Introduction
Pompe disease (PD), also known as type II glycogen
storage disease, is a neuromuscular disease resulting from acid
alpha glucosidase (GAA) gene mutations located on chromosome
17q25. This gene encodes the enzyme GAA, which is responsible for
the breakdown of glycogen in lysosomes (1). The most commonly used classification
of PD is based on the age of onset and the disease severity, which
are associated with the residual activity of the enzyme. Minimal to
absent GAA activity results in the early manifestation of the
disease within the first few months of life; this early
manifestation of the disease has been termed infantile-onset PD
(IOPD). Children often present with hypotonia, muscle weakness,
hypertrophic cardiomyopathy and eventually, cardiorespiratory
failure if left untreated. In late-onset PD, the cardiac muscle is
usually spared, while skeletal muscle function progressively
deteriorates from limb-girdle weakness to respiratory insufficiency
(2).
Although enzyme replacement therapy (ERT) has
markedly improved the survival rate of patients, its impact on
long-term functional outcomes and quality of life remains limited.
Studies have demonstrated that despite the early initiation of ERT,
a number of children continue to experience marked gross motor
delays, feeding difficulties and dependency on caregivers for daily
activities (2-6).
These persistent challenges highlight the gap between clinical
stabilization and real-world functional recovery. The present study
therefore aimed to assess the developmental delay, nutritional
intake and the caregiving burden among children with PD receiving
long-term ERT, in order to better understand the residual impact of
IOPD on daily life and identify areas where supportive care remains
essential.
Patients and methods
Study design
The present single-center cross-sectional study was
conducted at the Endocrinology, Genetics, Metabolism and Molecular
Therapy Center of the Vietnam National Children's Hospital, Hanoi,
Vietnam from November, 2021 to August, 2022.
Study subjects
Children diagnosed with PD at the Vietnam National
Children's Hospital were enrolled based on the diagnostic criteria
outlined in the 2006 guideline by the American College of Medical
Genetics and Genomics (ACMG) Work Group on Management of Pompe
Disease (2).
Measurement of outcomes
Anthropometric measurements including weight and
length/height were obtained using standardized methods and
classified using the World Health Organization (WHO) growth
standards (7,8). Nutrient intake was assessed via 24-h
dietary recalls collected through caregiver interviews by a single
nutritionist doctor.
School attendance and caregiving difficulties were
evaluated using a structured caregiver questionnaire. Gross motor
age was defined as the age corresponding to the furthest milestone
based on the Denver Development Screening Test (9) successfully achieved by the child, at
which 50% of age-matched peers are expected to have attained that
skill. Gross motor delay was calculated by the difference between
the gross motor age and the chronological age.
Statistical analysis
Data were analyzed using Stata 15.0 software
(StataCorp LLC). Descriptive statistics were used to summarize
demographic and outcome variables. Associations between variables
were evaluated using linear regression for continuous outcomes, of
which the dependent variable was the delay of the gross motor
function based on the Denver Development Screening Test. The
independent variables were the duration of the disease, the protein
intake, the sex and the body mass index-for-age z-score. A P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
During the study period, 34 children with PD aged
from 1-82 months (mean age, 32.8±23.1 months; of which 55.9% were
male) were recruited. All patients had IOPD, and 33/34 patients
(97.1%) had cardiac involvements (Table I). All patients received regular
biweekly ERT, except during the Lunar New Year holiday and COVID-19
lockdown periods, when infusions were administered monthly. The
cost of the enzyme was covered by pharmaceutical companies, while
hospital bed charges and medical consumables were reimbursed by the
national health insurance. The families of the patient were
responsible for travel-related expenses, including transportation
and meals.
 | Table ICharacteristics of the study
participants. |
Table I
Characteristics of the study
participants.
