Contents
Introduction
What are Aurora kinases?
Clinical applications of Aurora kinase
inhibitors
Gynecologic applications of Aurora kinase
inhibitors
Conclusion
Introduction
Conventional anticancer agents reduce the size of
tumors by damaging cells. However, the targeted cells are not
specific, leading to damage to normal cells and adverse reactions.
Therefore, cytotoxic anticancer drugs are administered at a maximum
tolerated dose calculated based on the anticancer effect and
adverse effects. By contrast, molecular-targeted anticancer agents
are developed based on the analysis of differences between cancer
and normal cells at the whole genome or molecular level as drugs
that inhibit specific molecules and which cause cancer
proliferation or metastasis. Thus, these agents have more specific
effects on cancer cells and a much lower level of adverse reactions
than conventional anticancer drugs, permitting administration at
the minimum effective dose, rather than the maximum-tolerated dose.
This is a major advantage of molecular-targeted anticancer agents.
However, conventional anticancer drugs currently play a key role in
treatments such as neoadjuvant and adjuvant chemotherapy for
gynecologic malignant tumors including ovarian, cervical and
endometrial cancer. These approaches use monotherapy or multidrug
combination therapy, mainly with platinum-containing agents or
taxanes. However, many patients suffer from associated adverse
reactions, which has led to a search for more effective drug
combinations. Investigations of the antitumor effects of
molecular-targeted drugs for gynecologic tumors are also underway,
including for everolimus, an inhibitor of mammalian target of
rapamycin and bevacizumab, a vascular endothelial growth factor
inhibitor.
What are Aurora kinases?
Correct replication and distribution to daughter
cells are essential for genetic information to be correctly
inherited by the daughter cells. Normal cell division requires
regulation by a plurality of protein kinases that each act with
temporal and spatial correctness. These mitotic kinases include
checkpoint-associated kinases such as Cyclin B-CDK1, as well as
NimA, Polo, Aurora and WARTS kinases. The transition from G2 to M
phase is the major control point in cell division and loss of
function of these kinases causes chromosomal instability due to
failed division and subsequent oncogenesis (1–4).
Aurora kinase genes were first isolated in 1995 from
a Drosophila mutant that showed abnormal spindle formation
in M phase. Aurora kinases are serine/threonine kinases associated
with the regulation of cell division in the G2-M phases, and are
particularly associated with chromosomal aggluti nation and
segregation, functions of centromeres, centrosomal maturation,
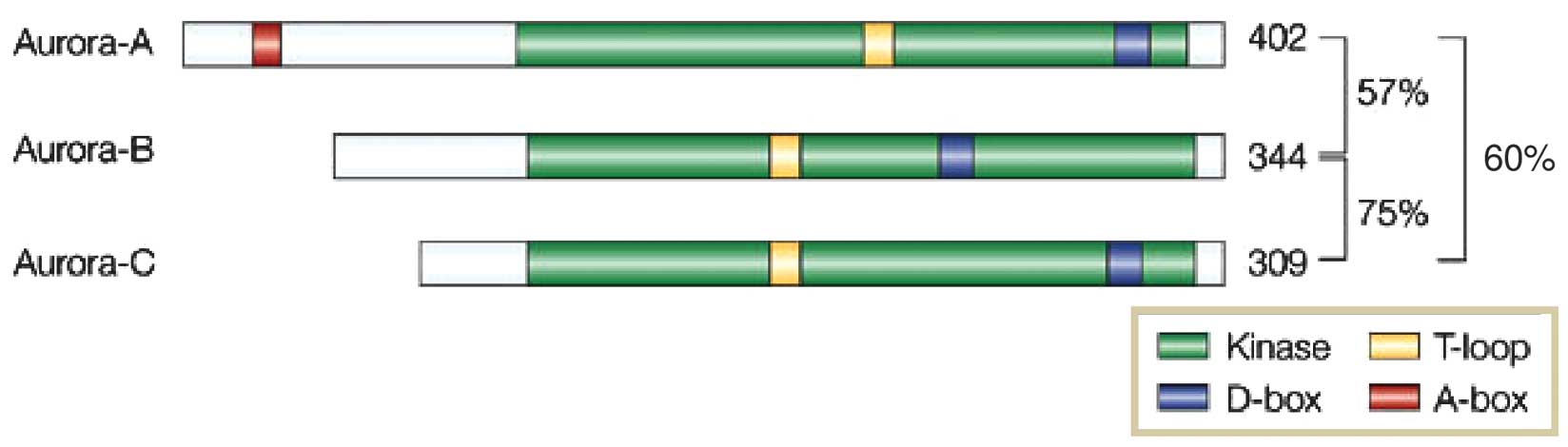
spindle formation and cytokinesis. The human Aurora kinase family
comprises the members Aurora kinase A (AURKA), B (AURKB) and C
(AURKC) (Table I). All three
members have a common N-terminal domain of various lengths, a
kinase domain and a short C-terminal domain. However, they have a
different location and timing of action and are considered to
contribute independently to the progression of M phase. AURKA
contains a catalytic kinase domain, an activating T-loop, a
destruction box (D-box) and a D-box-activating domain (DAD). The
D-box and DAD regions are degraded at the last stage of mitosis.
