during cancer propagation. Protein biosynthesis is
correctly regulated and requires essential and nonessential amino
acids. Cancer and non-cancer cells obtain these molecules through
growth factor signaling by providing surface transporters, which
allow them to direct these molecules from external sources and
maintain mTOR-signaling networks (81,82).
mTORC1 is mainly involved in stimulating protein
biosynthesis across its effector molecules (Fig. 2). mTORC1 phosphorylates the two
main effecters for protein translation, 4E-BP1- and S6K1. S6K1
phosphorylates a number of factors that are directly or indirectly
involved in protein biosynthesis, such as PDCD4, SKAR eIF4B and
eIF3 (83,84). Notably, mTORC1 is stimulated by
amino acids to promotes protein biosynthesis through its active
functions in translation and ribosome synthesis, phosphorylating
S6K and the initiation factor 4E-BP for eukaryotic translation
(85-87).
This phosphorylation cascade enhances the translation of mRNAs
involved in ribosome biogenesis and protein synthesis (88-91).
A crucial aspect of the role of mTORC1 is that it enhances
ribosomal protein synthesis, which is necessary for cellular growth
and proliferation. A previous study revealed that mTORC1 notably
affects ribosome biosynthesis in lung cancer cells, emphasizing its
function in supporting tumor cell growth through enhanced protein
biogenesis (92). With the
increase in ribosome biosynthesis, mTORC1 accelerates the total
protein biosynthesis rate, thus driving energetic tumor cell
metabolism towards progression and malignant behavior. Furthermore,
the interplay between mTOR roles and oncogenic cascade is crucial
for understanding the function of mTOR in cancer development
(93).
mTORC1 activation is related to the oncogenic action
of Myc. Myc enhances the expression of genes involved in protein
biogenesis, and its activity is mainly mTOR-dependent,
demonstrating that mTOR inhibitor drugs may be an efficient
therapeutic approach in Myc-dependent tumors (94,95).
In contrast to the above, the dysregulation of mTOR signaling
triggers the abnormal metabolism of protein, causative of tumor
cell persistence and drug challenge, as demonstrated in research
investigating the effects of mTOR inhibitors in several tumor
models, including renal adenocarcinoma (96). Recent developments in cancer
research biology have shown that the function of mTOR is not merely
the regulation of protein biogenesis. Still, it extends to
controlling the balance of the anabolic and catabolic pathways,
such as autophagy. Under high nutrient availability, mTORC1
stimulates protein production while hindering autophagy, a safe
process for degrading excessive or malfunctioning proteins and
damaged cellular components. Genomic data showed that treatment
with rapamycin was related to a decline in the transporters of
neutral amino acids (97).
Suppressing either Raptor or Rictor reduces, whereas their mutual
knockdown blocks transport action for alanine and leucine amino
acids (98). In addition, mTORC1
controls the transfer of the aforementioned transporters through
the inhibition of Nedd4-2 ubiquitin ligase activity, which
stimulates ubiquitination and diminished expression levels of the
mentioned transporters (99).
Moreover, the inhibition of mTOR activity with Torin 1 diminishes
the transcription of the gene for amino acid enzymes and
transporters essential for amino acid and protein metabolism
(100).
Furthermore, mTORC2 is directly engaged in the
metabolism and transport of amino acids through Ser26
phosphorylation of cysteine-glutamate antiporter, which impedes the
glutamate output and cysteine input (101). Therefore, the aforementioned data
support the critical roles of mTORC1 and mTORC2 in modulating
protein metabolism and synthesis by regulating the transporter
activity and/or level of amino acids. mTOR regulates asparagine
synthetase (ASNS) expression levels (102,103). mTORC1 controls the ASNS
expression level (104). In lung
adenocarcinoma, the elevated level of KRAS promotes ASNS activity
that occurs through AKT and Nrf2, facilitating the stimulation of
ATF4. Mutually AKT inhibition with the reduction of cellular
asparagine inhibits cancer growth (105). Rapamycin stimulates the gene
machinery of argininosuccinate synthetase-1 (ASS1), the crucial
enzyme for arginine synthesis (98). Arginine-deficient cancers, for
example hepatocellular carcinoma and melanoma, are resistant to
arginine deiminase therapy that reduces the cellular arginine and
increases ASS. These tumors become primarily sensitive to dual
inhibitors of PI3K and AKT (106); thus, mTORC2 is involved in
arginine biosynthesis. De novo serine and glycine synthesis
are also increased by mTORC1 via ATF4, the transcription factor
(107). Ultimately, the mTORCs,
through an indirect pathway, may be involved in protein and amino
acid synthesis via their activity in other metabolic signaling
cascades.
This paradox is critical for appreciating the
metabolic modification of tumor cells, as a high mTOR activity can
increase survival by inhibiting the autophagy pathway, permitting
an accumulation of the necessary proteins for fast cell progression
and proliferation (108).
Therefore, targeting mTORC1 and mTORC2 therapeutically interrupts
this balance, stimulating autophagy to fight carcinogenesis
efficiently.
The importance of the mTOR pathway in controlling FA
metabolism is a primary mechanism enabling tumor growth and
persistence Fig. 2. Cancer cells
create fatty acids for lipid membrane, lipid alterations, and
signaling cascade action (103-109).
FA synthesis requires acetyl-CoA and cytosolic form (NADPH) of the
reducing agents. Dynamic FA synthesis relies on the incorporation
of additional carbon metabolic signaling pathways and
oxidation-reduction reactions. Glucose is the main precursor of
acetyl-CoA for FA synthesis in the majority of culture media
(110-112).
In hypoxic and mitochondrial malfunction, acetate
glutamine and leucine breakdown are another resource for acetyl-CoA
(113-117).
Additionally, study revealed that FAs synthesis is controlled by
the sterol regulatory element-binding protein 1 (SREBP-1)
transcription factor (118).
SREBP-1 regulates the enzymes needed to transfer the acetyl-CoA
into fatty acid and the PPP enzymes needed to convert acetate and
glutamine to acetyl-CoA. Thus, SREBP-1 controls genes that help or
catalyze FA synthesis (119). In
tumor cells with an abnormal rate of FA synthesis, the mTOR
pathway, across its subsequent target S6 Kinase (S6K), stimulates
and promotes the transcription activity of both SREBP-1 and SREBP
2. The ultimate proteins regulate the transcription activity of
different genes that relate to sterol biosynthesis (120). Therefore, SREBP-1 and SREBP-2 are
essential for mTOR to promote and stimulate cell growth and
proliferation. Tumor and normal cells take FAs and lipids from the
extracellular sources to use them to maintain the synthesis of cell
membranes. In presence of growth factors, PI3K increases FAs and
lipid use and downregulates oxidation of lipid through
(β-oxidation) within the mitochondrial matrix to continue cell
growth and proliferation (121).
In tumor cells and under hypoxic conditions, the
mTOR pathway enhances endoplasmic reticulum action to sustain
protein biosynthesis via an extracellular resource of unsaturated
FAs. Moreover, ATP citrate lyase, the final enzyme that alters
acetate to acetyl-CoA, inhibits tumor cell growth and propagation
(122). Thus, targeting FA
synthesis and transport can reduce tumor cell growth and
proliferation. mTOR is a critical regulator of various metabolic
pathways in cancer cells, including fatty acid metabolism, which is
essential for understanding tumor biology and therapeutic
interventions. mTOR modulates metabolic processes by integrating
nutrient signals, growth factors, and stressors. Given that cancer
cells exhibit altered metabolism, often adapting to exploit various
substrates, the importance of mTOR in controlling the metabolism of
fatty acids stands out as a primary mechanism enabling tumor growth
and persistence.
Cancer cells have a reprogrammed metabolism to meet
the increased requirements for fuel and building blocks required
for fast proliferation. In this context, FAs function as primary
energy resources and synthesize different biomolecules, including
cell membranes and signaling biomolecules. mTOR plays a central
role in FA synthesis by controlling the expression and activity of
lipogenic enzymes. It has been demonstrated that mTORC1 directly
activates SREBP1, causing an increase in the expression of FA
synthase and subsequent FA synthesis (123). Consequently, the activating
mTORC1 pathway enhances lipogenesis, which is particularly relevant
in cancer types characterized by increased demand for lipids.
Moreover, PI3K/AKT/mTOR oncogenic cascade pathways
are frequently dysregulated in several types of cancer, further
enhancing lipogenic activities. For example, in thyroid gland
cancer, pyruvate carboxylase, an enzyme key for transferring
pyruvate to oxaloacetate, stimulates aggressive cancer action
partially through increased lipogenesis, which is controlled
through mTORC2 signaling (124).
This reveals that mTOR signaling not only accelerates the uptake of
FA, but also vigorously provides for their biosynthesis, providing
cancer cells with an adaptable energy source to acclimate under
unusual physiological stresses. Recent research suggests a growing
interest in how specific FAs influence mTOR signaling and metabolic
reprogramming in cancer cells through the high expression of
several enzymes that are essential for FAs metabolism, such as
carnitine transporter, CPT isoforms and CD36 in several types of
cancer, such as gastric and prostate cancer, and triple-negative
breast cancer (TNBC) (125). Some
researchers have investigated the function of omega-3 FAs that have
been shown to reduce mTORC1 signaling activity, theoretically
leading to diminished FA synthesis and boosting apoptosis in tumor
cells (126). This provides
prospective opportunities for a therapeutic approach, as combining
mTOR inhibitors with dietary FA variation could provide a
dual-target system to hinder tumor cell growth and stimulate
metabolic susceptibility.
mTORCs are key regulators of cell metabolism,
specifically from the perspective of nucleotide biosynthesis, which
is necessary for tumor cell development and proliferation (Fig. 2). Nucleotide biosynthesis (purine
and pyrimidine) is critical in rapidly-dividing cells,
particularly, in cancer cells with sufficient precursors for RNA
and DNA synthesis. The nucleotide biosynthesis is a complex
process, demanding enormous support from additional metabolic
signaling pathways in a specific manner. R5P is the intermediate
product of the PPP that provides the main sources for
phosphoribosylamine and glutamine. R5P also functions as an amide
precursor for nucleotide biosynthesis (127,128). In addition, one carbon cascade is
an additional resource for different non-methyl-group and
non-essential amino acids that are involved in pyrimidine purine
biosynthesis. Moreover, the TCA cycle supplies the oxaloacetate
that is transaminated to aspartate, and its vital element that is
necessary for nucleotides base synthesis (129-131).
Therefore, the inhibition of nucleotide biosynthesis by
pharmacological therapies may be an effective approach for
eradicating specific tumors, such as antifolates and nucleosides
that have been used in the treatment of cancer for a number of
years (132).
The activation of the mTORC1 pathway has been shown
to stimulate pyrimidine biosynthesis (132). Ribosomal K6S stimulates the
phosphorylation of carbamoyl-phosphate synthetase 2, aspartate
transcarbamylase, and dihydroorotase (CAD), a specific enzyme that
stimulates the first reaction of pyrimidine synthesis (133,134). mTORC1 actively stimulates
nucleotide breakdown by controlling the upregulation of central
enzymes in de novo nucleotide biosynthesis. Research has revealed
that mTORC1 stimulation boosts the biosynthesis of phosphoribosyl
pyrophosphate amidotransferase, a key enzyme in the purine
biosynthesis pathway. This control enhances the purine nucleotide
level that supports tumor cell growth and progression (135).
Moreover, mTORC1 incorporates signals from different
nutrient availability, mainly amino acids, to certify that
nucleotide synthesis is associated with the metabolic requests of
rapidly growing cancers. In addition to supporting nucleotide
biosynthesis, the mTORC1 pathway is highly linked to controlling
metabolic cascades that produce R5P, a key factor for the
biosynthesis of nucleotides. The PPP produces R5P from
glucose-6-phosphate, emphasizing the interconnection between the
glycolytic pathway, mTOR signaling, and metabolism of nucleotides
(136-138).
When glucose freely exists, mTORC1 triggers cellular propagation by
increasing glycolysis and nucleotide biosynthesis signaling
pathways, assisting tumor cells to proliferate quickly.
Furthermore, the communication between mTORC2 and
oncogenic cascades, such as the PI3K/AKT signaling pathway, further
highlights the importance of mTORC2 in nucleotide synthesis and
tumorigenesis. For example, PI3K/AKT pathway activation triggers
mTORC2, stimulating fat and nucleotide biosynthesis that is
critical for cancer cell growth. mTORC2 signaling is often
dysregulated in breast cancer; targeting this pathway can impede
nucleotide synthesis, impairing cancer growth (139-143).
