Introduction
Chronic myeloid leukemia (CML) is a clonal form of
chronic myeloproliferative neoplasm that is associated with the
presence of the BCR-ABL1 fusion gene in the Philadelphia chromosome
(1). The introduction of tyrosine
kinase inhibitors (TKIs) has markedly improved patient survival;
however, resistance remains a major clinical concern. Although ABL1
kinase domain mutations are the most well-studied resistance
mechanism, it is important to note that 40% of cases may occur
through BCR-ABL1-independent means (2,3),
often with chromosomal instability or mutation in other commonly
altered myeloid genes (4,5). Apart from the genetic mutations,
accumulating evidence indicates to epigenetic deregulation as a key
player in the pathogenesis of leukemia and response to treatment
(5,6). Epigenetic modifications (such as DNA
methylation and chromatin remodeling) are capable of impacting on
transcriptional programs independent of the underlying sequence
without altering the latter, enabling disease progression and
resistance to targeted therapies (7,8).
The de novo DNA methyltransferase encoded by
DNA methyltransferase 3 alpha (DNMT3A) is required for proper
differentiation of hematopoietic stem cells. Short variants,
present in 20-30% of acute myeloid leukemia (AML) cases can be
classed as truncating and impact DNA methylation observed after a
differentiation block and clonal outgrowth inducer (9-11).
However, limited information is available on the expression of the
DNMT3A gene in CML and its association with treatment outcomes.
Another key regulator, sex combs-like 1 (ASXL1), is a
chromatin-binding factor, which along with polycomb repressive
complexes, can directly affect the transcriptional programs that
are essential for stem cell function (12). ASXL1 mutations occur in 15-20% of
myeloid disorders and are uniformly associated with a poor
prognosis (12). In CML, mutations
in ASXL1 have been found to be associated with rapid transformation
and an inferior response to TKIs (13,14)
Nevertheless, the precise roles of DNMT3A and ASXL1 in CML have not
yet been fully elucidated, particularly in Middle Eastern countries
with limited molecular data.
Regional analyses have emphasized the critical
requirement for population-based investigations; for example, the
study by Sabir et al (8)
emphasized the importance of epigenetic regulation in patients with
CML who are treated with TKIs. Based on this background, the
primary objective of the present study was to explore whether
DNMT3A and ASXL1 expression, in addition to DNMT3A polymorphisms
are associated with CML in Iraqi patients and whether they may be
indicators for susceptibility and response towards therapy.
Patients and methods
Patients and study design
A cross-sectional analytical study was employed,
adhering to the Strengthening the Reporting of Observational
Studies in Epidemiology (STROBE) guidelines for cross-sectional
studies. The study was conducted between January, 2024 and July,
2025. The study included 140 patients with confirmed CML and 20
age- and sex-matched healthy controls (with no history of
hematological or other chronic diseases), for a total of 160
participants. All subjects were ≥18 years of age.
Patients were categorized according to their
molecular response to therapy with TKIs, following the European
Leukemia Net (ELN) 2020 guidelines (15). The study population was stratified
into three groups as follows: i) 20 TKI-naïve patients (newly
diagnosed, treatment-initiation group); ii) 60 responders; and iii)
60 non-responders/resistant cases. The molecular response was
assessed by measuring BCR-ABL1 transcript levels (expressed as
BCR-ABL1 on the international scale, BCR-ABL1^IS) at three
standardized time points: 3 months (≤10%), 6 months (≤1%) and 12
months (≤0.1%), in accordance with ELN 2020 criteria. Responders
were defined as patients who achieved ELN-recommended molecular
milestones, whereas non-responders were defined as those with
persistent failure to achieve ELN-defined response criteria across
follow-up time points, rather than a single time-point deviation.
To minimize potential confounding factors, patients with documented
non-adherence to TKI therapy were excluded. In addition, patients
who required treatment switching due to intolerance or adverse
effects were also excluded from the study.
TKI therapy
All patients received TKI therapy as part of their
standard clinical management for CML. The cohort comprised patients
receiving the following TKI regimens: i) Imatinib mesylate
(Gleevec®): A total of 60 patients (43%) received
imatinib at a standard dose of 400 mg daily as first-line therapy.
This group included 20 newly diagnosed patients, 20 responder
patients and 20 non-responder patients. ii) Nilotinib
(Tasigna®): A total of 40 patients (29%) received
nilotinib at a standard dose of 300 mg twice daily. This group
included 20 responder patients and 20 non-responder patients. iii)
Bosutinib (Bosulif®): A total of 40 patients (29%)
received bosutinib at a standard dose of 500 mg once daily as
second-line therapy following imatinib failure or intolerance. This
group comprised 20 responder patients and 20 non-responder
patients.
The distribution of TKI therapy across the study
groups was as follows: The TKI-naïve group (n=20) received no prior
TKI therapy; the responder group (n=60) comprised equal proportions
on imatinib (n=20; 33%), nilotinib (n=20; 33%) and bosutinib (n=20;
33%); the non-responder group (n=60) similarly received imatinib
(n=20; 33%), nilotinib (n=20; 33%) and bosutinib (n=20; 33%).
Treatment duration and follow-up
period
The duration of TKI treatment prior to enrollment
varied between the study groups. Patients who were responders had
received TKI therapy for a sufficient duration to achieve and
maintain optimal molecular response (BCR-ABL1IS ≤0.1%) by the
12-month assessment milestone as defined by the ELN 2020 criteria
(15). Patients who were
non-responders had received prolonged TKI therapy for a minimum of
12 months and failed to achieve ELN-defined molecular milestones,
with persistent BCR-ABL1IS levels >1% despite adequate adherence
and dose optimization. Molecular response assessment followed the
ELN 2020 guidelines, with BCR-ABL1 transcript levels monitored at
3, 6, and 12 months from the initiation of TKI therapy (15). Additional clinical follow-up and
molecular monitoring were conducted as clinically indicated beyond
the 12-month assessment point, with quarterly BCR-ABL1 transcript
monitoring performed in accordance with ELN recommendations.
Inclusion and exclusion criteria
Inclusion criteria comprised adult patients (aged
≥18 years) with a confirmed diagnosis of CML based on clinical,
hematological, cytogenetic and molecular analyses, including the
detection of the BCR-ABL1 fusion gene, and who were in chronic
phase at the time of enrollment. The healthy controls were age- and
sex-matched individuals with no history of hematological
malignancies or other chronic diseases, with normal complete blood
count (CBC) and differential counts.
Exclusion criteria included patients with concurrent
hematological malignancies other than CML, those with prior
chemotherapy or stem cell transplantation before enrollment,
patients with documented non-adherence to prescribed TKI therapy
(defined as <80% adherence over the treatment period), patients
who required treatment switching due to intolerance or severe
adverse effects during the active study observation period, and
individuals who declined to provide informed consent.
CML diagnosis and baseline
characterization
A confirmed diagnosis of CML was established through
integrated clinical, hematological, cytogenetic and molecular
analyses. All patients underwent CBC, bone marrow aspiration and
biopsy, cytogenetic analysis and quantitative polymerase chain
reaction (qPCR) for BCR-ABL1 transcript measurement. All patients
were confirmed to be in the chronic phase at diagnosis, with no
evidence of accelerated phase or blast crisis. The study sample
comprised all eligible patients who met the inclusion criteria and
were available at the study center during the recruitment period,
consistent with the exploratory nature of the present
investigation. Healthy controls were age- and sex-matched
individuals with no history of hematological malignancies or other
chronic diseases, with normal CBC and differential counts.