| Demographic
characteristics | |
|---|
| Age (months), mean ±
SD | 32.76±23.1 |
|
Infant and
young children (≤3 years), n (%) | 19 (55.9%) |
|
Preschool
(>3 to <6 years), n (%) | 13 (38.2%) |
|
School-aged
(from 6 years), n (%) | 2 (5.9%) |
| Sex, n (%) | |
|
Male | 19 (55.9%) |
|
Female | 15 (44.1%) |
| School attendance
rate, n (%) | |
|
Preschool
(>3 to <6 years) | 2 (15.4%) |
|
School-aged
(from 6 years) | 1 (50%) |
| Clinical
characteristics | |
| Clinical subtypes, n
(%) | |
|
Classical
IOPD | 33 (97.1%) |
|
Modified
IOPD | 1 (2.9%) |
| CK (IU/l), mean ±
SD | 1451.56±897.02 |
| AST (IU/l), mean ±
SD | 356.25±192.8 |
| ALT (IU/l) (mean ±
SD) | 183.76±78.57 |
| GAA activity essay
using acarbose (mean ± SD) | 93.4±2.4 |
| CRIM status, n
(%)a | |
|
Positive | 34 (100%) |
|
Negative | 0 |
| Disease duration
(months), mean ± SD | 27.4±20.5 |
Dietary intake analysis revealed that the median
energy intake among the participants was 101.5% of the recommended
value [interquartile range (IQR), 85.9-120.7%]. Of note, 29.4%
(10/34) of children consumed <90% of their requirements, while
41.2% (14/34) exceeded 110% (Table
II).
| Table IIAdequacy of nutrient intake amongst
children with IOPD. |
Table II
Adequacy of nutrient intake amongst
children with IOPD.
| Nutrient | Measure of central
tendency | % Below
recommendation | % Above
recommendation | P-value |
|---|
| Energy
adequacya | Median 101.5% (IQR,
85.9-120.7) | 29.4% (<90% of
EER) | 41.2% (>110% of
EER) | 0.0082 |
| Protein
intakeb | Median 3.5 g/kg/day
(IQR, 1.8-4.7) | 82.4% (<90% of
RNI) | 17.6% (>120% of
RNI) | <0.001 |
| Fiber
intakec | Mean 2.1±1.8 g/1,000
kcal | 100% (<14 g/1,000
kcal) | 0% | <0.001 |
Protein intake was insufficient in the majority of
patients, with a median intake of 3.5 g/kg/day (IQR, 1.8-4.7
g/kg/day). A total of 82.4% (28/34) of children consumed <90% of
their estimated needs, while 17.6% (6/34) consumed >120%
Table II). Fiber intake was low
across all participants, with a mean intake of 2.1±1.8 g/1,000
kcal. None of the children met the threshold of 14 g/1,000 kcal, as
recommended for an adequate fiber intake (Table II).
As regards food acceptance, 38.2% of the children
exhibited ‘picky’ eating behaviors related to texture or food
pieces. Among those aged >24 months, 66.7% still required food
to be blended or pureed. As regards supplementation, 55.9% (19/34)
were supplemented with vitamin D, with 78.9% of them receiving
>400 IU per day. Among the children aged >3 years, 60% (9/15)
used formula, of which 55.6% (5/9) were consuming growing-up
formula, while the remainder were consuming standard follow-up
formula.
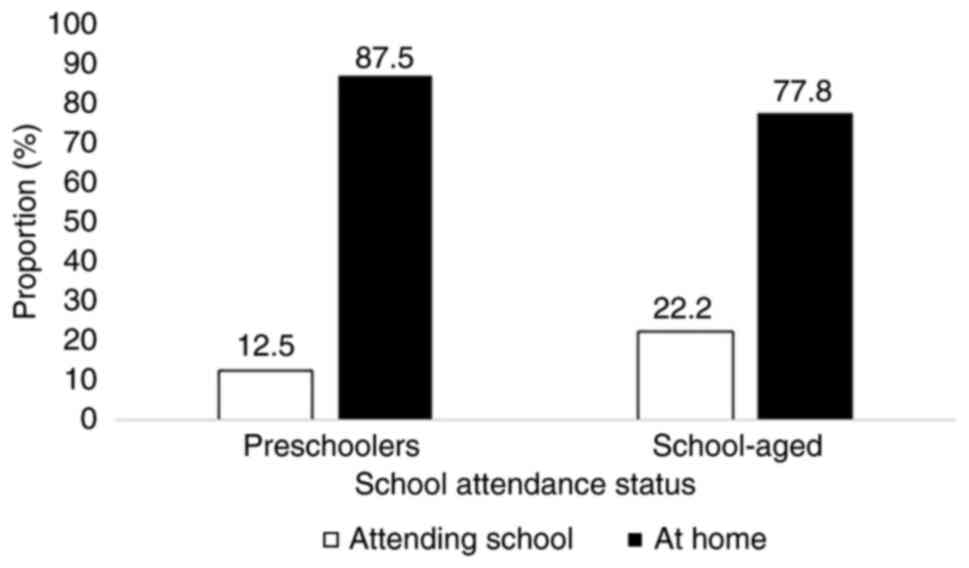
In terms of daily functioning, the majority of
children remained at home. Among the infants and toddlers, 15 of 17
were home-based, while 2 children remained hospitalized. Out of the
preschoolers, 87.5% stayed at home, and only 1 child attended
kindergarten. Similarly, 77.8% of the school-aged children were not
attending school (Fig. 1).