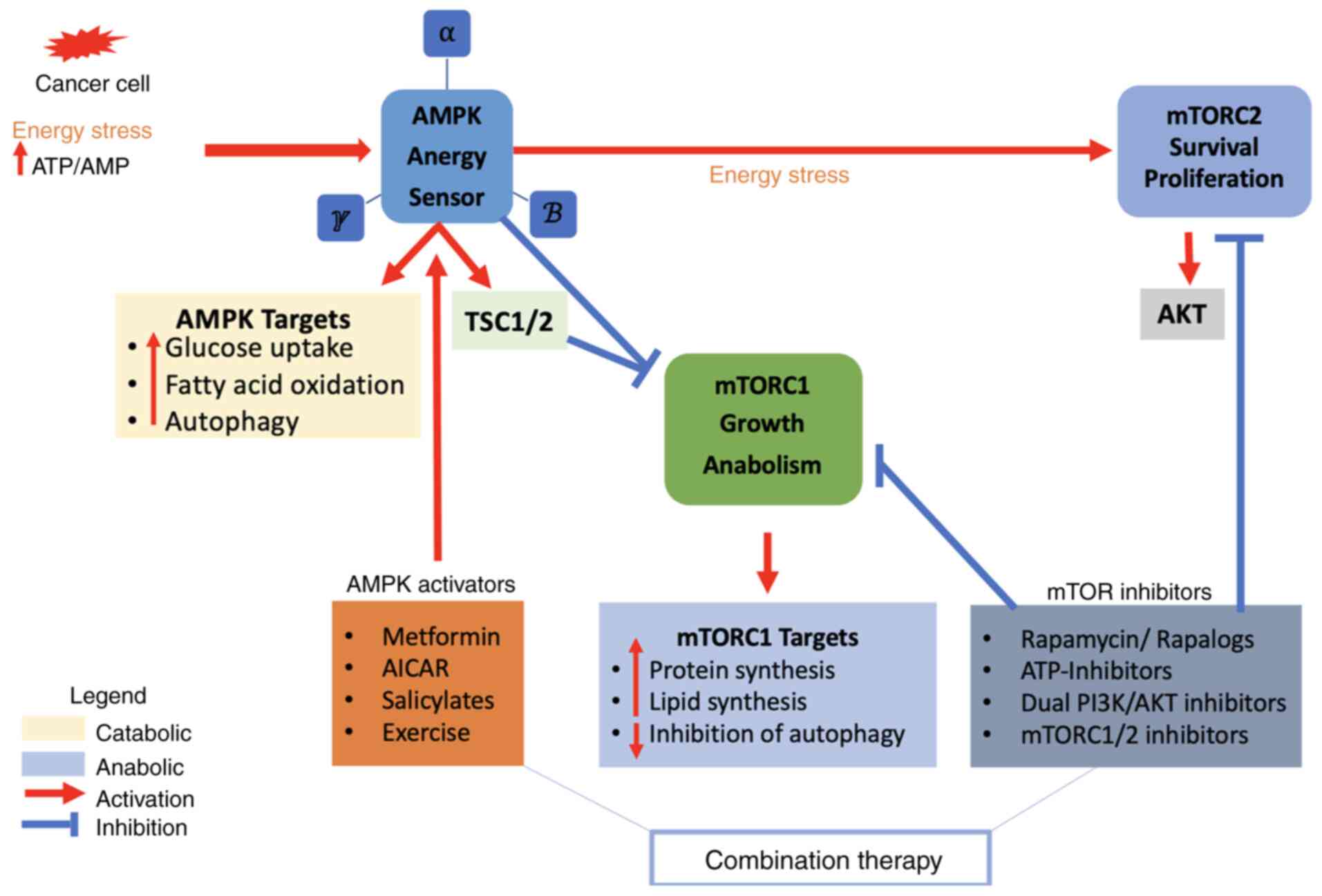
The mTOR complexes, mTORC1 and mTORC2, are pivotal
regulators of cancer cell metabolism by integrating nutrient
availability with signals that drive cell growth and proliferation
(Fig. 3). AMPK is an intracellular
sensor for energy equilibrium, which is presented in each
eukaryotic cell. AMPK consists of three subunits; (α) is the
enzymatic subunit and (β and γ) are the controlling subunits.
Different isoforms have been encoded by certain genes for each
submit in humans (144). AMP/ATP
and ADP/ATP ratios stimulate AMPK during severe energy stresses.
The activation of AMPK restores energy redox hemostasis by turning
on the catabolic cascade and turning off the anabolic cascades to
increase ATP production and maintain the energy equilibrium
(145,146). Moreover, AMPK stimulates glucose
consumption through the oxidation process in low proliferative
cells rather than rapid glucose utilization and glycolysis that are
employed in high proliferative cells (147,148). In yeast, the AMPK analog is
essential to shift the metabolism of yeast from fermentation
(glycolysis) to OXPHOS metabolism under severe starvation
situations. This rewired metabolism is similar to the reversal of
aerobic glycolysis (Warburg effect) that occurs in a number of
rapidly-growing cells, such as cancer cells (128). Salicylate and metformin are the
pharmacological activators of AMPK (149,150). They activate AMPK through
threonine residue (Thr172) phosphorylation and AMP binding
(151-153).
The actions of AMPK occur through three different mechanisms: i)
The enhanced phosphorylation of Thr 172 via LKB1; LKB1 is a tumor
suppressor, and it is the main upstream target of AMPK; ii) reduced
Thr172 dephosphorylation by a phosphatase; and iii) activated
allosteric stimulation. All AMPK activations occur due to AMP
binding to itself and not due to the upstream kinase outcome or
phosphatase (152-154).
Together, the complex interaction between mTORCs and AMPK signaling
cascades signifies a crucial metabolic step that guides cells to
anabolic processes that stimulate cancer development or turn to
catabolic process pathways that may defeat carcinogenesis.
AMPK redeems the energy balance following its
activation via energy stress by activating the catabolic signaling
cascade to produce sufficient ATP and by reducing the anabolic
cascades that use ATP (154,155). The loss of a single allele of
AMPK-α increases the development of B-cell lymphoma in mice with
c-Myc transgenic expression in B-cells, whereas the loss of both
alleles prompts a severe outcome (156). Therefore, this reveals that AMPK
may serve as a tumor suppressor, and any mutation in the genes of
AMPK subunits is uncommon in human tumors. Consequently, diminished
AMPK actions may be necessary for cancer cell survival and
malignancy by constraining the specific effects of AMPK on cell
development and propagation (157). The activation of AMPK is a
suitable approach to decrease tumor cell metabolism by hindering
the anabolic cascades and stimulating the catabolic cascades.
Histological slides from a human breast tumor study indicated that
the phosphorylation ratio of AMPK-α enzymatic subunits was less
than that of adjacent normal tissue (158). Another study revealed that the
phosphorylation of AMPK-α is a common occurrence in patients with
hepatocellular carcinoma (159).
Additionally, another study demonstrated that AMPK is a negative
switch of the Warburg effect in cell and animal models (160). Several mechanisms have been
suggested to elucidate the reduction of AMPK activity. For example,
the genetic damage of LKB1 is relatively common in cervical and
non-small cell lung tumors but not in other tumors (161-163).
The activation of insulin/IGF1 controls the protein kinase AKT/PKB
is additional machinery for downregulation of AMPK, gain function
genes mutations of AKT and loss-of-function mutations of PTEN
(tumor suppressor) that happen in various tumors (164-166).
Collectively, the regular arrangement of AMPK inactivation across
various tumor types through several biomolecular mechanisms such as
downregulation of LKB1, loss of PTEN and Akt/PKB upregulation,
highlights the basic roles of AMPK as an energy and metabolic
master, which represents a hopeful therapeutic approach for
correcting the unusual energy status that supports cancer cell
persistence and propagation.
In addition, AMPK has an essential pro-survival
function in hepatocellular carcinoma, pancreatic ductal
adenocarcinoma (PDAC) and AML through various mechanisms. A key
feature of AMPK is its role in buffering ROS, which are frequently
raised in tumor cells due to high metabolic requirements to support
tumorigenesis characters. The activation of AMPK enhances the
antioxidant ability by stimulating the main enzymes for antioxidant
activity, such as superoxide dismutase and glutathione (GSH;
specifically, by modifications of the regulatory metabolism of GSH
and regeneration pathways of NADPH), thus reducing oxidative stress
and supporting cell survival (167,168). Additionally, in the cancer
microenvironment, AMPK stimulates metabolic modifications, such as
increasing the oxidation of FA and downregulating the mTOR cascade
by phosphorylating acetyl-CoA carboxylase and fatty acid synthase,
allowing tumor cells to generate a sufficient amount of ATP under
different unfavorable and stressful conditions (169). Moreover, AMPK stimulates cell
cycle progression in hepatocellular carcinoma by modifying the
essential enzymes that are necessary for cell-cycle checkpoints
system, mainly the transition of G1/S (170). Under different stress conditions,
AMPK stimulates the phosphorylation of the p53-p21 axis, and
PERK-eIF2α in hepatocellular carcinoma, PDAC and AML respectively,
thus promoting cell cycle arrests, preventing DNA damage and
promoting cell survival (170-172).
PDAC, hepatocellular carcinoma and AML cells utilize the activation
of AMPK to support cancer progression under severe metabolic stress
by stimulating the autophagy regulators, such as beclin 1(BECN1),
unc-51 like autophagy activating kinase 1(ULK1) and transcription
factor EB (TFEB), promoting the degradation of protein aggregates
and damaged microgranules that would then stimulate the apoptotic
pathway (173). Recently, a
previous study have revealed that the activation of AMPK in AML
cells induces the downregulation of mTOR and increases autophagy,
generating a metabolically challenging phenotype that leads to drug
resistance (172). AMPK also
controls mitochondrial dynamics and biogenesis, confirming that
leukemic cells preserve adequate bioenergetic ability for survival
and cancer progression (174).
Taken together, these mechanisms emphasize the roles of AMPK in
regulating tumor cell survival, proposing its prospective approach
as a therapeutic strategy in hepatocellular carcinoma, PDAC and
AML.
Furthermore, the roles of AMPK depend on the
mechanistic foundation for this functional shift that present
inside cellular microenvironment such as nutrients levels and
hypoxia (175). Through the early
steps of cancer transformation, when cells preserve sufficient
nutrients and oxygen, AMPK stimulation functions as an energy check
that inhibits uncontrolled propagation by preventing main anabolic
cascade, such as FA biosynthesis, protein transformation and the
biosynthesis of nucleotides. The oppressive function of AMPK is
facilitated across the phosphorylation of acetyl-CoA carboxylase,
leading to reduced FA biosynthesis, and mTORC1 inhibition, which
diminishes protein biosynthesis and cell progression (176). Moreover, AMPK stimulates
mitochondrial genesis and autophagy processes that typically helps
the cellular microenvironment, but can ironically help established
cancers facing sever nutrient stress (177). Therefore, the shifting from
cancer suppressor to cancer promoter occurs when tumor cells face
metabolic restrictions through the cellular environment, where the
pro-survival roles of AMPK become beneficial for cancer growth and
development.
In breast cancer, AMPK displays perhaps the most
well-known dual roles, with its function determined mainly by tumor
stage, phenotype and microenvironmental situations (178). In hormone receptor-positive
breast cancers, AMPK stimulation is associated with improved
patient outcomes and minimized tumorigenesis (179). The tumor suppressive properties
are facilitated through the ability of AMPK to inhibit the
mevalonate cascade, decreasing cholesterol biosynthesis that is
critical for ER-receptor stimulation, and through the direct
suppression of FAS, constraining cell membrane biogenesis required
for rapid cell proliferation and division (180). Metformin, as indirect activator
of AMPK, exhibits significant anti-tumor roles in this situation by
stimulating metabolic stress and increasing insulin sensitivity
(181).
Conversely, in TNBC, AMPK mainly has an oncogenic
function and promotes metastasis. Under sever low O2
conditions, commonly observed in severe aggressive breast cancers,
AMPK enables metabolic switching to glycolysis and supports
autophagy-mediated persistence. This is mainly evident in
drug-resistant TNBC cells, where the activation of AMPK assists
survival during nutrients stress induced by cytotoxic compounds
(182). The protein biosynthesis
promotes cancer cell metastasis through controlling the dynamics of
cytoskeletal and stimulates blood vessels growth via HIF-1α
maintenance (183). Moreover, in
HER2+ breast cancers treated with trastuzumab,
AMPK-induced autophagy provides a resistance mechanism by reusing
cellular element to sustain energy equilibrium during targeted
drug-promoted stress (184).