Ethical considerations
The research protocol was approved by the Ethics
Committee of the College of Science, Al-Mustansiriyah University,
on December 30, 2023 (Ref. no. BCSMU/291/100477/2). The study was
conducted in accordance with the ethical principles outlined in the
Declaration of Helsinki and the guidelines of the approving
committee. Institutional Review Board (IRB) approval was obtained
prior to participant enrollment. Written informed consent was
obtained from all participants prior to enrollment. Participants
were informed of the study objectives, procedures, potential risks,
and their right to withdraw at any time without consequence.
Confidentiality of all personal and clinical data was ensured
throughout the study.
Type of sampling and reasons for
selection
A consecutive sampling strategy was employed,
whereby all eligible patients attending the National Center for
Research and Treatment of Hematology, Mustansiriyah University, and
the Center for Hematology and Bone Marrow Transplantation, Baghdad
Teaching Hospital, Medical City, Baghdad, Iraq, during the defined
recruitment period who satisfied the inclusion criteria were
systematically enrolled. This approach ensured that participant
selection was determined solely by eligibility and availability
rather than by subjective judgment, thereby minimizing selection
bias. The three clinical subgroups (TKI-naïve, responders and
non-responders) were defined exclusively on the basis of
objectively assessed molecular response criteria in accordance with
ELN 2020 guidelines, rather than by investigator discretion
(15).
Blood sample collection
Peripheral blood was obtained from each participant
under aseptic conditions. For gene expression analysis, whole blood
was mixed with TransZol Up reagent (TransGen Biotech) and processed
to extract total RNA, using the manufacturer's protocol, and then
subjected to reverse transcription-quantitative PCR (RT-qPCR)
analysis of the DNMT3A and ASXL1 genes. Samples were collected in
ethylenediaminetetraacetic acid (EDTA) tubes, stored at -20˚C, and
used for DNA extraction, PCR amplification and Sanger sequencing of
the DNMT3A target exons.
Study primers
Primers were designed using Primer 3Plus (version 4)
and verified through the UCSC and NCBI databases. Primers were
synthesized and lyophilized by Alpha DNA Ltd. The specific primer
sequences used for gene expression and sequencing analysis were
designed to yield distinct product sizes. For gene expression
analysis, the ASXL1 primers (forward, 5'-CGGCTTGAAGATCGTCAGTCCT-3';
reverse, 5'-GGCTGACCTTTAACCACCCAGG-3') generated a 146-bp product,
whereas the DNMT3A primers (forward, 5'-TATTGATGAGCGCACAAGAGAGC-3';
reverse, 5'-GGGTGTTCCAGGGTAACATTGAG-3') produced a 111-bp fragment.
The reference gene, glyceraldehyde-3-phosphate dehydrogenase
(GAPDH), was amplified using specific primers (forward,
5'-ACAACTTTGGTATCGTGGAAGG-3'; reverse, 5'-GCCATCACGCCACAGTTTC-3'),
resulting in a 101-bp product. For gene sequencing, the ASXL1
primers (forward, 5'-AGGTCAGATCACCCAGTCAGTT-3'; reverse,
5'-TAGCCCATCTGTGAGTCCAACTGT-3') yielded a 561-bp product, and the
DNMT3A primers (forward, 5'-TCCATATCTGGGAGGCTCAG-3'; reverse,
5'-CAGGAGGCGGTAGAACTCAA-3') produced a 738-bp fragment.
Gene expression analysis
Total RNA was isolated from peripheral blood using
the TransZol Up Plus RNA kit (TransGen Biotech) according to the
manufacturer's instructions. RNA concentration and purity were
assessed using a NanoDrop spectrophotometer (Thermo Fisher
Scientific, Inc.). Complementary DNA (cDNA) was synthesized by
adding 5 µl EasyScript Reverse Transcriptase (TransGen Biotech) to
the extracted RNA. qPCR was performed using a Rotor-Gene Q
Real-Time PCR system (Qiagen, Hilden, Germany) with SYBR Green
Master Mix (Qiagen GmbH). The cycling conditions included an
initial denaturation at 94˚C for 30 sec, followed by 40 cycles of
denaturation at 94˚C for 5 sec, annealing at 60˚C for DNMT3A and
58˚C for ASXL1 for 15 sec, and extension at 72˚C for 20 sec. Gene
expression levels were normalized using GAPDH as the reference
gene, based on its previously reported stability across different
tissues and experimental conditions. The 2-ΔΔCq method
was used to analyze relative gene expression levels, and the
2-ΔΔCq method was used to calculate fold changes between
groups. The healthy control group was used as the calibrator
(16).
Sequencing and single nucleotide
polymorphisms (SNP) genotyping
PCR amplification was performed using primers
specifically designed to amplify the clinically relevant hotspot
regions of DNMT3A exon 23 and ASXL1 exon 12. The rationale for
targeting exon 23 of DNMT3A stems from cumulative evidence
demonstrating that this exon harbors the most recurrently mutated
codon in myeloid malignancies arginine 882 (R882), which alone
accounts for >60% of all reported DNMT3A mutations (17). For ASXL1, exon 12 represents the
predominant site of pathogenic mutation across the spectrum of
myeloid disorders, most notably the frameshift variant G646WfsX12,
which has been identified as the canonical hotspot mutation in this
gene (18,19). While comprehensive full-gene
sequencing would have been ideal, resource limitations restricted
the analysis of these well-characterized hotspot regions.