The reasons for home care among the preschoolers and
school-aged children (82.4% collectively) included prolonged
mealtimes and special feeding (blended foods) requirements (58.8%),
non-ambulatory status (17.6%), and, in one case, challenges
involving 2 affected children in one household. The majority of
families (85.3%) had a full-time at-home caregiver, typically a
parent (93.1%). Gross motor ages were delayed by 22.5±20.5 months.
Amongst the preschoolers and school-age children (>3 years of
age), 82.4% were able to stand alone.
In the bivariate analysis, disease duration was
found to be significantly associated with gross motor delay
(adjusted r2=0.89, P<0.001), while there was no
statistically significant association between the protein intake
and the gross motor delay (r2=-0.0244, P=0.6472)
(Table III).
| Table IIIAssociation of factors with gross
motor delay. |
Table III
Association of factors with gross
motor delay.
| Associating
factors | β-coefficient | r2
value | P-value |
|---|
| Protein intake
(g/kg/day) | 0.78 | -0.0244 | 0.6472 |
| Disease duration
(months) | 0.83 | 0.8788 | <0.001 |
Discussion
The present study cohort exclusively comprised
children with IOPD, which is the most severe phenotype, with almost
all children presenting with cardiac involvement.
Dietary intakes and nutrition
practices in children with IOPD
Energy-wise, intake varied widely, with almost a
third of the children under-consuming and >40% exceeding
recommendations. This variability may reflect the challenges in
tailoring nutrition plans to individual clinical conditions, such
as varying degrees of disease progression, immobility, feeding
difficulties and metabolic demands. This was in accordance with the
recommendations of regular nutritional assessment along the disease
progression and individualized nutritional interventions (10-12).
Notably, >80% of children with IOPD in this
cohort consumed <90% of their estimated protein needs. On the
other hand, all participants failed to meet the fiber intake
threshold of 14 g/1,000 kcal. These findings may be explained by
the predominance of low-density liquid foods, such as formulas,
purees and restricted diets. In fact, a notable proportion of
children exhibited food refusal or required texture modification
beyond toddler age. Systematic supplementation and the prolonged
use of formula (to the extent of the high-energy growing formula)
were current strategies to prevent micronutrient deficiencies.
However, more global approaches including macronutrient
considerations (e.g. protein and energy requirements), food texture
sensitization in addition to oral motor function rehabilitation and
orthodontic treatments may prove to be beneficial (13) and should be considered.
Gross motor delay, limited
participation in school and care burden
In the present study cohort, 82.4% of the
preschoolers and school-aged children (aged >3 years) were able
to walk independently. This proportion is higher than that observed
in some previous studies (25-64%) (14-16),
which may be due to the fact that the participants in the present
study were younger (mean age, 32.8±23.1 months) and had a shorter
disease duration. Furthermore, 100% of the patients in the present
study were CRIM-positive, which is typically associated with a
better response to ERT. In a German-Austrian cohort, in which 86.7%
of the patients were CRIM-positive, the independent ambulation
status declined in the 7-year follow-up from 46.7% (median age of 2
years) to 33.3% (median age of 9.1 years) (16). In another study, in a UK
multicenter population, the mean ERT duration was 13 years 9 months
and the CRIM-positive rate was 55%; in that study, 30% of patients
achieved independent ambulation (17).