In addition, lung adenocarcinoma presents a special
model for acknowledging the dual roles of AMPK due to the high
mutation rate pf LKB1, the main upstream signaling kinase that
controls the activation of AMPK (185). In the presence of wild-type LKB1,
AMPK sustains its tumor suppressive roles across traditional
mechanisms, such as the activation of TSC2 and the final inhibition
of mTORC1(186). Lung cancer
reacts positively to severe metabolic stress and exhibits a
response to AMPK activators, such as metformin and AICAR (187). The tumor inhibition features are
highlighted through the inhibition of glycolysis flux, increased
oxidative metabolism, and sustained cell cycle checkpoints that
stimulate apoptosis in response to oncogenic burden and stress
(188). Nevertheless, ~30% of
non-small lung cancer (NSLCS) cases harbor LKB1 mutations,
primarily shifting the functions of AMPK and removing its tumor
suppressive ability. In these cancers, different signaling cascades
compensate for the damage of AMPK-facilitated metabolic
controlling, leading to increase in addiction on glutamine and
glucose metabolism (189).
Paradoxically, while AMPK is activated in LKB1-mutant lung
adenocarcinoma, it can stimulate tumor propagation by accelerating
metabolic modification to nutrient restriction and oxidative stress
(190). This is mainly
significant in KRAS-dependent lung adenocarcinoma, where AMPK
supports maintain redox balance during oncogene-mediated metabolic
stress (191). Moreover, hypoxic
areas within lung cancers manipulate AMPK to stimulate VEGF
upregulation and increase angiogenesis, promoting cancer
vascularization and progression (192).
Moreover, AMPK displays a dual role in colorectal
cancer progression through its complex connections with different
inflammatory and metabolic signaling pathways (193). In the initial stages of
colorectal cancer, mainly in the stage of inflammatory intestine
disease and abnormal polyposis, AMPK plays a protective role by
inhibiting the chronic inflammatory reaction inside bowel and
decreasing oxidative damage by inhibiting the NF-κB pathway
activity, thus decreasing assembly of pro-inflammatory elements
such as cytokines that trigger cancer initiation and development
(194). AMPK also inhibits the
Wnt/β-catenin pathway in APC-mutant colorectal tumors, reducing one
of the main drivers of colorectal cancer growth (195). As colorectal cancer proceeds to
more advanced phases, the function of AMPK changes towards cancer
promotion, specifically in the mediating of hepatic metastases
where metabolic needs are enhanced. In the advanced stages of
colorectal cancer, cells use AMPK-drive metabolic reprograming to
modify to the liver microenvironment that differs drastically from
the colonic tissue in terms of metabolic availability and nutrients
requirements (196). Hypoxic
areas within colorectal tumors employ AMPK to sustain glycolysis
bioproducts and stimulate autophagy-promote survival during periods
of hypoxic angiogenesis. Additionally, AMPK accelerates resistance
to regular drugs such as 5-fluorouracil by increasing nucleotide
salvage cascades and supporting cellular energy equilibrium through
treatment-mediated metabolic stress (197).
Additionally, hepatocellular carcinoma provides a
convincing model of how AMPK functions under different metabolic
stress conditions. In the pre-carcinogenesis stage of fatty liver
and steatohepatitis, AMPK functions as an essential tumor
suppressor by inhibiting fatty liver accumulation and decreasing
oxidative damage. AMPK inhibits SREBP-1C to trigger lipogenesis and
β-oxidation, thus diminishing the inflammatory environment that
prompts the progression of hepatocellular carcinoma (198). This protective role is mainly
evident in patients with diabetes mellitus, when AMPK is activated
through metformin and provides hepatoprotective benefits,
decreasing the risk of developing hepatocellular carcinoma
(199). Once hepatocellular
carcinoma develops, particularly in conditions of chronic hepatic
diseases and cirrhosis, AMPK shifts to a tumor-accelerator role. In
the microenvironment with severe metabolic deprivation and hypoxia,
the metabolic modifications of AMPK become beneficial for
carcinogenesis (200). In
hepatocellular carcinoma, AMPK reprograms glutamine metabolism as
the main energy source when glucose sources become limited,
stimulating autophagy-enhanced persistence during sorafenib
treatment (201). AMPK also
accelerates modification to the unique metabolic difficulties of
the liver microenvironment, where competition for metabolic
elements with adjacent hepatocytes and immune cells generates
selective pressure for metabolically adaptable cancer cells
(202,203).
The mechanistic interaction between the redox
metabolic roles of AMPK and hormonal signaling pathways to regulate
cancer behavior has also been observed in prostate cancer (PC). In
hormone-sensitive prostate cancer, AMPK displays well-defined tumor
suppressive roles through its connections with the AR signaling
pathway in androgen-sensitive PC (204). The activation of AMPK decreases
AR transcription directly and indirectly through
phosphorylation-trigger inhibition and via the suppression of
mTORC1 activity, respectively, which is necessary for AR protein
biosynthesis (205). AMPK also
reduces lipid biosynthesis cascade, which are necessary for
building blocks to support high PC proliferation and arrest the
cell cycle through p21 stimulation (206). These results provide good
outcomes for patients with identified PC who sustain usual
metabolic function. During the transition to castration resistance,
PC marks an important shift in the function of AMPK towards a tumor
developer (207). AMPK become an
essential player in metabolic reprograming and tumor survival in
the AR-depleted milieu formed by AR deprivation chemotherapy. In
the androgen-depleted environment created by androgen deprivation
therapy, AMPK becomes essential for metabolic adaptation and
survival. PC cells manipulate AMPK to stimulate AR-VS splicing
(alternative splicing of AR variants) that maintain main activity
in the absence of AR, accelerating hormone-independent progression
(208). Under the low
O2 conditions normally established in advanced-stage PC,
AMPK stimulates metabolic switching from OXPHOS to glycolysis and
increases autophagy-facilitated survival in docetaxel resistance PC
patients. Survival function is particularly evident in docetaxel
resistant (207,208).
A number of anabolic signaling pathways are
activated by the mTORC1 pathway, which, in the end, inhibits AMPK
activation (209). This
inhibition occurs through various mechanisms, such as TSCs
phosphorylation or the phosphorylation of Raptor, a regulatory part
of mTORC1 (210,211). Thus, AMPK activation inhibits the
biosynthesis of lipids, nucleotide, and proteins, reducing cell
growth and proliferation. Moreover, it arrests the cell cycle at
the G0/G1 phase by stimulating p53 phosphorylation, thus preventing
DNA biosynthesis (212-214).
Conversely, it has been demonstrated that AMPK can directly
stimulate mTORC2 by phosphorylating its parts, supporting cell
growth and survival during severe energetic stress (215,216). Therefore, both roles of the AMPK
regulator with the inhibition of mTORC1 stimulate anabolic
pathways; the potential stimulation of mTORC2 highlights a
multifaceted association that should be cautiously modified to
accomplish efficient tumor therapy.
In various types of malignancies, such as
colorectal, PDAC and glioblastoma, the association between mTORC1
and AMPK is necessary for cell progression under metabolic stress
environments. Data from hepatocellular carcinoma study have
established that the deprivation of amino acids and the later
re-addition of leucine lead to a specific alteration in mTORC1
activity, with the consequent stimulation of AMPK during glutamine
deficiency, promoting the inhibition of the mTORC1 cascade
(217). This suggests that the
glutamine level and nitrogen source, which modify the metabolic
environment, can significantly regulate mTORC1 stimulation across
the AMPK signaling pathway, suggesting a feasible therapeutic area
for tumor glutamine-dependent metabolism. Furthermore, data from
T-cell acute lymphoblastic leukemia studies have revealed that AMPK
stimulation is crucial for glycolytic intermediates
ratio/mitochondrial function and metabolism, consequently
accelerating the downregulation of mTORC1 in situations of severe
metabolic stress (218,219). Additionally, emerging data from
an AML study have revealed a complex association between the role
of AMPK and mTORC1; these appear to function through two parallel
essential cascades that connect the activation of mTORC1 and
glucose metabolism to control the complete cellular homeostasis
(220). The co-activation of AMPK
and mTORC1 within specific leukemia cells demonstrates the
paradoxical landscape of these metabolic pathways, with AMPK
fostering OXPHOS, whereas mTORC1 mainly triggers glycolysis. This
indicates that controlling AMPK activity could produce distinctive
effects in metabolic reprogramming, conditional on the specific
cancer environment (221,222). Furthermore, the link between
mTORC2 and AMPK is progressively acknowledged as a critical factor
in regulating the metabolism of several cancers and treatment
responses. While mTORC1 has been significantly studied for its
function in growth signaling, mTORC2 is more strictly connected
with cell survival and metabolism through switching, as for
example, metabolic deprivation and the withdrawal of growth
factors. Data from recent project on colorectal cancer have
demonstrated that AMPK stimulation can boost mTORC2 signaling,
stimulating cell growth and survival under different nutrient
deprivations (223). This
association suggests a complex response mechanism. At the same
time, AMPK usually serves to impede mTORC1 activities in a low
energy environment. Still, it can also maintain mTORC2, thus
permitting the stimulation of pro-survival metabolic pathways
throughout AKT phosphorylation, finally modifying cancer
aggressiveness (224). Study on
lung cancer that are resistant to EGFR inhibitor therapy have
demonstrated that mTORC2 is necessary for mediating resistance
against the aforementioned inhibitors. Upregulated mTORC2 helps to
sustain lung cancer cell propagation despite EGFR inhibitors
(225). AMPK shapes cellular
reactions in such models, theoretically stimulating a shift towards
an aerobic glycolysis pathway to meet energy demands under
therapy-induced stress. The role of AMPK not only affects mTORC2,
but also overlaps with additional oncogenic cascades, encouraging
metabolic switches that maintain cell growth and proliferation
(226). These data mutually
demonstrated that the AMPK/mTORC1 signaling pathway functions as a
in adynamic and metabolic control, where the stability between
these two contrasting cascades regulate tumor cell outcome and
metabolic elasticity, emphasizing the essential for accuracy
therapeutic methods that need for cancer-specific metabolic
addictions and microenvironmental restrictions.
Moreover, from the perspective of pancreatic cancer,
the mTORC2 pathway has been connected to modifying metabolic
flexibility and response to different therapies, such as PARP
inhibitors. A study in this area has confirmed that mTORC2
signaling controls DNA repair by regulating BRCA1(227). The precise interaction between
AMPK and BRCA1-dependent processes is still being disclosed.
However, it highlights the possibility of the AMPK-regulated
modulation of mTORC2 increasing therapeutic sensitivity, possibly
providing dual therapies that control these signaling pathways.
Furthermore, the notion that mTORC2 provides glycolytic switching
in response to elevated glutamine levels underlines the necessity
to appreciate the dynamic interaction between metabolic
availability and mTORC2 action through AMPK (228). In a study on glioblastoma, the
AMPK/mTORC2 axis displayed its functions in stimulating cell
survival during nutrient deprivation, which is critical as these
tumor cells frequently encounter shifting metabolic environments
within tumors. The upregulation of mTORC2 by AMPK increased AKT
signaling, which helps support cellular stability and functions and
escape apoptosis when the growth factor is diminished (229). This role of AMPK may appear
paradoxical given its established function in inhibiting mTORC1;
however, it affirms the importance of context-dependent signaling
where AMPK activation can favor survival by engaging mTORC2 while
simultaneously restraining excessive mTORC1 activity.
The clinical efficiency of mTOR inhibitors is
dependent on the progress of developed resistance; there are
numerous connected molecular mechanisms that have been
significantly described. The PI3K/AKT/mTOR signaling pathway is a
highly conserved signal transduction pathway in eukaryotic cells
that stimulates cell survival, cell progression, and cell cycle
events, and any malfunction of this signaling pathway can prompt to
tumor development (230). Tumor
cells can modify their response against mTOR inhibitors by
stimulating compensatory persistence pathways that overcome the
therapeutic blockade through various key processes.
The stimulation of receptor tyrosine kinases, such
as IGF-1R, HER2/neu and EGFR is one of the highest prominent
resistance mechanisms against mTOR inhibitors. These receptors can
be overactivated in response to mTOR inhibitors, generating other
signaling pathways that maintain cellular growth and development
(231). Another study has
confirmed that resistance to PI3Kβ/AKT inhibition in PTEN-null
breast cancer cells is conferred by the loss of specific genes in
the PI3K/AKT/mTOR pathway, including TSC1/2, INPPL1 and PIK3R2
relatively than genes connected with different signaling cascades
(232). This result emphasizes
there is an intrinsic pathway of resistance.
Moreover, mutations of S6K1 and 4E-BP1 can reduce
cell dependence on the mTOR pathway for protein biosynthesis and
development regulation. Cancer cells have a special metabolic
shifting that represents another critical feature of developed
resistance, where persistent mTOR inhibition pushes tumor cells to
increase their dependance on different metabolic signaling
pathways, such as GLUT1 stimulation to increase glucose usage
through, aerobic glycolysis, and stimulation of autophagy for
prorogation purpose (233).
Additionally, histone modification, DNA methylation are epigenetic
modifications that significantly contribute to the secure
resistance for several phenotypes, making these modifications
mainly challenging to available chemotherapy (234).