The PCR amplification protocol comprised of an
initial denaturation at 94˚C for 5 min, 35 cycles including a
denaturation step at 94˚C for 30 sec, annealing at 58˚C for 30 sec,
and extension at 72˚C for 50 sec, with a final elongation step at
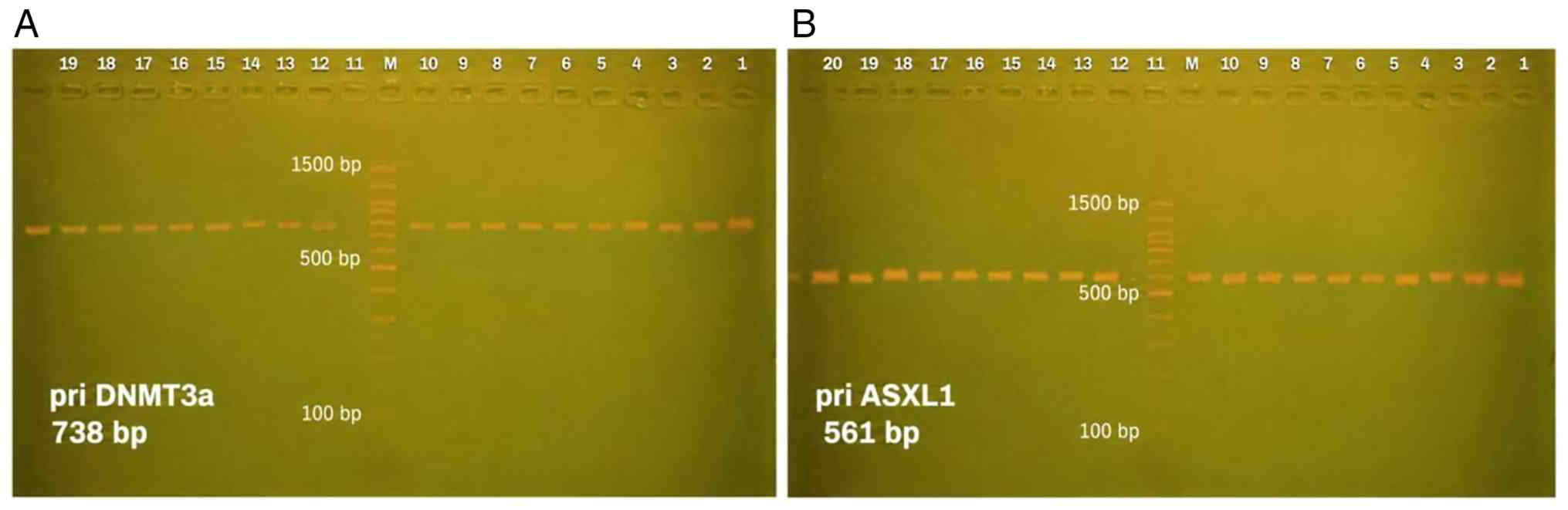
72˚C for 5 min. The product were confirmed by electrophoresis on a
2% agarose gel prepared in 1X TBE buffer and stained with ethidium
bromide. A 100-bp DNA ladder was used as a molecular size marker,
and the electrophoresis was performed at 90 V for 60 min. The bands
were visualized under UV illumination using a gel documentation
system, and the expected amplicon size was confirmed by comparison
with the DNA ladder. Representative gel images for DNMT3A and ASXL1
PCR amplification are presented in Fig. 1. No densitometric quantification
was performed, as agarose gel electrophoresis was used only for
qualitative confirmation of PCR amplification prior to sequencing.
Sequencing analysis of the purified PCR products was performed
using an ABI 3730XL Genetic Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) at Macrogen Inc. The sequences were
analyzed using Chromas v2. 6 (Technelysium Pty Ltd.) and mapped to
the human reference genome (GRCh38). Gene-specific reference
sequences were retrieved from NCBI GenBank (ASXL1: NG_027868.1;
DNMT3A: NG_029465.2). Variants were also confirmed using NCBI BLAST
and annotated by both the dbSNP and ClinVar databases.
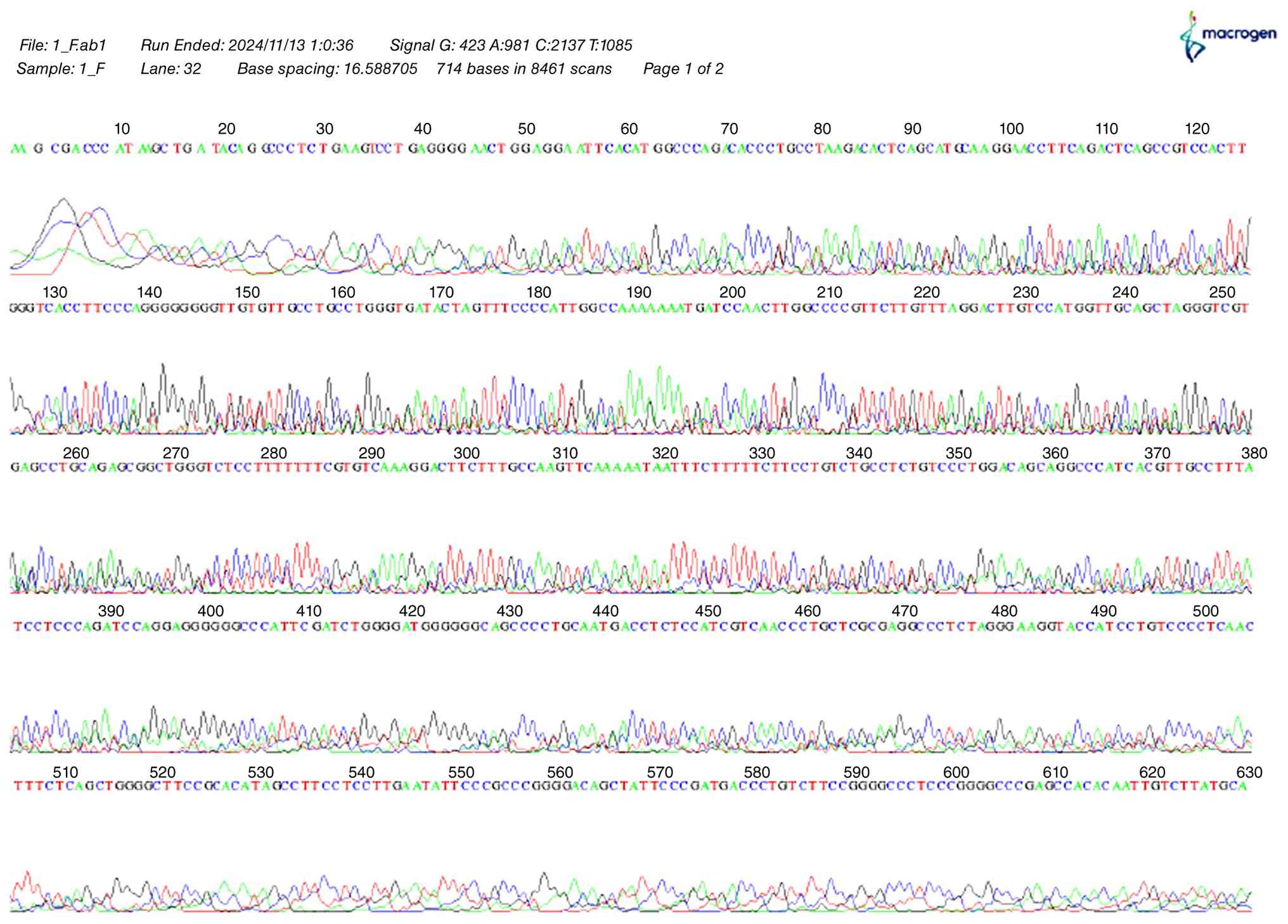
Accuracy, reproducibility and quality
control
Several measures were implemented to ensure data
accuracy and reproducibility. RNA and DNA concentrations and purity
were assessed using a NanoDrop spectrophotometer (Thermo Fisher
Scientific, Inc.) prior to downstream applications. PCR
amplification products were confirmed by agarose gel
electrophoresis before sequencing. Sanger sequencing was performed
by an accredited external facility (Macrogen Inc.) using an ABI
3730XL Genetic Analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Representative Sanger sequencing chromatograms
were reviewed to confirm sequencing quality. A representative
chromatogram demonstrating clear peak resolution and reliable base
calling is presented in Fig. 2.
Sequence analysis was conducted using Chromas v2.6 (Technelysium
Pty Ltd.) and variants were cross-validated against the human
reference genome (GRCh38) using NCBI BLAST and annotated through
both the dbSNP and ClinVar databases. Gene expression analyses were
performed in accordance with standardized RT-qPCR protocols, and
the reference gene, GAPDH, was used as an internal control to
normalize expression data.