Of note, a significant proportion (17.2%) of the
present study subgroup (>3 years of age) with a median ERT
duration of 40.5 months remained non-ambulant, suggesting that ERT
alone may not be sufficient to prevent long-term motor
deterioration. This highlights the need for early intervention and
additional therapeutic strategies beyond ERT in order to improve
motor outcomes and preserve function over time.
The present study also demonstrated that the care
burden on families was considerable, with 85.3% of households
having a full-time at-home caregiver, most often a parent. This
reflects the substantial demands of daily care, including prolonged
mealtimes, specialized feeding practices and physical assistance
due to limited mobility. Similar findings have been reported in
previous studies, where caregivers of children with IOPD experience
high levels of emotional, physical and financial stress (18,19).
The requirement for continuous care may limit parental employment
and social participation, underscoring the need for structured
psychosocial support and access to community-based services.
The present study has certain limitations, which
should be mentioned. The present study was limited by the
cross-sectional design, small sample size and reliance on
caregiver-reported intake, which may be subject to recall bias.
Moreover, the cross-sectional design cannot infer causal
associations; therefore, further studies with larger sample sizes
and a longitudinal design are warranted. Nonetheless, the present
study provides valuable insight into the real-world challenges of
managing nutrition and development in children with IOPD.
In conclusion, children with IOPD face significant
nutritional and motor challenges despite ERT. Suboptimal protein
and fiber intake, along with high variabilities in energy needs,
highlight the importance of tailored nutritional care. Although the
ambulation rates in the present study cohort were higher than those
in other studies, motor delays remain common, reinforcing the need
for early and multidisciplinary interventions. The high caregiving
burden further emphasizes the need for broader family support.
Acknowledgements
The authors are also grateful to the medical and
administrative staff at Vietnam National Children's Hospital
(Hanoi, Vietnam) for their instutional support.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are not
publicly available due to ethical restrictions, but may be
requested from the corresponding author.
Authors' contributions
TMTL conceptualized and designed the study,
collected and analyzed the data, performed a literature search,
interpreted the results and drafted the manuscript. ATP was
responsible for dietary recalls, anthropometric measurements,
nutritional interventions and was also involved in drafting the
manuscript. CDV was responsible for clinical examinations,
interpreted the results and performed a literature search. PNT
interpreted the imaging results (interpretation of chest X-rays to
assess cardiomegaly), and performed the literature search. TTHN
contributed to the nutritional interventions, patient management,
was involved in drafting the manuscript. NKN conducted the clinical
examinations, and critically revised the manuscript for important
intellectual content. TMTL and ATP confirm the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Vietnam National Children's Hospital, Hanoi,
Vietnam (2176-BVNTW-HĐĐĐ). Written informed consent was obtained
from the caregivers of all participants prior to enrollment.
Participation was entirely voluntary, and caregivers were informed
that they could withdraw from the study at any time without any
impact on the patient's care. All collected data were anonymized
and used exclusively for research purposes.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, the authors did
use artificial intelligence assisted tools in order to improve the
readability and language of the manuscript. After using this tool
service, the authors did review and edit the content as needed,
taking full responsibility for the content of the publication.
References
|
1
|
Hers HG: alpha-Glucosidase deficiency in
generalized glycogenstorage disease (Pompe's disease). Biochem J.
86:11–16. 1963.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kishnani PS, Steiner RD, Bali D, Berger K,
Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, et
al: Pompe disease diagnosis and management guideline. Genet Med.
8:267–288. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kishnani PS, Corzo D, Leslie ND, Gruskin
D, Van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, Bauer MS,
et al: Early treatment with alglucosidase alpha prolongs long-term
survival of infants with Pompe disease. Pediatr Res. 66:329–335.
2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chakrapani A, Vellodi A, Robinson P, Jones
S and Wraith JE: Treatment of infantile Pompe disease with
alglucosidase alpha: The UK experience. J Inherit Metab Dis.