PI3K/AKT rebound activation can be the highest
clinically important limitation of mTOR inhibitors. This phenomenon
occurs through the disturbance of negative-feedback loops in the
PI3K/AKT/mTOR axis (235). In
addition, the key mechanism has been recorded in the recent
literature, and it is described as the basic challenge for the
single mTOR inhibitor chemotherapy (236). The activation of mTOR causes a
negative feedback-loop control through S6K1 that decreases the
activity of PI3K and S6K1 phosphorylation deactivates IRS-1 that is
required for insulin signaling through PI3K (237). The degradation of IRS-1is
required for the subsequent and constant attenuation of PI3K/AKT
signaling under normal homeostasis conditions (238). Conversely, once mTOR is inhibited
using a single mTOR inhibitor, this negative loop is perturbed,
causing IRS-1 stabilization and accumulation. In addition, use of
mTOR inhibitor will prompt IRS-1stimulation releasing the negative
loop, resulting in paradoxical cascade restimulation (239).
Mechanistically, recent studies have specified more
details about the dynamic of the rebound activation phenomenon.
Inhibition of PI3K cascade blocks phosphorylation of 4E-BP1 through
mTOR which then inhibits translation of PTEN, causing a rebound of
AKT phosphorylation 2-4 h after-treatment (240). This rapid reactivation displays
that the compensatory reaction occurs within hours of treatment
initiation, efficiently reducing the therapeutic margin of mTOR
inhibitors. The accumulated IRS-1 then serves as an effective
upstream activator for PI3K, causing a strong phosphorylation and
activation for PI3K that can surpass the initial level detected
pretreatment (241). The
effective stimulation of PI3K helps to avoid the pro-apoptotic
action of mTOR inhibition, promote cell survival, propagation and
metabolic switching. The consequences of this challenge phenomena
it modifies the cytotoxic or cytostatic intervention effect into a
mysterious progression stimulating signal (242).
Imminent studies aim to employ the connections
between AMPK and mTORC2, mainly focusing on understanding how this
connection can be manipulated to improve treatment efficiency and
cope with resistance mechanisms in tumor cells (243). These adverse dual roles of AMPK
concurrently restraining mTORC1 whereas hypothetically boosting
mTORC2 cascades discloses a new metabolic persistence way that
supports tumor cells to sustain the cellular homeostasis under
metabolic stress, suggesting that efficient therapeutic approach
must hit both mTORCs in combination with AMPK signaling to stop
redeeming persistence mechanisms and shifting metabolic routes.
Targeting the AMPK/mTORC2 signaling cascade opens a
new avenue for groundbreaking tumor therapies and emphasizes
clarifying these complexes' signaling pathways to develop
wide-ranging therapy approaches. The double role of AMPK in
controlling both mTORCs clarifies new opportunities for targeted
treatment approaches in several cancers is illustrated in (Fig. 3).
The complex metabolic switching of cancer cells is
one of the most basic features of tumorigenesis, with AMPK and mTOR
complexes serving as the primary regulators of this pathological
alteration. The metabolism of cancer cells is well-known for its
notable flexibility, enabling tumor cells to survive and
proliferate under different microenvironmental situations through a
metabolic rewiring strategy. The dysregulation of the AMPK/mTOR
crosstalk orchestrates a wide-ranging metabolic renovation in tumor
cells, endorsing enhanced glucose utilization, modified lipid
metabolism, enlarged amino acid uptake, and adjusted nucleotide
biosynthesis to meet the eminent biosynthetic requirements of
rapidly dividing cells. This metabolic elasticity enables tumor
cells to manipulate the Warburg effect, preferring glycolysis even
in oxygen-rich environment, but concurrently sustaining the ability
to use alternative energy sources during nutrient paucity.
The paradoxical role of AMPK in cancer cell
metabolism, acting as both a tumor suppressor through metabolic
control and a survival helper under a severe stress environment,
highlights the complex metabolic modification mechanisms that tumor
cells utilize. Moreover, the metabolic divergence and heterogeneity
detected across different tumor types and even across specific
tumors reveal complex interactions between AMPK/mTOR complexes
signaling pathways and tissue-specific metabolic demands.
Understanding and clarifying these metabolic
complexities is critical for formulating precision innovative
oncology strategies to efficiently eradicate tumor cell metabolism
without damaging normal cell function. As research advances towards
more sophisticated, creative therapeutic approaches, manipulating
the distinctive metabolism of cancer cells across a targeted
variety of AMPK/mTOR pathways provides immense promise for
inventing metabolically based therapy strategies that can subdue
therapeutic challenges and advance patient outcomes against
cancer.
Not applicable.
Funding: No funding was received.
Not applicable.
AA conceived and supervised the review. AA wrote the
draft of the review. SC reviewed and edited the review. AA and NF
revised the review and prepared the figures. All the authors have
read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Fan H, Wu Y, Yu S, Li X, Wang A, Wang S,
Chen W and Lu Y: Critical role of mTOR in regulating aerobic
glycolysis in carcinogenesis (review). Int J Oncol. 58:9–19.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hua H, Kong Q, Zhang H, Wang J, Luo T and
Jiang Y: Targeting mTOR for cancer therapy. J Hematol Oncol.
12(71)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zarogoulidis P, Lampaki S, Turner JF,
Huang H, Kakolyris S, Syrigos K and Zarogoulidis K: mTOR pathway: A
current, up-to-date mini-review (review). Oncol Lett. 8:2367–2370.
2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lien EC, Lyssiotis CA and Cantley LC:
Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer.
Recent Results Cancer Res. 207:39–72. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mossmann D, Park S and Hall MN: mTOR
signaling and cellular metabolism are mutual determinants in
cancer. Nat Rev Cancer. 18:744–757. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ramanathan A and Schreiber SL: Direct
control of mitochondrial function by mTOR. Proc Natl Acad Sci USA.
106:22229–22232. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang N, Wang B, Maswikiti EP, Yu Y, Song
K, Ma C, Han X, Ma H, Deng X, Yu R and Chen H: AMPK-a key factor in
crosstalk between tumor cell energy metabolism and immune
microenvironment? Cell Death Discov. 10(237)2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Torres Acosta MA, Gurkan JK, Liu Q,
Mambetsariev N, Reyes Flores C, Helmin KA, Joudi AM,
Morales-Nebreda L, Cheng K, Abdala-Valencia H, et al: AMPK is
necessary for Treg functional adaptation to microenvironmental
stress during malignancy and viral pneumonia. J Clin Invest.
135(e179572)2025.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Marafie SK, Al-Mulla F and Abubaker J:
mTOR: Its critical role in metabolic diseases, cancer, and the
aging process. Int J Mol Sci. 25(6141)2024.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Smiles WJ, Ovens AJ, Kemp BE, Galic S,
Petersen J and Oakhill JS: New developments in AMPK and mTORC1
cross-talk. Essays Biochem. 68:321–336. 2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Keerthana CK, Rayginia TP, Shifana SC,
Anto NP, Kalimuthu K, Isakov N and Anto RJ: The role of AMPK in
cancer metabolism and its impact on the immunomodulation of the
tumor microenvironment. Front Immunol. 14(1114582)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhang T, Wang X, Alexander PG, Feng P and
Zhang J: An analysis of AMPK and ferroptosis in cancer: A potential
regulatory axis. Front Biosci (Landmark Ed).
30(36618)2025.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liao H, Wang Y, Zou L, Fan Y, Wang X, Tu
X, Zhu Q, Wang J, Liu X and Dong C: Relationship of mTORC1 and
ferroptosis in tumors. Discov Oncol. 15(107)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sun Z, Liu L, Liang H and Zhang L:
Nicotinamide mononucleotide induces autophagy and ferroptosis via
AMPK/mTOR pathway in hepatocellular carcinoma. Mol Carcinog.
63:577–588. 2024.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang X, Tan X, Zhang J, Wu J and Shi H:
The emerging roles of MAPK-AMPK in ferroptosis regulatory network.
Cell Commun Signal. 21(200)2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lee H, Zandkarimi F, Zhang Y, Meena JK,
Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al:
Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat
Cell Biol. 22:225–234. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sun Q, Zhen P, Li D, Liu X, Ding X and Liu
H: Amentoflavone promotes ferroptosis by regulating reactive oxygen
species (ROS)/5'AMP-activated protein kinase (AMPK)/mammalian
target of rapamycin (mTOR) to inhibit the malignant progression of
endometrial carcinoma cells. Bioengineered. 13:13269–13279.
2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chen W, Zhao H and Li Y: Mitochondrial
dynamics in health and disease: Mechanisms and potential targets.
Signal Transduct Target Ther. 8(333)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Crosas-Molist E, Graziani V, Maiques O,
Pandya P, Monger J, Samain R, George SL, Malik S, Salise J, Morales
V, et al: AMPK is a mechano-metabolic sensor linking cell adhesion
and mitochondrial dynamics to Myosin-dependent cell migration. Nat
Commun. 14(2740)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu Q, Zhang L, Zou Y, Tao Y, Wang B, Li
B, Liu R, Wang B, Ding L, Cui Q, et al: Modulating p-AMPK/mTOR
pathway of mitochondrial dysfunction caused by MTERF1 abnormal
expression in colorectal cancer cells. Int J Mol Sci.
23(12354)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu A, Lu X, Song Y, Pei J and Wei R: ACK1
condensates promote STAT5 signaling in lung squamous cell
carcinoma. Cancer Cell Int. 25(237)2025.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Mehta S and Zhang J: Liquid-liquid phase
separation drives cellular function and dysfunction in cancer. Nat
Rev Cancer. 22:239–252. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Peng Q, Tan S, Xia L, Wu N, Oyang L, Tang
Y, Su M, Luo X, Wang Y, Sheng X, et al: Phase separation in cancer:
From the impacts and mechanisms to treatment potentials. Int J Biol
Sci. 18:5103–5122. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Maqsood Q, Sumrin A, Saleem MZ, Perveen R,
Hussain N, Mahnoor M, Akhtar MW, Wajid A and Ameen E: An insight
into cancer from biomolecular condensates. Cancer Screen Prev.
2:177–190. 2023.
|
|
26
|
Li Y, Feng Y, Geng S, Xu F and Guo H: The
role of liquid-liquid phase separation in defining cancer EMT. Life
Sci. 353(122931)2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Warburg O and Minami S: Versuche an
Überlebendem carcinom-gewebe. Klin Wochenschr. 2:776–777. 1923.
|
|
29
|
Lunt SY and Vander Heiden MG: Aerobic
glycolysis: Meeting the metabolic requirements of cell
proliferation. Annu Rev Cell Dev Biol. 27:441–464. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Koppenol WH, Bounds PL and Dang CV: Otto
Warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tochigi T, Shuto K, Kono T, Ohira G, Tohma
T, Gunji H, Hayano K, Narushima K, Fujishiro T, Hanaoka T, et al:
Heterogeneity of glucose metabolism in esophageal cancer measured
by fractal analysis of fluorodeoxyglucose positron emission
tomography image: Correlation between metabolic heterogeneity and
survival. Dig Surg. 34:186–191. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Larson SM: Positron emission
tomography-based molecular imaging in human cancer: Exploring the
link between hypoxia and accelerated glucose metabolism. Clin
Cancer Res. 10:2203–2204. 2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv.
2(e1600200)2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even Warburg did not anticipate.
Cancer Cell. 21:297–308. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li ZY and Zhang HF: Reprogramming of
glucose, fatty acid and amino acid metabolism for cancer
progression. Cell Mol Life Sci. 73:377–392. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kim SY: Cancer energy metabolism: Shutting
power off cancer factory. Biomol Ther (Seoul). 26:39–44.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Corbet C and Feron O: Cancer cell
metabolism and mitochondria: Nutrient plasticity for TCA cycle
fueling. Biochim Biophys Acta Rev Cancer. 1868:7–15.
2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Weinberg F and Chandel NS: Mitochondrial
metabolism and cancer. Ann NY Acad Sci. 1177:66–73. 2009.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhao L, Mao Y, Zhao Y, Cao Y and Chen X:
Role of multifaceted regulators in cancer glucose metabolism and
their clinical significance. Oncotarget. 7:31572–31585.
2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Finley LWS: What is cancer metabolism?
Cell. 186:1670–1688. 2023.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Stine ZE, Schug ZT, Salvino JM and Dang
CV: Targeting cancer metabolism in the era of precision oncology.
Nat Rev Drug Discov. 21:141–162. 2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Vander Heiden MG, Locasale JW, Swanson KD,
Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G,
Rabinowitz JD, Asara JM and Cantley LC: Evidence for an alternative
glycolytic pathway in rapidly proliferating cells. Science.