Statistical analysis
Statistical analyses were performed using IBM SPSS
Statistics version 29 (IBM Corp.). Quantitative data for gene
expression are expressed as the mean ± standard deviation (SD) and
compared using one-way analysis of variance (ANOVA) followed by
Tukey's HSD post hoc test for pairwise comparisons where
appropriate. Genotype and allele frequencies of DNMT3A
polymorphisms were calculated by direct counting and analyzed using
Pearson's Chi-squared test or Fisher's exact test, reporting odds
ratios (ORs) and 95% confidence intervals (CIs). Haplotype
construction and linkage disequilibrium (LD) were assessed using
the SHEsisPlus online platform (20), and haplotype distributions between
patients and controls were compared using the Chi-squared test with
ORs and 95% CIs. A two-tailed P-value of <0.05 was considered to
indicate a statistically significant difference.
Results
The demographic and clinical characteristics of the
study participants are summarized in Table I. There were no significant
differences in age distribution among the study groups (P=0.292).
However, a significant difference in sex distribution was observed
(P=0.039).
 | Table IDemographic characteristics of the
study population. |
Table I
Demographic characteristics of the
study population.
| | Groups | Mean | Std. Deviation | Std. Error | P-value |
|---|
| Age, years | | | | | |
| | Control (n=20) | 42.66 | 13.17 | 5.37 | 0.292 (NS) |
| | Newly (n=20) | 45.11 | 11.93 | 2.89 | |
| | Response to therapy
(n=60) | 48.72 | 12.72 | 1.73 | |
| | Non-response to
therapy (n=60) | 50.57 | 10.41 | 1.812 | |
| | Sex, n (%) | Male | Female | Chi-squared test
value | P-value |
| | Control (n=20) | 14 (70%) | 6 (30%) | 8.350 | 0.039a |
| | Newly (n=20) | 9 (45%) | 11 (55%) | | |
| | Response to therapy
(n=60) | 26 (43.33%) | 34 (56.67%) | | |
| | Non-response to
therapy (n=60) | 39 (65%) | 21 (35%) | | |
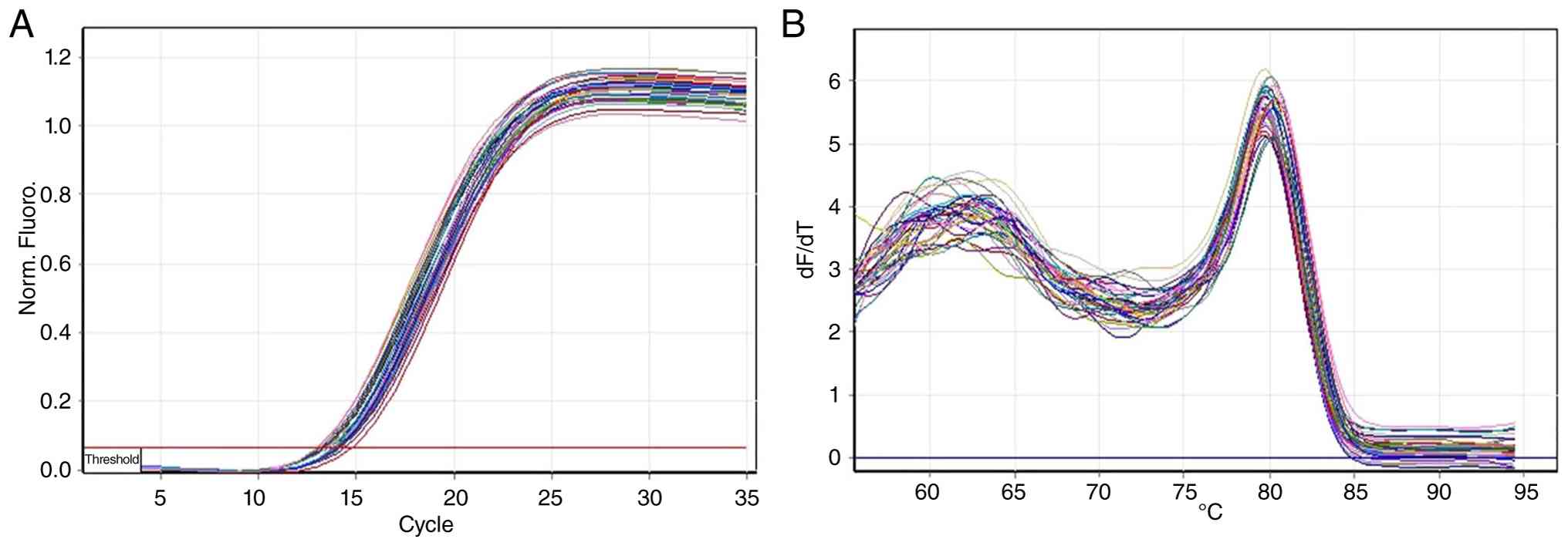
qPCR amplification and the melt curves of GAPDH,
DNMT3A and ASXL1 confirmed efficient and specific amplification
(Figs. 3, 4 and 5).
DNMT3A expression was significantly downregulated in
all CML subgroups compared with the healthy controls (P=0.004),
with the greatest reduction observed in the newly diagnosed and
responder patients (Table II and
Fig. 6A). By contrast, ASXL1
expression was significantly upregulated, exhibiting a progressive
increase across disease stages and peaking in non-responders
(P=0.01) (Table III and Fig. 6B).