33:747–750. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hahn A, Praetorius S, Karabul N, Dießel J,
Schmidt D, Motz R, Haase C, Baethmann M, Hennermann JB, Smitka M,
et al: Outcome of patients with classical infantile pompe disease
receiving enzyme replacement therapy in Germany. In: JIMD Reports.
Volume 20. Zschocke J, Baumgartner M, Morava E, Patterson M, Rahman
S and Peters V (eds). Springer, Berlin, Heidelberg, pp65-75,
2015.
|
|
6
|
Hahn A and Schänzer A: Long-term outcome
and unmet needs in infantile-onset Pompe disease. Ann Transl Med.
7(283)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
World Health Organization (WHO): WHO child
growth standards: length/height-for-age, weight-for-age,
weight-for-length, weight-for-height and body mass index-for-age:
Methods and development. World Health Organization, 2006.
|
|
8
|
de Onis M, Onyango AW, Borghi E, Siyam A,
Nishida C and Siekmann J: Development of a WHO growth reference for
school-aged children and adolescents. Bull World Health Organ.
85:660–667. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Frankenburg WK, Dodds J, Archer P, Shapiro
H and Bresnick B: The denver II: A major revision and
restandardization of the denver developmental screening test.
Pediatrics. 89:91–97. 1992.PubMed/NCBI
|
|
10
|
Fatehi F, Ashrafi MR, Babaee M, Ansari B,
Beiraghi Toosi M, Boostani R, Eshraghi P, Fakharian A, Hadipour Z,
Haghi Ashtiani B, et al: Recommendations for infantile-onset and
late-onset pompe disease: An Iranian consensus. Front Neurol.
12(739931)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Llerena Junior JC, Nascimento OJ, Oliveira
ASB, Dourado Junior ME, Marrone CD, Siqueira HH, Sobreira CF,
Dias-Tosta E and Werneck LC: Guidelines for the diagnosis,
treatment and clinical monitoring of patients with juvenile and
adult Pompe disease. Arq Neuropsiquiatr. 74:166–176.
2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tarnopolsky MA and Nilsson MI: Nutrition
and exercise in Pompe disease. Ann Transl Med.
7(282)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Galeotti A, De Rosa S, Uomo R,
Dionisi-Vici C, Deodato F, Taurisano R, Olivieri G and Festa P:
Orofacial features and pediatric dentistry in the long-term
management of infantile Pompe disease children. Orphanet J Rare
Dis. 15(329)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Prater SN, Banugaria SG, DeArmey SM, Botha
EG, Stege EM, Case LE, Jones HN, Phornphutkul C, Wang RY, Young SP
and Kishnani PS: The emerging phenotype of long-term survivors with
infantile Pompe disease. Genet Med. 14:800–810. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Parini R, De Lorenzo P, Dardis A, Burlina
A, Cassio A, Cavarzere P, Concolino D, Della Casa R, Deodato F,
Donati MA, et al: Long term clinical history of an Italian cohort
of infantile onset Pompe disease treated with enzyme replacement
therapy. Orphanet J Rare Dis. 13(32)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pfrimmer C, Smitka M, Muschol N, Husain
RA, Huemer M, Hennermann JB, Schuler R and Hahn A: Long-term
outcome of infantile onset pompe disease patients treated with
enzyme replacement therapy-data from a german-austrian cohort. J
Neuromuscul Dis. 11:167–177. 2024.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Broomfield A, Fletcher J, Davison J,
Finnegan N, Fenton M, Chikermane A, Beesley C, Harvey K, Cullen E,
Stewart C, et al: Response of 33 UK patients with infantile-onset
Pompe disease to enzyme replacement therapy. J Inherit Metab Dis.
39:261–271. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Benedetto L, Musumeci O, Giordano A,
Porcino M and Ingrassia M: Assessment of parental needs and quality
of life in children with a rare neuromuscular disease (Pompe
disease): A quantitative-qualitative study. Behav Sci (Basel).
13(956)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Schoser B, Bilder DA, Dimmock D, Gupta D,
James ES and Prasad S: The humanistic burden of Pompe disease: are
there still unmet needs? A systematic review. BMC Neurol.
17(202)2017.PubMed/NCBI View Article : Google Scholar
|