329:1492–1499. 2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kalyanaraman B, Cheng G, Hardy M, Ouari O,
Lopez M, Joseph J, Zielonka J and Dwinell MB: A review of the
basics of mitochondrial bioenergetics, metabolism, and related
signaling pathways in cancer cells: Therapeutic targeting of tumor
mitochondria with lipophilic cationic compounds. Redox Biol.
14:316–327. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Stepien M, Duarte-Salles T, Fedirko V,
Floegel A, Barupal DK, Rinaldi S, Achaintre D, Assi N, Tjønneland
A, Overvad K, et al: Alteration of amino acid and biogenic amine
metabolism in hepatobiliary cancers: Findings from a prospective
cohort study. Int J Cancer. 138:348–360. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Frezza C: Cancer metabolism: Addicted to
serine. Nat Chem Biol. 12:389–390. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Gordan JD, Thompson CB and Simon MC: HIF
and c-Myc: Sibling rivals for control of cancer cell metabolism and
proliferation. Cancer Cell. 12:108–113. 2007.PubMed/NCBI View Article : Google Scholar
|
|
48
|
West MJ, Stoneley M and Willis AE:
Translational induction of the c-myc oncogene via activation of the
FRAP/TOR signalling pathway. Oncogene. 17:769–780. 1998.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Kalkat M, De Melo J, Hickman KA, Lourenco
C, Redel C, Resetca D, Tamachi A, Tu WB and Penn LZ: MYC
deregulation in primary human cancers. Genes (Basel).
8(151)2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Goetzman ES and Prochownik EV: The role
for Myc in coordinating glycolysis, oxidative phosphorylation,
glutaminolysis, and fatty acid metabolism in normal and neoplastic
tissues. Front Endocrinol (Lausanne). 9(129)2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Pourdehnad M, Truitt ML, Siddiqi IN,
Ducker GS, Shokat KM and Ruggero D: Myc and mTOR converge on a
common node in protein synthesis control that confers synthetic
lethality in Myc-driven cancers. Proc Natl Acad Sci USA.
110:11988–11993. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Sukumaran A, Choi K and Dasgupta B:
Insight on transcriptional regulation of the energy sensing AMPK
and biosynthetic mTOR pathway genes. Front Cell Dev Biol.
8(671)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Khan MW, Biswas D, Ghosh M, Mandloi S,
Chakrabarti S and Chakrabarti P: mTORC2 controls cancer cell
survival by modulating gluconeogenesis. Cell Death Discov.
1(15016)2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Sun Q, Chen X, Ma J, Peng H, Wang F, Zha
X, Wang Y, Jing Y, Yang H, Chen R, et al: Mammalian target of
rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is
critical for aerobic glycolysis and tumor growth. Proc Natl Acad
Sci USA. 108:4129–4134. 2011.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Wei H, Dong C and Shen Z:
Kallikrein-related peptidase (KLK10) cessation blunts colorectal
cancer cell growth and glucose metabolism by regulating the
PI3K/Akt/mTOR pathway. Neoplasma. 67:889–897. 2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Iqbal MA, Siddiqui FA, Gupta V,
Chattopadhyay S, Gopinath P, Kumar B, Manvati S, Chaman N and
Bamezai RN: Insulin enhances metabolic capacities of cancer cells
by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol
Cancer. 12(72)2013.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Buller CL, Loberg RD, Fan MH, Zhu Q, Park
JL, Vesely E, Inoki K, Guan KL and Brosius FC III: A
GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose
transporter expression. Am J Physiol Cell Physiol. 295:C836–C843.
2008.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Shi LZ, Wang R, Huang G, Vogel P, Neale G,
Green DR and Chi H: HIF1alpha-dependent glycolytic pathway
orchestrates a metabolic checkpoint for the differentiation of TH17
and Treg cells. J Exp Med. 208:1367–1376. 2011.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Siska PJ, van der Windt GJW, Kishton RJ,
Cohen S, Eisner W, MacIver NJ, Kater AP, Weinberg JB and Rathmell
JC: Suppression of Glut1 and glucose metabolism by decreased
Akt/mTORC1 signaling drives T cell impairment in B cell leukemia. J
Immunol. 197:2532–2540. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Feng Y and Wu L: mTOR up-regulation of
PFKFB3 is essential for acute myeloid leukemia cell survival.
Biochem Biophys Res Commun. 483:897–903. 2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Finlay DK, Rosenzweig E, Sinclair LV,
Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K
and Cantrell DA: PDK1 regulation of mTOR and hypoxia-inducible
factor 1 integrate metabolism and migration of CD8+ T cells. J Exp
Med. 209:2441–2453. 2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Chen X, Zhu Y, Wang Z, Zhu H, Pan Q, Su S,
Dong Y, Li L, Zhang H, Wu L, et al: mTORC1 alters the expression of
glycolytic genes by regulating KPNA2 abundances. J Proteomics.
136:13–24. 2016.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Li Y, He ZC, Liu Q, Zhou K, Shi Y, Yao XH,
Zhang X, Kung HF, Ping YF and Bian XW: Large intergenic non-coding
RNA-RoR inhibits aerobic glycolysis of glioblastoma cells via Akt
pathway. J Cancer. 9:880–889. 2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Holloway RW and Marignani PA: Targeting
mTOR and glycolysis in HER2-positive breast cancer. Cancers
(Basel). 13(2922)2021.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Dibble CC and Manning BD: Signal
integration by mTORC1 coordinates nutrient input with biosynthetic
output. Nat Cell Biol. 15:555–564. 2013.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Kullmann L and Krahn MP: Controlling the
master-upstream regulation of the tumor suppressor LKB1. Oncogene.
37:3045–3057. 2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Zhang J, Pavlova NN and Thompson CB:
Cancer cell metabolism: The essential role of the nonessential
amino acid, glutamine. EMBO J. 36:1302–1315. 2017.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:619–634. 2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Choi YK and Park KG: Targeting glutamine
metabolism for cancer treatment. Biomol Ther (Seoul). 26:19–28.
2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Masui K, Tanaka K, Akhavan D, Babic I,
Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, et al: mTOR
complex 2 controls glycolytic metabolism in glioblastoma through
FoxO acetylation and upregulation of c-Myc. Cell Metab. 18:726–739.
2013.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Prieto J, García-Cañaveras JC, León M,
Sendra R, Ponsoda X, Izpisúa Belmonte JC, Lahoz A and Torres J:
c-MYC triggers lipid remodelling during early somatic cell
reprogramming to pluripotency. Stem Cell Rev Rep. 17:2245–2261.
2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Khan MW, Layden BT and Chakrabarti P:
Inhibition of mTOR complexes protects cancer cells from glutamine
starvation induced cell death by restoring Akt stability. Biochim
Biophys Acta Mol Basis Dis. 1864:2040–2052. 2018.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Csibi A, Lee G, Yoon SO, Tong H, Ilter D,
Elia I, Fendt SM, Roberts TM and Blenis J: The mTORC1/S6K1 pathway
regulates glutamine metabolism through the eIF4B-dependent control
of c-Myc translation. Curr Biol. 24:2274–2280. 2014.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Shaw E, Talwadekar M, Rashida Z, Mohan N,
Acharya A, Khatri S, Laxman S and Kolthur-Seetharam U: Anabolic
SIRT4 exerts retrograde control over TORC1 signaling by glutamine
sparing in the mitochondria. Mol Cell Biol. 40:e00212–19.
2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Wang D, He J, Huang B, Liu S, Zhu H and Xu
T: Emerging role of the Hippo pathway in autophagy. Cell Death Dis.
11(880)2020.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Trejo-Solís C, Serrano-García N,
Castillo-Rodríguez RA, Robledo-Cadena DX, Jimenez-Farfan D,
Marín-Hernández Á, Silva-Adaya D, Rodríguez-Pérez CE and
Gallardo-Pérez JC: Metabolic dysregulation of tricarboxylic acid
cycle and oxidative phosphorylation in glioblastoma. Rev Neurosci.
35:813–838. 2024.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Morita M, Gravel SP, Chénard V, Sikström
K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C,
et al: mTORC1 controls mitochondrial activity and biogenesis
through 4E-BP-dependent translational regulation. Cell Metab.
18:698–711. 2013.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18787. 2008.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Byun JK, Choi YK, Kim JH, Jeong JY, Jeon
HJ, Kim MK, Hwang I, Lee SY, Lee YM, Lee IK and Park KG: A positive
feedback loop between Sestrin2 and mTORC2 is required for the
survival of glutamine-depleted lung cancer cells. Cell Rep.
20:586–599. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Durán RV, Oppliger W, Robitaille AM,
Heiserich L, Skendaj R, Gottlieb E and Hall MN: Glutaminolysis
activates Rag-mTORC1 signaling. Mol Cell. 47:349–358.
2012.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Chiu M, Tardito S, Barilli A, Bianchi MG,
Dall'Asta V and Bussolati O: Glutamine stimulates mTORC1
independent of the cell content of essential amino acids. Amino
Acids. 43:2561–2567. 2012.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Bodineau C, Tomé M, Courtois S, Costa ASH,
Sciacovelli M, Rousseau B, Richard E, Vacher P, Parejo-Pérez C,
Bessede E, et al: Two parallel pathways connect glutamine
metabolism and mTORC1 activity to regulate glutamoptosis. Nat
Commun. 12(4814)2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Dennis MD, Jefferson LS and Kimball SR:
Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4
proteins in the regulation of protein synthesis. J Biol Chem.
287:42890–42899. 2012.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Dalle Pezze P, Ruf S, Sonntag AG,
Langelaar-Makkinje M, Hall P, Heberle AM, Razquin Navas P, van
Eunen K, Tölle RC, Schwarz JJ, et al: A systems study reveals
concurrent activation of AMPK and mTOR by amino acids. Nat Commun.
7(13254)2016.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Nicklin P, Bergman P, Zhang B,
Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson
C, et al: Bidirectional transport of amino acids regulates mTOR and
autophagy. Cell. 136:521–534. 2009.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Selwan EM and Edinger AL: Branched chain
amino acid metabolism and cancer: The importance of keeping things
in context. Transl Cancer Res. 6 (Suppl 3):S578–S584.
2017.PubMed/NCBI View Article : Google Scholar
|
|
87
|
McCracken AN and Edinger AL: Nutrient
transporters: The Achilles' heel of anabolism. Trends Endocrin Met.
24:200–208. 2013.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Kawasome H, Papst P, Webb S, Keller GM,
Johnson GL, Gelfand EW and Terada N: Targeted disruption of
p70(s6k) defines its role in protein synthesis and rapamycin
sensitivity. Proc Natl Acad Sci USA. 95:5033–5038. 1998.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Martineau Y, Wang X, Alain T, Petroulakis
E, Shahbazian D, Fabre B, Bousquet-Dubouch MP, Monsarrat B,
Pyronnet S and Sonenberg N: Control of Paip1-eukaryotic translation
initiation factor 3 interaction by amino acids through S6 kinase.
Mol Cell Biol. 34:1046–1053. 2014.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Iadevaia V, Liu R and Proud CG: mTORC1
signaling controls multiple steps in ribosome biogenesis. Semin
Cell Dev Biol. 36:113–120. 2014.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Abetov DA, Kiyan VS, Zhylkibayev AA,
Sarbassova DA, Alybayev SD, Spooner E, Song MS, Bersimbaev RI and
Sarbassov DD: Formation of mammalian preribosomes proceeds from
intermediate to composed state during ribosome maturation. J Biol
Chem. 294:10746–10757. 2019.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Gentilella A, Kozma SC and Thomas G: A
liaison between mTOR signaling, ribosome biogenesis and cancer.
Biochim Biophys Acta. 1849:812–820. 2015.PubMed/NCBI View Article : Google Scholar
|
|
93
|
He J, Yang Y, Zhang J, Chen J, Wei X, He J
and Luo L: Ribosome biogenesis protein Urb1 acts downstream of mTOR
complex 1 to modulate digestive organ development in zebrafish. J
Genet Genomics. 44:567–576. 2017.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Rad E, Murray JT and Tee AR: Oncogenic
signalling through mechanistic target of rapamycin (mTOR): A driver
of metabolic transformation and cancer progression. Cancers
(Basel). 10(5)2018.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Qin L, Cheng X, Wang S, Gong G, Su H,
Huang H, Chen T, Damdinjav D, Dorjsuren B, Li Z, et al: Discovery
of novel aminobutanoic acid-based ASCT2 inhibitors for the
treatment of non-small-cell lung cancer. J Med Chem. 67:988–1007.
2024.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Leibowitz BJ, Zhao G, Xia W, Wang Y, Ruan
H, Zhang L and Yu J: mTOR inhibition suppresses Myc-driven
polyposis by inducing immunogenic cell death. Oncogene.
42:2007–2016. 2023.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Elkon R, Loayza-Puch F, Korkmaz G, Lopes
R, van Breugel PC, Bleijerveld OB, Altelaar AF, Wolf E, Lorenzin F,
Eilers M and Agami R: Myc coordinates transcription and translation
to enhance transformation and suppress invasiveness. EMBO Rep.