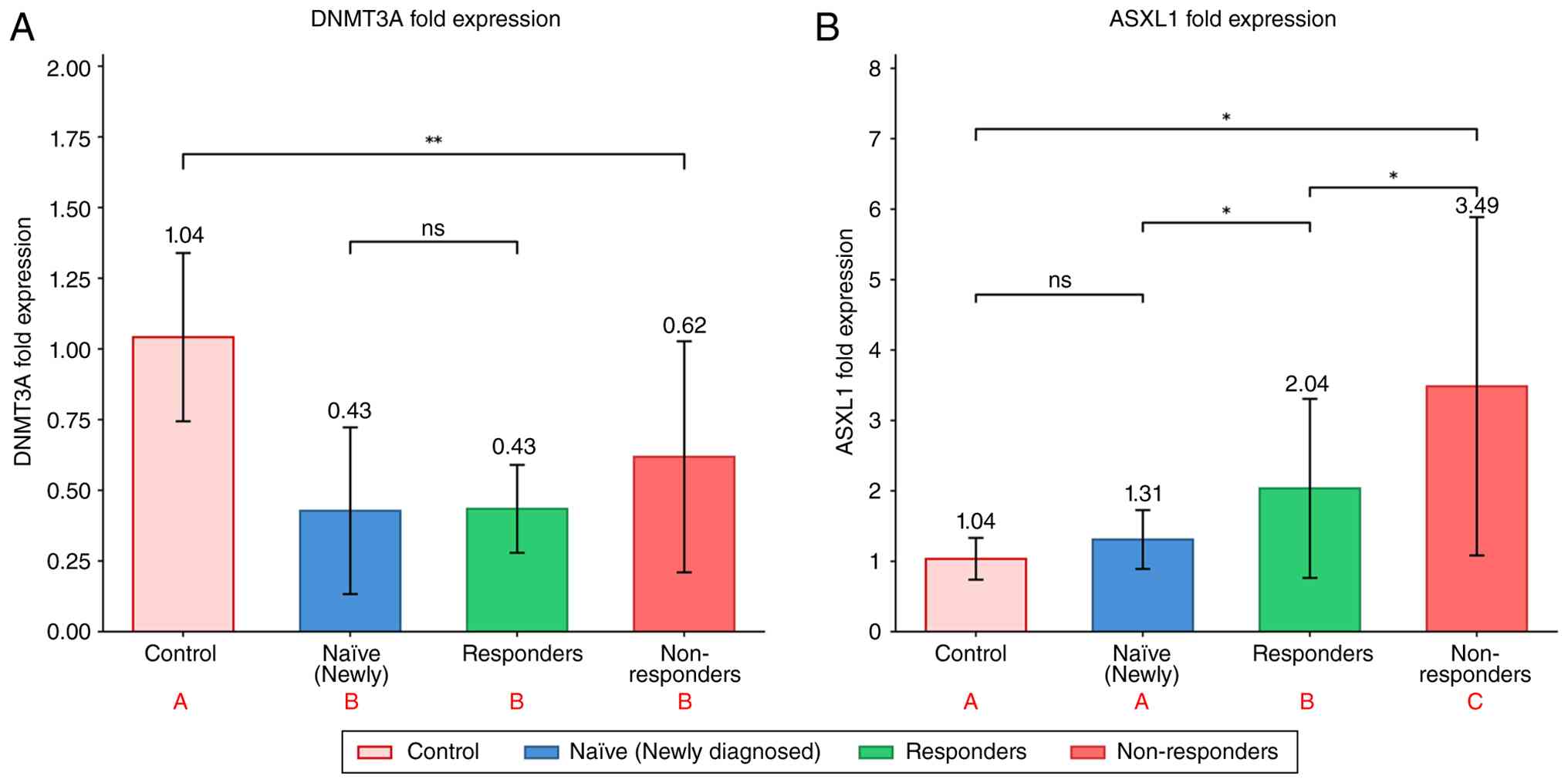 | Figure 6Fold expression of DNMT3A and ASXL1
genes across patient groups and controls. (A) DNMT3A fold
expression levels in control subjects, naïve (newly diagnosed)
patients, responders to therapy, and non-responders to therapy. All
three patient groups exhibited significantly decreased DNMT3A
expression compared to the control group (P=0.004, one-way ANOVA).
No significant difference was observed among the three patient
subgroups). (B) ASXL1 fold expression levels in control subjects,
naïve (newly diagnosed) patients, responders to therapy, and
non-responders to therapy. ASXL1 expression was progressively
upregulated across groups, with non-responders, exhibiting showing
the highest expression (3.49±2.40), followed by responders
(2.04±1.27), naïve patients (1.31±0.42) and controls (1.04±0.30);
overall ANOVA, P=0.015. Data are presented as the mean ± standard
deviation. Statistical analysis was performed using one-way ANOVA
followed by Tukey's HSD post hoc test. Different letters in red (A,
B and C) denote homogeneous subsets; groups sharing the same letter
are not significantly different from each other.
*P<0.05 and **P<0.01, significant
difference; ns, not significant. |
| Table IIDNMT3A gene expression of patient and
control groups. |
Table II
DNMT3A gene expression of patient and
control groups.
| DNMT3A fold
expression | Mean | Std. Deviation | Std. Error | P-value |
|---|
| Control | 1.042b | 0.29727 | 0.13294 | 0.004a |
| Newly | 0.428c | 0.29482 | 0.13185 | |
| Response to
therapy | 0.435c | 0.15537 | 0.04012 | |
| Non-response to
therapy | 0.619c | 0.40855 | 0.10549 | |
| Table IIIASXL1 gene expression of patient and
control groups. |
Table III
ASXL1 gene expression of patient and
control groups.
| ASXL1 fold
expression | Mean | Std. Deviation | Std. Error | P-value |
|---|
| Control | 1.0360b | 0.29670 | 0.13269 | 0.015a |
| Newly | 1.3100b | 0.41755 | 0.18674 | |
| Response to
therapy | 2.0360c | 1.27139 | 0.32827 | |
| Non-response to
therapy | 3.4860d | 2.40202 | 0.62020 | |
DNMT3A and ASXL1 genotyping was performed by PCR
amplification and Sanger sequencing. A representative Sanger
sequencing chromatogram confirming the sequencing quality is
presented in Fig. 2. The
investigation of DNMT3A polymorphisms revealed significant
variations in genotype and allele frequencies between the patients
with CML and the controls. In total, four SNPs (rs2149275435,
rs2149275458, rs25240928 and rs25240958) exhibited a strong
association with CML susceptibility, whereas no significant
association was found for rs734693. Homozygous mutant genotypes
(AA) at rs2149275435 and rs2149275458 were identified exclusively
in patients and were associated with markedly elevated disease risk
(OR, >20; P<0.01). The CC genotype of rs25240928 and the AC
genotype of rs25240958 were similarly enriched in cases, suggesting
potential pathogenic relevance (Table
IV).
| Table IVSignificant genotype and allele
associations. |
Table IV
Significant genotype and allele
associations.
| SNP |
Genotype/allele | Odds ratio
(OR) | 95% CI | P-value |
|---|
| rs2149275435 | AA | 21.67 | 3.14-484.86 | <0.01 |
| rs2149275458 | AA | 41.82 | 5.72-931.15 | <0.01 |
| rs25240928 | CC | 33.85 | 4.74-754.74 | <0.01 |
| rs25240958 | AC | 11.92 | 1.77-268.65 | <0.01 |
| rs734693 | - | 1.57 | 0.35-6.43 | 0.6 (NS) |
In addition, significant deviations from the
Hardy-Weinberg Equilibrium (HWE) were observed for rs734693,
rs2149275458, rs2149275435 and rs25240928 in the control group,
whereas rs25240958 was monomorphic (Table V).
| Table VHardy-Weinberg equilibrium analysis
for DNMT3A SNPs in the control group. |
Table V
Hardy-Weinberg equilibrium analysis
for DNMT3A SNPs in the control group.
| SNP | Observed
(Wild/Het/Mut) | Expected
(Wild/Het/Mut) | χ² | P-value |
|---|
| rs734693 | 0/20/0 | 5/10/5 | 20.000 | <0.0001 |
| rs2149275458 | 0/20/0 | 5/10/5 | 20.000 | <0.0001 |
| rs2149275435 | 0/20/0 | 5/10/5 | 20.000 | <0.0001 |
| rs25240958 | 20/0/0 | 20/0/0 | - | Monomorphic |
| rs25240928 | 0/16/4 | 3.2/9.6/7.2 | 8.889 | 0.003 |
Linkage disequilibrium analysis revealed a strong
non-random association between rs2149275435 and rs2149275458
(D'=1.0, r²=0.758), supporting the presence of a haplotype block.
Haplotype analysis revealed significant associations of specific
DNMT3A haplotypes with CML susceptibility and treatment response.