16:1723–1736. 2015.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Peng T, Golub TR and Sabatini DM: The
immunosuppressant rapamycin mimics a starvation-like signal
distinct from amino acid and glucose deprivation. Mol Cell Biol.
22:5575–5584. 2002.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Rosario FJ, Kanai Y, Powell TL and Jansson
T: Mammalian target of rapamycin signalling modulates amino acid
uptake by regulating transporter cell surface abundance in primary
human trophoblast cells. J Physiol. 591:609–625. 2013.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Rosario FJ, Dimasuay KG, Kanai Y, Powell
TL and Jansson T: Regulation of amino acid transporter trafficking
by mTORC1 in primary human trophoblast cells is mediated by the
ubiquitin ligase Nedd4-2. Clin Sci (Lond). 130:499–512.
2016.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Park Y, Reyna-Neyra A, Philippe L and
Thoreen CC: mTORC1 balances cellular amino acid supply with demand
for protein synthesis through post-transcriptional control of ATF4.
Cell Rep. 19:1083–1090. 2017.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Gu Y, Albuquerque CP, Braas D, Zhang W,
Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H, et al: mTORC2
regulates amino acid metabolism in cancer by phosphorylation of the
cystine-glutamate antiporter xCT. Mol Cell. 67:128–138.e7.
2017.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Toda K, Kawada K, Iwamoto M, Inamoto S,
Sasazuki T, Shirasawa S, Hasegawa S and Sakai Y: Metabolic
alterations caused by KRAS mutations in colorectal cancer
contribute to cell adaptation to glutamine depletion by
upregulation of asparagine synthetase. Neoplasia. 18:654–665.
2016.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Pupo E, Avanzato D, Middonti E, Bussolino
F and Lanzetti L: KRAS-driven metabolic rewiring reveals novel
actionable targets in cancer. Front Oncol. 9(848)2019.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Gwinn DM, Lee AG, Briones-Martin-Del-Campo
M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D and
Sweet-Cordero EA: Oncogenic KRAS regulates amino acid homeostasis
and asparagine biosynthesis via ATF4 and alters sensitivity to
L-asparaginase. Cancer Cell. 33:91–107.e6. 2018.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Long Y, Tsai WB, Wangpaichitr M, Tsukamoto
T, Savaraj N, Feun LG and Kuo MT: Arginine deiminase resistance in
melanoma cells is associated with metabolic reprogramming, glucose
dependence, and glutamine addiction. Mol Cancer Ther. 12:2581–2590.
2013.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Selvarajah B, Azuelos I, Platé M,
Guillotin D, Forty EJ, Contento G, Woodcock HV, Redding M, Taylor
A, Brunori G, et al: mTORC1 amplifies the ATF4-dependent de novo
serine-glycine pathway to supply glycine during
TGF-β1-induced collagen biosynthesis. Sci Signal.
12(eaav3048)2019.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Kma L and Baruah TJ: The interplay of ROS
and the PI3K/Akt pathway in autophagy regulation. Biotechnol Appl
Biochem. 69:248–264. 2022.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131.
2002.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Randle PJ: Regulatory interactions between
lipids and carbohydrates: The glucose fatty acid cycle after 35
years. Diabetes Metab Rev. 14:263–283. 1998.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Chen Y and Li P: Fatty acid metabolism and
cancer development. Sci Bull. 61:1473–1479. 2016.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Alasadi A, Humaish HH and Al-hraishawi H:
Evaluation the predictors of non-alcoholic fatty liver disease
(NAFLD) in type 2 diabetes mellitus (T2DM) patients. Syst Rev
Pharm. 11:421–430. 2020.
|
|
113
|
Mullen AR, Wheaton WW, Jin ES, Chen PH,
Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS and
DeBerardinis RJ: Reductive carboxylation supports growth in tumour
cells with defective mitochondria. Nature. 481:385–388.
2011.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Metallo CM, Gameiro PA, Bell EL, Mattaini
KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L,
et al: Reductive glutamine metabolism by IDH1 mediates lipogenesis
under hypoxia. Nature. 481:380–384. 2011.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Wise DR, Ward PS, Shay JE, Cross JR,
Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC and
Thompson CB: Hypoxia promotes isocitrate dehydrogenase-dependent
carboxylation of α-ketoglutarate to citrate to support cell growth
and viability. Proc Natl Acad Sci USA. 108:19611–19616.
2011.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Schug ZT, Peck B, Jones DT, Zhang Q,
Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K,
et al: Acetyl-CoA synthetase 2 promotes acetate utilization and
maintains cancer cell growth under metabolic stress. Cancer Cell.
27:57–71. 2015.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Green CR, Wallace M, Divakaruni AS,
Phillips SA, Murphy AN, Ciaraldi TP and Metallo CM: Branched-chain
amino acid catabolism fuels adipocyte differentiation and
lipogenesis. Nat Chem Biol. 12:15–21. 2016.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Eberlé D, Hegarty B, Bossard P, Ferré P
and Foufelle F: SREBP transcription factors: master regulators of
lipid homeostasis. Biochimie. 86:839–848. 2004.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Nakamura MT, Cheon Y, Li Y and Nara TY:
Mechanisms of regulation of gene expression by fatty acids. Lipids.
39:1077–1083. 2004.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Pégorier JP, Le May C and Girard J:
Control of gene expression by fatty acids. J Nutr. 134:2444S–2449S.
2004.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Düvel K, Yecies JL, Menon S, Raman P,
Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S,
et al: Activation of a metabolic gene regulatory network downstream
of mTOR complex 1. Mol Cell. 39:171–183. 2010.PubMed/NCBI View Article : Google Scholar
|
|
122
|
DeBerardinis RJ, Lum JJ and Thompson CB:
Phosphatidylinositol 3-kinase-dependent modulation of carnitine
palmitoyltransferase 1A expression regulates lipid metabolism
during hematopoietic cell growth. J Biol Chem. 281:37372–37380.
2006.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Hao YJ, Samuels Y, Li QL, Krokowski D,
Brunengraber H, Hatzoglou M, Zhang GF, Vogelstein B and Wang Z:
Abstract 1125: Oncogenic PIK3CA mutations reprogram glutamine
metabolism in colorectal cancers. Cancer Res. 75 (15
Suppl)(S1125)2015.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Liu C, Zhou X, Pan Y, Liu Y and Zhang Y:
Pyruvate carboxylase promotes thyroid cancer aggressiveness through
fatty acid synthesis. BMC Cancer. 21(722)2021.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Schiliro C and Firestein BL: Mechanisms of
metabolic reprogramming in cancer cells supporting enhanced growth
and proliferation. Cells. 10(1056)2021.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Andrade-Vieira R, Han JH and Marignani PA:
Omega-3 polyunsaturated fatty acid promotes the inhibition of
glycolytic enzymes and mTOR signaling by regulating the tumor
suppressor LKB1. Cancer Biol Ther. 14:1050–1058. 2013.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Kuma A, Hatano M, Matsui M, Yamamoto A,
Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T and Mizushima N: The
role of autophagy during the early neonatal starvation period.
Nature. 432:1032–1036. 2004.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Alasadi AH, Guo J, Tao H and Jin S:
Preventing and treating hepatic metastatic colon and pancreatic
cancers by targeting cell metabolism. Cancer Res. 76 (14
Suppl)(S45)2016.
|
|
129
|
Aird KM and Zhang R: Nucleotide
metabolism, oncogene-induced senescence and cancer. Cancer Lett.
356:204–210. 2015.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Cantor JR and Sabatini DM: Cancer cell
metabolism: One hallmark, many faces. Cancer Discov. 2:881–898.
2012.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Vander Heiden MG: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Ben-Sahra I, Howell JJ, Asara JM and
Manning BD: Stimulation of de novo pyrimidine synthesis by growth
signaling through mTOR and S6K1. Science. 339:1323–1328.
2013.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Robitaille AM, Christen S, Shimobayashi M,
Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P
and Hall MN: Quantitative phosphoproteomics reveal mTORC1 activates
de novo pyrimidine synthesis. Science. 339:1320–1323.
2013.PubMed/NCBI View Article : Google Scholar
|
|
134
|
He L, Wei X, Ma X, Yin X, Song M,
Donninger H, Yaddanapudi K, McClain CJ and Zhang X: Simultaneous
quantification of nucleosides and nucleotides from biological
samples. J Am Soc Mass Spectrom. 30:987–1000. 2019.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Qian X, Li X, Tan L, Lee JH, Xia Y, Cai Q,
Zheng Y, Wang H, Lorenzi PL and Lu Z: Conversion of PRPS hexamer to
monomer by AMPK-mediated phosphorylation inhibits nucleotide
synthesis in response to energy stress. Cancer Discov. 8:94–107.
2018.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Yecies JL, Zhang HH, Menon S, Liu S,
Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS,
Lee CH and Manning BD: Akt stimulates hepatic SREBP1c and
lipogenesis through parallel mTORC1-dependent and independent
pathways. Cell Metab. 14:21–32. 2011.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Chen J, Yang S, Li Y, Ziwen X, Zhang P,
Song Q, Yao Y and Pei H: De novo nucleotide biosynthetic pathway
and cancer. Genes Dis. 10:2331–2338. 2022.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Qian J, Chen Y, Meng T, Ma L, Meng L, Wang
X, Yu T, Zask A, Shen J and Yu K: Molecular regulation of apoptotic
machinery and lipid metabolism by mTORC1/mTORC2 dual inhibitors in
preclinical models of HER2+/PIK3CAmut breast cancer. Oncotarget.
7:67071–67086. 2016.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Evert M, Calvisi DF, Evert K, De Murtas V,
Gasparetti G, Mattu S, Destefanis G, Ladu S, Zimmermann A, Delogu
S, et al: V-AKT murine thymoma viral oncogene homolog/mammalian
target of rapamycin activation induces a module of metabolic
changes contributing to growth in insulin-induced
hepatocarcinogenesis. Hepatology. 55:1473–1484. 2012.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Cheng J, Huang Y, Zhang X, Yu Y, Wu S,
Jiao J, Tran L, Zhang W, Liu R, Zhang L, et al: TRIM21 and PHLDA3
negatively regulate the crosstalk between the PI3K/AKT pathway and
PPP metabolism. Nat Commun. 11(1880)2020.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Saha A, Connelly S, Jiang J, Zhuang S,
Amador DT, Phan T, Pilz RB and Boss GR: Akt phosphorylation and
regulation of transketolase is a nodal point for amino acid control
of purine synthesis. Mol Cell. 55:264–276. 2014.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Wang W and Guan KL: AMP-activated protein
kinase and cancer. Acta Physiol (Oxf). 196:55–63. 2009.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Cantó C and Auwerx J: Calorie restriction:
Is AMPK a key sensor and effector? Physiology (Bethesda).
26:214–224. 2011.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Klaus S, Keipert S, Rossmeisl M and
Kopecky J: Augmenting energy expenditure by mitochondrial
uncoupling: A role of AMP-activated protein kinase. Genes Nutr.
7:369–386. 2012.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Fujii N, Aschenbach WG, Musi N, Hirshman
MF and Goodyear LJ: Regulation of glucose transport by the
AMP-activated protein kinase. Proc Nutr Soc. 63:205–210.
2004.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Haurie V, Boucherie H and Sagliocco F: The
Snf1 protein kinase controls the induction of genes of the iron
uptake pathway at the diauxic shift in Saccharomyces cerevisiae. J
Biol Chem. 278:45391–45396. 2003.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Hawley SA, Fullerton MD, Ross FA,
Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green
KA, Mustard KJ, et al: The ancient drug salicylate directly
activates AMP-activated protein kinase. Science. 336:918–922.
2012.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Daurio NA, Tuttle SW, Worth AJ, Song EY,
Davis JM, Snyder NW, Blair IA and Koumenis C: AMPK activation and
metabolic reprogramming by tamoxifen through estrogen
receptor-independent mechanisms suggests new uses for this
therapeutic modality in cancer treatment. Cancer Res. 76:3295–3306.
2016.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Xiao B, Heath R, Saiu P, Leiper FC, Leone
P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, et al:
Structural basis for AMP binding to mammalian AMP-activated protein
kinase. Nature. 449:496–500. 2007.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Scott JW, Hawley SA, Green KA, Anis M,
Stewart G, Scullion GA, Norman DG and Hardie DG: CBS domains form
energy-sensing modules whose binding of adenosine ligands is
disrupted by disease mutations. J Clin Invest. 113:274–284.
2004.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Gowans GJ, Hawley SA, Ross FA and Hardie
DG: AMP is a true physiological regulator of AMP-activated protein
kinase by both allosteric activation and enhancing net
phosphorylation. Cell Metab. 18:556–566. 2013.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Hardie DG: Molecular pathways: Is AMPK a
friend or a foe in cancer? Clin Cancer Res. 21:3836–3840.