The C A A A C haplotype was overrepresented in newly diagnosed
patients and non-responders, while the T A A A C haplotype was more
frequent in responder and non-responder groups compared with
controls. In addition, the T G A A C haplotype was strongly
associated with newly diagnosed CML patients. These haplotype
distributions and their corresponding OR and P-values are presented
in Table VI).
| Table VIComplete linkage disequilibrium
analysis across DNMT3A SNP pairs in CML cohorts and controls. |
Table VI
Complete linkage disequilibrium
analysis across DNMT3A SNP pairs in CML cohorts and controls.
| SNP pair | Newly vs. control
(D', r²) | Response vs.
control (D', r²) | Non-response vs.
control (D', r²) |
|---|
|
rs734693-rs2149275458 | 0.388, 0.058 | 0.259, 0.031 | 0.058, 0.001 |
|
rs734693-rs2149275435 | 0.235, 0.010 | 0.281, 0.039 | 0.282, 0.016 |
|
rs734693-rs25240958 | 1.000, 0.076 | 1.000, 0.041 | 1.000, 0.059 |
|
rs734693-rs25240928 | 0.389, 0.035 | 0.212, 0.019 | 0.348, 0.027 |
|
rs2149275458-rs2149275435 | 1.000, 0.474 | 1.000, 0.943 | 1.000, 0.758 |
|
rs2149275458-rs25240958 | 0.999, 0.195 | 0.999, 0.088 | 0.998, 0.080 |
|
rs2149275458-rs25240928 | 0.564, 0.189 | 0.871, 0.676 | 1.000, 0.682 |
|
rs2149275435-rs25240958 | 0.999, 0.075 | 0.999, 0.083 | 0.495, 0.072 |
|
rs2149275435-rs25240928 | 1.000, 0.796 | 0.933, 0.732 | 0.815, 0.598 |
|
rs25240958-rs25240928 | 0.999, 0.095 | 1.000, 0.099 | 0.458, 0.025 |
Furthermore, there is a significant increase in
values of the AAT and CAAAC haplotype frequencies among the patient
groups compared to the controls (Table VII).
| Table VIIHaplotype analysis across CML patient
groups compared with controls. |
Table VII
Haplotype analysis across CML patient
groups compared with controls.
| Haplotype | Newly vs. control
(freq %, OR, P-value) | Response vs.
control (freq %, OR, P-value) | Non-response vs.
control (freq %, OR, P-value) |
|---|
| C A A A C | 25.0%, OR 7.31,
P=0.009 | 5.0%, OR 1.21,
P=0.85 | 20.0%, OR 5.48,
P=0.033 |
| T G A A C | 45.0%, OR 13.69,
P<0.001 | - | - |
| T A A A C | - | 62.5%, OR 32.19,
P<0.001 | 47.0%, OR 14.84,
P<0.001 |
| T A A C C | 15.0%, not
estimable, P=0.011 | 20.0%, not
estimable, P=0.002 | 5.0%, not
estimable, P=0.15 |
| T A G C T | 10.0%, not
estimable, P=0.040 | 7.5%, not
estimable, P=0.070 | 8.0%, not
estimable, P=0.068 |
Discussion
In the present study, there was no difference in the
GAPDH mRNA levels between each of the groups investigated which
confirms its adequacy as an internal reference gene for CML
expression profiling. This is in line with previous evidence
supporting the suitability of using GAPDH as a reference gene in
different tissues and pathologies (21). DNMT3A encodes a de novo DNA
methyltransferase that is required for silencing, hematopoietic
differentiation and the maintenance of genomic stability. There is
a general downregulation of DNMT3A in numerous hematological
malignancies, particularly AML, which is frequently attributed to
loss-of-function mutations or epigenetic silencing. Such reduction
results in global DNA hypomethylation, abnormal activation of
oncogenic signaling, and compromised lineage fidelity (22-24).
DNMT3A inhibition is associated with an unfavorable prognosis and
impaired stem cell differentiation (25), as well as resistance to apoptosis
and self-renewal capability in DNMT3A-null cells (26). These findings underscore the
tumor-suppressive contribution of DNMT3A and point to its silencing
as a potential mechanism facilitating leukemic escape during
treatment with TKIs. In the present study, ASXL1 expression,
however, was markedly higher in the CML groups than the controls
and also gradually increased between the CML groups; ASXL1
expression exhibited a progressive increase, increasing from
1.31-fold in the newly diagnosed patients to 2.04-fold in the
responders (Table III). ASXL1
deregulation and mutations have been widely implicated in the
transformation of myeloid malignancies, including myelodysplastic
syndromes (MDS), AML and CML (12,27).
Recent clinical studies consistently position ASXL1 among the most
relevant adverse genetic markers in CML, particularly concerning
treatment refractoriness. For instance, Bidikian et al
(13) identified ASXL1 as the most
frequently mutated gene in chronic-phase CML and the sole
independent predictor of inferior event-free survival. Similarly,
the TIGER trial by Schönfeld et al (14) demonstrated that newly diagnosed
patients harboring ASXL1 mutations exhibited inferior molecular
responses to nilotinib, confirming that its adverse prognostic
impact extends beyond imatinib-treated cohorts. These findings
align with broader evidence that variants in epigenetic modifier
genes predict response failure to TKIs and maintain their
prognostic significance even under proactive therapeutic strategies
(28). Consistent with this
adverse role, the present study observed a progressive increase in
ASXL1 expression from newly diagnosed to resistant patients,
further underscoring its strong link to treatment failure. However,
no pathogenic mutations were identified within the sequenced region
of ASXL1 exon 12 in the present study cohort. This absence suggests
that the observed upregulation is likely driven by alternative,
non-mutational mechanisms, such as transcriptional or epigenetic
deregulation. Ultimately, these findings support the emerging
consensus that whether through genetic mutation or alternative
overexpression mechanisms, ASXL1 contributes to TKI resistance by
reprogramming and activating alternative pro-survival signaling
pathways (12,14,29).
As regards the genotype distributions, HWE testing
in the control group revealed significant deviations for rs734693,
rs2149275458 and rs2149275435 (all P<0.0001) and rs25240928
(P=0.003), which may reflect population stratification or small
sample size effects. Notably, rs25240958 was monomorphic in the
control group, precluding HWE calculation. Therefore, association
results for these SNPs should be interpreted with caution and
require replication in larger, independently validated cohorts.
Sequencing data of DNMT3A indicated that four
variants (rs2149275435, rs2149275458, rs25240928 and rs25240958)
were associated with CML risk, whereas no significant association
was found for rs734693. Notably, rs2149275435 and rs2149275458 have
not been previously reported in the context of CML or myeloid
malignancies in any published literature, at least to the best of
our knowledge, representing potential population-enriched variants
in the Iraqi cohort that merit further functional characterization.
The enrichment of homozygous mutant genotypes, such as AA at
rs2149275435 and rs2149275458, and CC or AC at rs25240928 and
rs25240958, suggests pathogenic effects. Consistent observations in
AML and MDS have associated DNMT3A polymorphisms with reduced DNA
methylation, clonal hematopoiesis and adverse clinical outcomes
(23,25). In the present study, linkage
disequilibrium analysis indicated that rs2149275435 and
rs2149275458 were in significant non-random association (D'=1.0,
r²=0.758), forming a risk haplotype block. Haplotype analyses
demonstrated that the CAAAC and TGAAC haplotypes were significantly
enriched in newly diagnosed and non-responder patients, suggesting
that the additive effects of allele combinations promote leukemia
persistence rather than individual alleles acting in isolation
(30). Analogous haplotype-level
associations have been observed in myeloid malignancies, with
DNMT3A haplotypes implicated in the modification of DNA methylation
and disease susceptibility (31-33).
Recent literature has further strengthened the
clinical importance of somatic mutation profiling beyond canonical
kinase-domain analysis. Contemporary reports indicate that ASXL1
abnormalities at diagnosis continue to be associated with inferior
outcomes and may even be associated with a higher risk of acquiring
ABL1 kinase domain mutations during therapy, suggesting that
epigenetic dysregulation may create a permissive background for
subsequent evolutionary adaptation (34). In parallel, broader outcome
analyses across age groups have confirmed that ASXL1, DNMT3A and
TET2 remain among the most recurrently altered epigenetic
regulators in adolescent, young adult, and older adult CML
populations (35). Taken together,
these studies reinforce the interpretation that the expression
abnormalities and DNMT3A variants detected in the present study
cohort are clinically meaningful and fit within the current
understanding of CML as a genetically and epigenetically
heterogeneous disease.
Overall, the results of the present study support a
model in which a reduced DNMT3A activity and an increased ASXL1
expression contribute to an epigenetic state that favors leukemic
persistence, attenuated therapeutic response, and possible clonal
selection under TKI pressure. Although BCR-ABL1 remains the
defining lesion in CML, accumulating evidence indicates that
additional epigenetic abnormalities strongly influence the disease
trajectory (13,23,28,36).
Consequently, the incorporation of epigenetic biomarkers into
future prognostic algorithms may improve risk stratification and
guide individualized treatment decisions, especially in patients
with unexpected resistance or those considering treatment
discontinuation. These findings await confirmation in larger,
ethnically diverse populations and should be integrated with
sequencing of regulatory regions to establish true prognostic value
and therapeutic opportunities.
Given the exploratory nature and modest sample size
of the present study, the findings should be regarded as
preliminary. The elevated ORs observed for certain genotypes should
be interpreted with caution, as these estimates may be influenced
by the limited sample size and warrant confirmation in larger,
prospective, and ethnically diverse independent cohorts.
Several limitations should be acknowledged when
interpreting the findings of the present study. First, the
relatively small sample size, including a limited control group
(n=20), and the single-center design reduce statistical power and
limit generalizability, underscoring the need for external
validation in independent, ethnically diverse cohorts. Second,
although the cross-sectional design precludes formal establishment
of temporal associations, the consistent and statistically
significant expression patterns observed across all patient groups
strongly support the biological relevance of the findings. Third,
molecular profiling was necessarily restricted to the highest-yield
hotspot regions of DNMT3A (exon 23) and ASXL1 (exon 12); while
comprehensive next-generation sequencing would further enrich these
findings, the associated costs rendered this approach unfeasible in
the context of the present self-funded investigation. Additionally,
BCR-ABL1 kinase domain mutation analysis was not performed;
however, the differential expression patterns identified herein
represent independent epigenetic alterations contributing insights
beyond canonical resistance pathways. The use of GAPDH as a sole
reference gene, although widely adopted, may introduce
normalization bias and should be considered when interpreting
fold-change values. Finally, ROC curve analysis was not performed,
as the primary objective was to characterize molecular expression
patterns rather than establish diagnostic thresholds, and such
analysis would be more appropriately conducted in future
prospective studies. Notwithstanding these limitations, the present
study provides novel insight into the epigenetic dysregulation of
CML in an underrepresented population, contributing to the growing
evidence implicating DNMT3A and ASXL1 in disease susceptibility and
response to TKIs.
In conclusion, the downregulation of DNMT3A and the
upregulation of ASXL1 expression, along with specific DNMT3A
polymorphisms, present a combined epigenetic and genetic signature
associated with CML susceptibility and resistance to TKIs.
Integrating these biomarkers into prognostic models may enhance
risk stratification and guide personalized therapeutic strategies
in CML management.
Acknowledgements
The authors are grateful to Al-Mustansiriyah
University (College of Science) and the National Center of
Hematology, Baghdad, Iraq, for the laboratory facilities and
technical support.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to patient
confidentiality considerations, but are available from the
corresponding author upon reasonable request. All sequence data
were mapped to the human reference genome (GRCh38), and
gene-specific reference sequences were retrieved from NCBI GenBank
(ASXL1: NG_027868.1; DNMT3A: NG_029465.2).
Authors' contributions
AMA conceptualized the study. AMA and ANAR were
involved in the study methodology, and in the writing, reviewing
and editing of the manuscript. All authors (AMA, ANAR and AFA) were
involved in data validation, investigation and in the writing and
preparation of the original draft of the manuscript. AFA
contributed to the formal analysis, data curation and figure
preparation. AMA provided laboratory facilities, reagents,
instruments and technical support. AFA supervised the study. AMA
was involved in project administration. All authors have read and
agreed to the published version of the manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Institutional
Review Board (IRB) of the College of Science, Al-Mustansiriyah
University (Ref. No. BCSMU/291/100477/2). The study was conducted
in accordance with the ethical principles of the Declaration of
Helsinki and the guidelines of the approving committee.
Participants provided written informed consent before being
recruited into the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jabbour E and Kantarjian H: Chronic
myeloid leukemia: A review. JAMA. 333:1618–1629. 2025.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pamuk GE and Ehrlich LA: An overview of
myeloid blast-phase chronic myeloid leukemia. Cancers (Basel).
16(3615)2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yohanan B and George B: Current management
of chronic myeloid leukemia myeloid blast phase. Clin Med Insights
Oncol. 16(11795549221139357)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Issa GC, Kantarjian HM, Gonzalez GN,
Borthakur G, Tang G, Wierda W, Sasaki K, Short NJ, Ravandi F, Kadia
T, et al: Clonal chromosomal abnormalities appearing in
Philadelphia chromosome-negative metaphases during CML treatment.
Blood. 130:2084–2091. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Alves R, Gonçalves AC, Rutella S, Almeida
AM, De Las Rivas J, Trougakos IP and Sarmento Ribeiro ABS:
Resistance to tyrosine kinase inhibitors in chronic myeloid
leukemia-from molecular mechanisms to clinical relevance. Cancers
(Basel). 13(4820)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Za'ror YSMA, Zulkafli Z, Al-Eitan LN,
Elsalem L, Al-Husein BA and Azlan M: The expression of BCL11A,
KLF1, and ERK of mitogen-activated protein kinase pathway on stem
cell factor and erythropoietin-treated K562 cells. Biomed
Biotechnol Res J. 6:563–568. 2022.
|
|
7
|
Song J, Yang P, Chen C, Ding W, Tillement
O, Bai H and Zhang S: Targeting epigenetic regulators as a
promising avenue to overcome cancer therapy resistance. Signal
Transduct Target Ther. 10(219)2025.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sabir SF, Matti BF and Alwatar WMA:
Assessment of regulatory T cells (Tregs) and Foxp3 methylation
level in chronic myeloid leukemia patients on tyrosine kinase
inhibitor therapy. Immunogenetics. 75:145–153. 2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Park DJ, Kwon A, Cho BS, Kim HJ, Hwang KA,
Kim M and Kim Y: Characteristics of DNMT3A mutations in acute
myeloid leukemia. Blood Res. 55:17–26. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kalal AA, Shetty VV, Shetty KP, Arumugam
M, Shetty RA, Kulkarni NV and Shetty DP: Correlation between
platelet-to-lymphocyte ratio and neutrophil-to-lymphocyte ratio
with hematological parameters in multiple myeloma patients. Biomed
Biotechnol Res J. 6:132–137. 2022.
|
|
11
|
Zhao A, Zhou H, Yang J, Li M and Niu T:
Epigenetic regulation in hematopoiesis and its implications in the
targeted therapy of hematologic malignancies. Signal Transduct
Target Ther. 8(71)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Medina EA, Delma CR and Yang FC: ASXL1/2
mutations and myeloid malignancies. J Hematol Oncol.