2015.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Faubert B, Vincent EE, Griss T, Samborska
B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB,
Shaw RJ and Jones RG: Loss of the tumor suppressor LKB1 promotes
metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad
Sci USA. 111:2554–2559. 2014.PubMed/NCBI View Article : Google Scholar
|
|
155
|
El-Masry OS, Brown BL and Dobson PRM:
Effects of activation of AMPK on human breast cancer cell lines
with different genetic backgrounds. Oncol Lett. 3:224–228.
2012.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Hadad SM, Baker L, Quinlan PR, Robertson
KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG,
et al: Histological evaluation of AMPK signaling in primary breast
cancer. BMC Cancer. 9(307)2009.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Lee CW, Wong LLY, Tse EYT, Liu HF, Leong
VY, Lee JM, Hardie DG, Ng IO and Ching YP: AMPK promotes p53
acetylation via phosphorylation and inactivation of SIRT1 in liver
cancer cells. Cancer Res. 72:4394–4404. 2012.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Laderoute KR, Calaoagan JM, Chao WR, Dinh
D, Denko N, Duellman S, Kalra J, Liu X, Papandreou I, Sambucetti L
and Boros LG: 5'-AMP-activated protein kinase (AMPK) supports the
growth of aggressive experimental human breast cancer tumors. J
Biol Chem. 289:22850–22864. 2014.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Zheng L, Yang W, Wu F, Wang C, Yu L, Tang
L, Qiu B, Li Y, Guo L, Wu M, et al: Prognostic significance of AMPK
activation and therapeutic effects of metformin in hepatocellular
carcinoma. Clin Cancer Res. 19:5372–5380. 2013.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Faubert B, Boily G, Izreig S, Griss T,
Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et
al: AMPK is a negative regulator of the Warburg effect and
suppresses tumor growth in vivo. Cell Metab. 17:113–124.
2013.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Sanchez-Cespedes M, Parrella P, Esteller
M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG and Sidransky
D: Inactivation of LKB1/STK11 is a common event in adenocarcinomas
of the lung. Cancer Res. 62:3659–3662. 2002.PubMed/NCBI
|
|
162
|
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara
K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al:
LKB1 modulates lung cancer differentiation and metastasis. Nature.
448:807–810. 2007.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Wingo SN, Gallardo TD, Akbay EA, Liang MC,
Contreras CM, Boren T, Shimamura T, Miller DS, Sharpless NE,
Bardeesy N, et al: Somatic LKB1 mutations promote cervical cancer
progression. PLoS One. 4(e5137)2009.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Hawley SA, Ross FA, Gowans GJ, Tibarewal
P, Leslie NR and Hardie DG: Phosphorylation by Akt within the ST
loop of AMPK-α1 down-regulates its activation in tumour cells.
Biochem J. 459:275–287. 2014.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Horman S, Vertommen D, Heath R, Neumann D,
Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L and
Rider MH: Insulin antagonizes ischemia-induced Thr172
phosphorylation of AMP-activated protein kinase alpha-subunits in
heart via hierarchical phosphorylation of Ser485/491. J Biol Chem.
281:5335–5340. 2006.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Zhang Y, Li W, Niu J, Fan Z, Li X and
Zhang H: Reprogramming of glucose metabolism in pancreatic cancer:
Mechanisms, implications, and therapeutic perspectives. Front
Immunol. 16(1586959)2025.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Du W, Xu A, Huang Y, Cao J, Zhu H, Yang B,
Shao X, He Q and Ying M: The role of autophagy in targeted therapy
for acute myeloid leukemia. Autophagy. 17:2665–2679.
2021.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Mohanty SS, Warrier S and Rangarajan A:
Rethinking AMPK: A reversible switch fortifying cancer cell
stress-resilience. Yale J Biol Med. 98:33–52. 2025.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Baek SY, Hwang UW, Suk HY and Kim YW:
Hemistepsin A inhibits cell proliferation and induces G0/G1-phase
arrest, cellular senescence and apoptosis via the AMPK and p53/p21
signals in human hepatocellular carcinoma. Biomolecules.
10(713)2020.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Wang EM, Akasaka H, Zhao J, Varadhachary
GR, Lee JE, Maitra A, Fleming JB, Hung MC, Wang H and Katz MHG:
Expression and clinical significance of protein kinase RNA-like
endoplasmic reticulum kinase and phosphorylated eukaryotic
initiation factor 2α in pancreatic ductal adenocarcinoma. Pancreas.
48:323–328. 2019.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Grenier A, Poulain L, Mondesir J, Jacquel
A, Bosc C, Stuani L, Mouche S, Larrue C, Sahal A, Birsen R, et al:
AMPK-PERK axis represses oxidative metabolism and enhances
apoptotic priming of mitochondria in acute myeloid leukemia. Cell
Rep. 38(110197)2022.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Wang S, Li H, Yuan M, Fan H and Cai Z:
Role of AMPK in autophagy. Front Physiol.
13(1015500)2022.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Panina SB, Pei J and Kirienko NV:
Mitochondrial metabolism as a target for acute myeloid leukemia
treatment. Cancer Metab. 9(17)2021.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Liu Y and Ma Z: Leukemia and mitophagy: A
novel perspective for understanding oncogenesis and resistance. Ann
Hematol. 103:2185–2196. 2024.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Li Y, Sun R, Zou J, Ying Y and Luo Z: Dual
roles of the AMP-activated protein kinase pathway in angiogenesis.
Cells. 8(752)2019.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Carling D: AMPK hierarchy: A matter of
space and time. Cell Res. 29:425–426. 2019.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Uprety B and Abrahamse H: Targeting breast
cancer and their stem cell population through AMPK activation:
Novel insights. Cells. 11(576)2022.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Jhaveri TZ, Woo J, Shang X, Park BH and
Gabrielson E: AMP-activated kinase (AMPK) regulates activity of
HER2 and EGFR in breast cancer. Oncotarget. 6:14754–14765.
2015.PubMed/NCBI View Article : Google Scholar
|
|
180
|
Göbel A, Riffel RM, Hofbauer LC and
Rachner TD: The mevalonate pathway in breast cancer biology. Cancer
Lett. 542(215761)2022.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Amengual-Cladera E, Morla-Barcelo PM,
Morán-Costoya A, Sastre-Serra J, Pons DG, Valle A, Roca P and
Nadal-Serrano M: Metformin: From diabetes to cancer-unveiling
molecular mechanisms and therapeutic strategies. Biology (Basel).
13(302)2024.PubMed/NCBI View Article : Google Scholar
|
|
182
|
Pei W, Dai L, Li M, Cao S, Xiao Y, Yang Y,
Ma M, Deng M, Mo Y and Liu M: Targeting mitochondrial quality
control for the treatment of triple-negative breast cancer: From
molecular mechanisms to precision therapy. Biomolecules.
15(970)2025.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Lim JS, Kim E, Song JS and Ahn S:
Energy-stress-mediated activation of AMPK sensitizes MPS1 kinase
inhibition in triple-negative breast cancer. Oncol Rep.
52(101)2024.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Mustafa M, Abbas K, Alam M, Ahmad W,
Moinuddin Usmani N, Siddiqui SA and Habib S: Molecular pathways and
therapeutic targets linked to triple-negative breast cancer (TNBC).
Mol Cell Biochem. 479:895–913. 2024.PubMed/NCBI View Article : Google Scholar
|
|
185
|
Li C, Syed MU, Nimbalkar A, Shen Y, Vieira
MD, Fraser C, Inde Z, Qin X, Ouyang J, Kreuzer J, et al: LKB1
regulates JNK-dependent stress signaling and apoptotic dependency
of KRAS-mutant lung cancers. Nat Commun. 16(4112)2025.PubMed/NCBI View Article : Google Scholar
|
|
186
|
Huang Q, Ren Y, Yuan P, Huang M, Liu G,
Shi Y, Jia G and Chen M: Targeting the AMPK/Nrf2 pathway: A novel
therapeutic approach for acute lung injury. J Inflamm Res.
17:4683–4700. 2024.PubMed/NCBI View Article : Google Scholar
|
|
187
|
Fatehi Hassanabad A and MacQueen KT:
Molecular mechanisms underlining the role of metformin as a
therapeutic agent in lung cancer. Cell Oncol (Dordr). 44:1–18.
2021.PubMed/NCBI View Article : Google Scholar
|
|
188
|
Bernasconi R, Soodla K, Sirp A, Zovo K,
Kuhtinskaja M, Lukk T, Vendelin M and Birkedal R: Higher AMPK
activation in mouse oxidative compared with glycolytic muscle does
not correlate with LKB1 or CaMKKβ expression. Am J Physiol
Endocrinol Metab. 328:E21–E33. 2025.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Sumbly V and Landry I: Unraveling the role
of STK11/LKB1 in non-small cell lung cancer. Cureus.
14(e21078)2022.PubMed/NCBI View Article : Google Scholar
|
|
190
|
Shi Y, Zheng H, Wang T, Zhou S, Zhao S, Li
M and Cao B: Targeting KRAS: From metabolic regulation to cancer
treatment. Mol Cancer. 24(9)2025.PubMed/NCBI View Article : Google Scholar
|
|
191
|
Liu H, Zhou D, Ou Y, Chen S, Long Y, Yuan
T, Li Y and Tan Y: Multiple signaling pathways in the frontiers of
lung cancer progression. Front Immunol. 16(1593793)2025.PubMed/NCBI View Article : Google Scholar
|
|
192
|
Yibcharoenporn C, Muanprasat C,
Moonwiriyakit A, Satitsri S and Pathomthongtaweechai N: AMPK in
intestinal health and disease: A multifaceted therapeutic target
for metabolic and inflammatory disorders. Drug Des Devel Ther.
19:3029–3058. 2025.PubMed/NCBI View Article : Google Scholar
|
|
193
|
Li S, Wang Y, Zhang D, Wang H, Cui X,
Zhang C and Xin Y: Gliclazide reduces colitis-associated colorectal
cancer formation by deceasing colonic inflammation and regulating
AMPK-NF-κB signaling pathway. Dig Dis Sci. 69:453–462.
2024.PubMed/NCBI View Article : Google Scholar
|
|
194
|
Su G, Wang D, Yang Q, Kong L, Ju X, Yang
Q, Zhu Y, Zhang S and Li Y: Cepharanthine suppresses APC-mutant
colorectal cancers by down-regulating the expression of β-catenin.
Nat Prod Bioprospect. 14(18)2024.PubMed/NCBI View Article : Google Scholar
|
|
195
|
Liu JY, Liu JX, Li R, Zhang ZQ, Zhang XH,
Xing SJ, Sui BD, Jin F, Ma B and Zheng CX: AMPK, a hub for the
microenvironmental regulation of bone homeostasis and diseases. J
Cell Physiol. 239(e31393)2024.PubMed/NCBI View Article : Google Scholar
|
|
196
|
Fakhri S, Moradi SZ, Moradi SY, Piri S,
Shiri Varnamkhasti B, Piri S, Khirehgesh MR and Bishayee A,
Casarcia N and Bishayee A: Phytochemicals regulate cancer
metabolism through modulation of the AMPK/PGC-1α signaling pathway.
BMC Cancer. 24(1079)2024.PubMed/NCBI View Article : Google Scholar
|
|
197
|
Cheng D, Zhang M, Zheng Y, Wang M, Gao Y,
Wang X, Liu X, Lv W, Zeng X, Belosludtsev KN, et al:
α-Ketoglutarate prevents hyperlipidemia-induced fatty liver
mitochondrial dysfunction and oxidative stress by activating the
AMPK-pgc-1α/Nrf2 pathway. Redox Biol. 74(103230)2024.PubMed/NCBI View Article : Google Scholar
|
|
198
|
Naiini MR, Shahouzehi B, Azizi S, Shafiei
B and Nazari-Robati M: Trehalose-induced SIRT1/AMPK activation
regulates SREBP-1c/PPAR-α to alleviate lipid accumulation in aged
liver. Naunyn Schmiedebergs Arch Pharmacol. 397:1061–1070.
2024.PubMed/NCBI View Article : Google Scholar
|
|
199
|
Cunha V, Cotrim HP, Rocha R, Carvalho K
and Lins-Kusterer L: Metformin in the prevention of hepatocellular
carcinoma in diabetic patients: A systematic review. Ann Hepatol.
19:232–237. 2020.PubMed/NCBI View Article : Google Scholar
|
|
200
|
Tufail M, Jiang CH and Li N: Altered
metabolism in cancer: Insights into energy pathways and therapeutic
targets. Mol Cancer. 23(203)2024.PubMed/NCBI View Article : Google Scholar
|
|
201
|
Liu B, Liu L and Liu Y: Targeting cell
death mechanisms: The potential of autophagy and ferroptosis in
hepatocellular carcinoma therapy. Front Immunol.
15(1450487)2024.PubMed/NCBI View Article : Google Scholar
|
|
202
|
Li A, Wang R, Zhao Y, Zhao P and Yang J:
Crosstalk between epigenetics and metabolic reprogramming in
metabolic dysfunction-associated steatotic liver disease-induced
hepatocellular carcinoma: A new sight. Metabolites.
14(325)2024.PubMed/NCBI View Article : Google Scholar
|
|
203
|
Nadile M, Sze NS, Fajardo VA and Tsiani E:
Inhibition of prostate cancer cell survival and proliferation by
carnosic acid is associated with inhibition of Akt and activation
of AMPK signaling. Nutrients. 16(1257)2024.PubMed/NCBI View Article : Google Scholar
|
|
204
|
Schooling CM, Yang G, Soliman GA and Leung
GM: A hypothesis that glucagon-like peptide-1 receptor agonists
exert immediate and multifaceted effects by activating adenosine
monophosphate-activate protein kinase (AMPK). Life (Basel).
15(253)2025.PubMed/NCBI View Article : Google Scholar
|
|
205
|
Manoharan R: Salt-inducible kinases (SIKs)
in cancer: Mechanisms of action and therapeutic prospects. Drug
Discov Today. 30(104279)2025.PubMed/NCBI View Article : Google Scholar
|
|
206
|
Espitia-Pérez PJ, Espitia-Perez LM and
Negrette-Guzmán M: Targeting Prostate cancer metabolism through
transcriptional and epigenetic modulation: A multi-target approach
to therapeutic innovation. Int J Mol Sci. 26(6013)2025.PubMed/NCBI View Article : Google Scholar
|
|
207
|
Pujana-Vaquerizo M, Bozal-Basterra L and
Carracedo A: Metabolic adaptations in prostate cancer. Br J Cancer.
131:1250–1262. 2024.PubMed/NCBI View Article : Google Scholar
|
|
208
|
Feng Y, Zhang Y, Li H, Wang T, Lu F, Liu
R, Xie G, Song L, Huang B, Li X, et al: Enzalutamide inhibits PEX10
function and sensitizes prostate cancer cells to ROS activators.
Cell Death Dis. 15(559)2024.PubMed/NCBI View Article : Google Scholar
|
|
209
|
Lemos G, Fernandes CMADS, Silva FH and
Calmasini FB: The role of autophagy in prostate cancer and
prostatic diseases: A new therapeutic strategy. Prostate Cancer
Prostatic Dis. 27:230–238. 2024.PubMed/NCBI View Article : Google Scholar
|
|
210
|
Inoki K, Kim J and Guan KL: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012.PubMed/NCBI View Article : Google Scholar
|
|
211
|
Choi BH and Coloff JL: The diverse
functions of non-essential amino acids in cancer. Cancers (Basel).
11(675)2019.PubMed/NCBI View Article : Google Scholar
|
|
212
|
Jin J, Byun JK, Choi YK and Park KG:
Targeting glutamine metabolism as a therapeutic strategy for
cancer. Exp Mol Med. 55:706–715. 2023.PubMed/NCBI View Article : Google Scholar
|
|
213
|
Halama A and Suhre K: Advancing cancer
treatment by targeting glutamine metabolism-A roadmap. Cancers
(Basel). 14(553)2022.PubMed/NCBI View Article : Google Scholar
|
|
214
|
Linke M, Fritsch SD, Sukhbaatar N,
Hengstschläger M and Weichhart T: mTORC1 and mTORC2 as regulators
of cell metabolism in immunity. FEBS Lett. 591:3089–3103.
2017.PubMed/NCBI View Article : Google Scholar
|
|
215
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J and Green DR: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011.PubMed/NCBI View Article : Google Scholar
|
|
216
|
Valvezan AJ and Manning BD: Molecular
logic of mTORC1 signalling as a metabolic rheostat. Nat Metab.
1:321–333. 2019.PubMed/NCBI View Article : Google Scholar
|
|
217
|
Nishitani S, Horie M, Ishizaki S and Yano
H: Branched chain amino acid suppresses hepatocellular cancer stem
cells through the activation of mammalian target of rapamycin. PLoS
One. 8(e82346)2013.PubMed/NCBI View Article : Google Scholar
|
|
218
|
Kazyken D, Magnuson B, Bodur C,
Acosta-Jaquez HA, Zhang D, Tong X, Barnes TM, Steinl GK, Patterson
NE, Altheim CH, et al: AMPK directly activates mTORC2 to promote
cell survival during acute energetic stress. Sci Signal.
12(eaav3249)2019.PubMed/NCBI View Article : Google Scholar
|
|
219
|
Sadria M and Layton AT: Interactions among
mTORC, AMPK and SIRT: A computational model for cell energy balance
and metabolism. Cell Commun Signal. 19(57)2021.PubMed/NCBI View Article : Google Scholar
|
|
220
|
Sujobert P, Poulain L, Paubelle E,
Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM,
Nicodeme E, Decrooqc J, et al: Co-activation of AMPK and mTORC1
induces cytotoxicity in acute myeloid leukemia. Cell Rep.
11:1446–1457. 2015.PubMed/NCBI View Article : Google Scholar
|
|
221
|
Davie E, Forte GMA and Petersen J:
Nitrogen regulates AMPK to control TORC1 signaling. Curr Biol.
25:445–454. 2015.PubMed/NCBI View Article : Google Scholar
|
|
222
|
Lie S, Wang T, Forbes B, Proud CG and
Petersen J: The ability to utilise ammonia as nitrogen source is
cell type specific and intricately linked to GDH, AMPK and mTORC1.
Sci Rep. 9(1461)2019.PubMed/NCBI View Article : Google Scholar
|
|
223
|
Lu T, Sun L, Wang Z, Zhang Y, He Z and Xu
C: Fatty acid synthase enhances colorectal cancer cell
proliferation and metastasis via regulating AMPK/mTOR pathway. Onco
Targets Ther. 12:3339–3347. 2019.PubMed/NCBI View Article : Google Scholar
|
|
224
|
Pereira O, Teixeira A, Sampaio-Marques B,
Castro I, Girão H and Ludovico P: Signalling mechanisms that
regulate metabolic profile and autophagy of acute myeloid leukaemia
cells. J Cell Mol Med. 22:4807–4817. 2018.PubMed/NCBI View Article : Google Scholar
|
|
225
|
Chiang CT, Demetriou AN, Ung N, Choudhury
N, Ghaffarian K, Ruderman DL and Mumenthaler SM: mTORC2 contributes
to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor
resistant cells in non-small cell lung cancer. Cancer Lett.
434:152–159. 2018.PubMed/NCBI View Article : Google Scholar
|
|
226
|
Zhang P, Fu HJ, Lv LX, Liu CF, Han C, Zhao
XF and Wang JX: WSSV exploits AMPK to activate mTORC2 signaling for
proliferation by enhancing aerobic glycolysis. Commun Biol.
6(361)2023.PubMed/NCBI View Article : Google Scholar
|
|
227
|
He Z, Houghton PJ, Williams TM and Shen C:
Regulation of DNA duplication by the mTOR signaling pathway. Cell
Cycle. 20:742–751. 2021.PubMed/NCBI View Article : Google Scholar
|
|
228
|
Bu C, Zhao L, Wang L, Yu Z and Zhou J:
mTORC2 promotes pancreatic cancer progression and PARP inhibitor
resistance. Oncol Res. 31:495–503. 2023.PubMed/NCBI View Article : Google Scholar
|
|
229
|
Pournajaf S and Pourgholami MH: The mTOR
pathway in Gliomas: From molecular insights to targeted therapies.
Biomed Pharmacother. 189(118237)2025.PubMed/NCBI View Article : Google Scholar
|
|
230
|
Glaviano A, Foo ASC, Lam HY, Yap KCH,
Jacot W, Jones RH, Eng H, Nair MG, Makvandi P, Geoerger B, et al:
PI3K/AKT/mTOR signaling transduction pathway and targeted therapies
in cancer. Mol Cancer. 22(138)2023.PubMed/NCBI View Article : Google Scholar
|
|
231
|
Carew JS, Kelly KR and Nawrocki ST:
Mechanisms of mTOR inhibitor resistance in cancer therapy. Target
Oncol. 6:17–27. 2011.PubMed/NCBI View Article : Google Scholar
|
|
232
|
Dunn S, Eberlein C, Yu J, Gris-Oliver A,
Ong SH, Yelland U, Cureton N, Staniszewska A, McEwen R, Fox M, et
al: AKT-mTORC1 reactivation is the dominant resistance driver for
PI3Kβ/AKT inhibitors in PTEN-null breast cancer and can be overcome
by combining with Mcl-1 inhibitors. Oncogene. 41:5046–5060.
2022.PubMed/NCBI View Article : Google Scholar
|
|
233
|
Basnet R, Basnet BB, Gupta R, Basnet T,
Khadka S and Alam MS: Mammalian target of rapamycin (mTOR)
signalling pathway-a potential target for cancer intervention: A
short overview. Curr Mol Pharmacol.
17(e310323215268)2024.PubMed/NCBI View Article : Google Scholar
|
|
234
|
Kim H, Lebeau B, Papadopoli D, Jovanovic
P, Russo M, Avizonis D, Morita M, Afzali F, Ursini-Siegel J,
Postovit LM, et al: MTOR modulation induces selective perturbations
in histone methylation which influence the anti-proliferative
effects of mTOR inhibitors. iScience. 27(109188)2024.PubMed/NCBI View Article : Google Scholar
|
|
235
|
Faes S, Santoro T, Troquier L, Silva OD
and Dormond O: Rebound pathway overactivation by cancer cells
following discontinuation of PI3K or mTOR inhibition promotes
cancer cell growth. Biochem Biophys Res Commun. 513:546–552.
2019.PubMed/NCBI View Article : Google Scholar
|
|
236
|
Khorasani ABS, Hafezi N, Sanaei MJ,
Jafari-Raddani F, Pourbagheri-Sigaroodi A and Bashash D: The
PI3K/AKT/mTOR signaling pathway in breast cancer: Review of
clinical trials and latest advances. Cell Biochem Funct.
42(e3998)2024.PubMed/NCBI View Article : Google Scholar
|
|
237
|
Napolitano F, Lin CC, Ahuja K, Ye D,
Chica-Parrado MR, Uemoto Y, Luna P, Matsunaga Y, Mendiratta S, Unni
N, et al: Abstract 3003: Targeting mechanisms of adaptive
resistance to the PI3Kαmutant selective inhibitor RLY-2608 in
HR+/PIK3CA mutant breast cancer. Cancer Res. 85 (8 Suppl
1)(S3003)2025.
|
|
238
|
Elkanawati RY, Sumiwi SA and Levita J:
Impact of lipids on insulin resistance: Insights from human and
animal studies. Drug Des Devel Ther. 18:3337–3360. 2024.PubMed/NCBI View Article : Google Scholar
|
|
239
|
Martínez Báez A, Ayala G, Pedroza-Saavedra
A, González-Sánchez HM and Chihu Amparan L: Phosphorylation codes
in IRS-1 and IRS-2 are associated with the activation/inhibition of
insulin canonical signaling pathways. Curr Issues Mol Biol.
46:634–649. 2024.PubMed/NCBI View Article : Google Scholar
|
|
240
|
Banjac K, Obradovic M, Zafirovic S, Essack
M, Gluvic Z, Sunderic M, Nedic O and Isenovic ER: The involvement
of Akt, mTOR, and S6K in the in vivo effect of IGF-1 on the
regulation of rat cardiac Na+/K+-ATPase. Mol
Biol Rep. 51(517)2024.PubMed/NCBI View Article : Google Scholar
|
|
241
|
Wright SCE, Vasilevski N, Serra V, Rodon J
and Eichhorn PJA: Mechanisms of resistance to PI3K inhibitors in
cancer: Adaptive responses, drug tolerance and cellular plasticity.
Cancers (Basel). 13(1538)2021.PubMed/NCBI View Article : Google Scholar
|
|
242
|
Pourbarkhordar V, Rahmani S, Roohbakhsh A,
Hayes AW and Karimi G: Melatonin effect on breast and ovarian
cancers by targeting the PI3K/Akt/mTOR pathway. IUBMB Life.
76:1035–1049. 2024.PubMed/NCBI View Article : Google Scholar
|
|
243
|
Vadla R and Haldar D: Mammalian target of
rapamycin complex 2 (mTORC2) controls glycolytic gene expression by
regulating Histone H3 Lysine 56 acetylation. Cell Cycle.
17:110–123. 2018.PubMed/NCBI View Article : Google Scholar
|