15(127)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bidikian A, Kantarjian H, Jabbour E, Short
NJ, Patel K, Ravandi F, Sasaki K and Issa GC: Prognostic impact of
ASXL1 mutations in chronic phase chronic myeloid leukemia. Blood
Cancer J. 12(144)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Schönfeld L, Rinke J, Hinze A, Nagel SN,
Schäfer V, Schenk T, Fabisch C, Brümmendorf TH, Burchert A, le
Coutre P, et al: ASXL1 mutations predict inferior molecular
response to nilotinib treatment in chronic myeloid leukemia.
Leukemia. 36:2242–2249. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hochhaus A, Baccarani M, Silver RT,
Schiffer C, Apperley JF, Cervantes F, Clark RE, Cortes JE,
Deininger MW, Guilhot F, et al: European LeukemiaNet 2020
recommendations for treating chronic myeloid leukemia. Leukemia.
34:966–984. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ley TJ, Ding L, Walter MJ, McLellan MD,
Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et
al: DNMT3A mutations in acute myeloid leukemia. N Engl J Med.
363:2424–2433. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Grossmann V, Kohlmann A, Haferlach C,
Alpermann T, Wild M, Weissmann S, Eder C, Dicker F, Kern W,
Schnittger S and Haferlach T: Landmark analyses of DNMT3A mutations
in hematological malignancies. Blood. 118(407)2011.
|
|
19
|
Medina EA, Delma CR and Yang FC: ASXL1/2
mutations and myeloid malignancies. J Hematol Oncol. BioMed
Central. 15:1–18. 2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Shen J, Li Z, Chen J, Song Z, Zhou Z and
Shi Y: SHEsisPlus, a toolset for genetic studies on polyploid
species. Sci Rep. 6(24095)2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Caracausi M, Piovesan A, Antonaros F,
Strippoli P, Vitale L and Pelleri MC: Systematic identification of
human housekeeping genes possibly useful as references in gene
expression studies. Mol Med Rep. 16:2397–2410. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nteliopoulos G, Bazeos A, Claudiani S,
Gerrard G, Curry E, Szydlo R, Alikian M, Foong HE, Nikolakopoulou
Z, Loaiza S, et al: Somatic variants in epigenetic modifiers can
predict failure of response to imatinib but not to
second-generation tyrosine kinase inhibitors. Haematologica.
104:2400–2409. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Adnan Awad S, Brück O, Shanmuganathan N,
Jarvinen T, Lähteenmäki H, Klievink J, Ibrahim H, Kytölä S,
Koskenvesa P, Hughes TP, et al: Epigenetic modifier gene mutations
in chronic myeloid leukemia (CML) at diagnosis are associated with
risk of relapse upon treatment discontinuation. Blood Cancer J.
12(69)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wu W, Xu N, Zhou X, Liu L, Tan Y, Luo J,
Huang J, Qin J, Wang J, Li Z, et al: Integrative genomic analysis
reveals cancer-associated gene mutations in chronic myeloid
leukemia patients with resistance or intolerance to tyrosine kinase
inhibitor. Onco Targets Ther. 13:8581–8591. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Challen GA, Sun D, Jeong M, Luo M, Jelinek
J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al: Dnmt3a is
essential for hematopoietic stem cell differentiation. Nat Genet.
44:23–31. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Jeong M, Sun D, Luo M, Huang Y, Challen
GA, Rodriguez B, Zhang X, Chavez L, Wang H, Hannah R, et al: Large
conserved domains of low DNA methylation maintained by Dnmt3a. Nat
Genet. 46:17–23. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
McCurry D, Ge Z, Lee J, Raparla P, Koehnke
T, Pasumarthi R, Leng X, Pasvolsky O, Maurer K, Li S, et al: ASXL1
truncating mutations drive leukemic resistance to T cell attack.
Blood. 142(364)2023.
|
|
28
|
Shanmuganathan N, Wadham C, Shahrin N,
Feng J, Thomson D, Wang P, Saunders V, Kok CH, King RM, Kenyon RR,
et al: Impact of additional genetic abnormalities at diagnosis of
chronic myeloid leukemia for first-line imatinib-treated patients
receiving proactive treatment intervention. Haematologica.
108:2380–2395. 2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Miyashita N, Onozawa M, Kasahara K,
Matsukawa T, Onodera Y, Suzuki K, Takaku T, Teshima T and Kondo T:
CML with mutant ASXL1 showed decreased sensitivity to TKI treatment
via upregulation of the ALOX5-BLTR signaling pathway. Cancer Sci.
116:1115–1125. 2025.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yuan XQ, Zhang DY, Yan H, Yang YL, Zhu KW,
Chen YH, Li X, Yin JY, Li XL, Zeng H and Chen XP: Evaluation of
DNMT3A genetic polymorphisms as outcome predictors in AML patients.
Oncotarget. 7:60555–60574. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Venugopal K, Feng Y, Shabashvili D and
Guryanova OA: Alterations to DNMT3A in hematologic malignancies.
Cancer Res. 81:254–263. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Do C, Lang CF, Lin J, Darbary H, Krupska
I, Gaba A, Petukhova L, Vonsattel JP, Gallagher MP, Goland RS, et
al: Mechanisms and disease associations of haplotype-dependent
allele-specific DNA methylation. Am J Hum Genet. 98:934–955.
2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yang Y, Dai Y, Yang X, Wu S and Wang Y:
DNMT3A mutation-induced CDK1 overexpression promotes leukemogenesis
by modulating the interaction between EZH2 and DNMT3A.
Biomolecules. 11(781)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Shanmuganathan N, Yeung DT, Wadham C,
Fernandes A, Maqsood M, Shahrin N, Saunders V, Kenyon RR, Lin M,
Toubia J, et al: Impact of ASXL1 at diagnosis in patients with CML
receiving frontline potent TKIs: High risk of kinase domain
mutations. Blood. 146:2821–2832. 2025.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Krizkova J, Polivkova V, Laznicka A, Curik
N, Benesova A, Suchankova P, Smazik T, Vysinova V, Mikulenkova D,
Klamova H, et al: Somatic mutations and outcomes in chronic myeloid
leukemia adolescent and young adults compared to children, adults,
and BCR::ABL1-positive acute lymphoblastic leukemia. Leukemia.
39:1670–1677. 2025.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Perusini MA, Žáčková D, Kim T, Pagnano K,
Pavlovsky C, Ježíšková I, Kvetková A, Jurček T, Kim J, Yoo Y, et
al: Mutations in myeloid transcription factors and activated
signaling genes predict chronic myeloid leukemia outcomes. Blood
Adv. 8:2361–2372. 2024.PubMed/NCBI View Article : Google Scholar
|