AURKB and AURKC do not contain a DAD and the D-box is not degraded
(Fig. 1) (1).
 | Table IProperties of Aurora kinases. |
Table I
Properties of Aurora kinases.
| Kinases | Chromosomal
location | Expression in
cancer | Function/role |
|---|
| Aurora A | 20q13.2 | Amplification | Targeting to
microtubules |
| | Overexpression | Suppression of
transformation potential |
| | | Inhibition of kinase
activity |
| | | Positive and negative
regulation of kinase activity |
| | | Control of
microtubule dynamics at the spindle poles |
| Aurora B | 17p13.1 | Overexpression | Targeting to
centromere/midzone |
| | | Elevation of kinase
activity |
| | | Chromosome
segregation? |
| | | Cytokinesis? |
| | | Inhibition of kinase
activity |
| Aurora C | 19q13.43 | Overexpression | Unclear |
AURKA accumulates at centrosomes during the late G2
phase and partially moves into the nucleus before the cell enters M
phase, and before accumulating in the nucleus during M phase.
Subsequent to the breakdown of the nuclear envelope, AURKA
accumulates at spindle poles and on spindles due to segregation of
the centrosomes replicated during S phase. Targeting protein for
Xenopus kinesin-like protein 2 (TPX2) is an important
activator of AURKA. AURKA activated by TPX2 is introduced into
microtubules and is associated with centrosomal functions such as
maturation and the formation of microtubules through replication,
formation and stabilization of spindles in the metaphase. In late M
phase, the amount of AURKA is downregulated by ubiquitin-mediated
degradation and AURKA moves to the central spindle present between
two nuclei after segregation and disappears (1–4).
Inhibition of the expression or activity of AURKA
causes delayed entry into M phase and failed centrosomal
segregation, resulting in the spindle not being formed. By
contrast, the overexpression of AURKA causes increased centrosomal
numbers in the cell or formation of aneuploid cells and poses a
major risk of carcinogenesis. The gene encoding AURKA is localized
to chromosome 20q13, which is amplified in malignant tumors such as
breast and ovarian cancer and is considered to be an oncogene
(5). The spindle formation
checkpoint, which inhibits entry into the anaphase until the
chromosomes are arranged in the equatorial plane, is also disrupted
by the overexpression of AURKA. Bub1-3 and Mad1-3 play an important
role at this checkpoint by managing the number of micro tubules
extending from the two poles at the centrosomes and attaching to
daughter centromeres and pulling equally on both sides, as well as
by stopping cell division when an abnormal event occurs. If
activity of Bub1-3 or Mad1-3 is inhibited, cell division passes the
checkpoint and enters anaphase. In cells overexpressing AURKA and
with the expression of a spindle formation checkpoint protein,
Mad2, Anand et al(6)
reported the improper entry into anaphase as well as the
progression of cell division, with the emergence of aneuploid
cells. Overexpression of AURKA also causes downregulation of p53
through its kinase activity. Thus, tumor cells avoid the induction
of p53-dependent apoptosis and extinction of aneuploid cells
through the Rb/p16 cellular aging pathway (7). If DNA damage or aberrant cell division
occurs, p53 interacts with a transcriptional coactivator, the
heterogeneous nuclear ribonucleoprotein K (hnRNPK) and induces
transcription of the p53 gene. AURKA deactivates hnRNPK by
phosphorylation of Ser379 in hnRNPK and indirectly inhibits p53
activity (8).
AURKB emerges in the nucleus in the early S phase;
localizes in centromeres with three proteins, inner centromere
protein, survivin and borealin, in metaphase; moves to the center
of the spindle in anaphase; and regulates cytokinesis in the median
zone domain of a cell undergoing mitosis. Proteins that are
transported from the chromosome to the center of the spindle are
referred to as chromosomal passenger proteins and have key roles in
centromeres. These proteins include deletion of improper
attachments that interrupt even tension on spindle fibers on both
sides and induction of correct bipolar attachment and arranging of
chromosomes in the equatorial plane. In cases in which microtubules
are unevenly attached to a centromere, the spindle formation
checkpoint is activated and transition to anaphase is inhibited.
The mechanism by which AURKB enhances bipolar attachment is
unclear. AURKB has also been associated with chromosomal
stabilization, such as chromosomal agglutination (1–3,9).
The AURKC gene is located on human chromosome
19q13.43. Recombination and deficiency are often noted in this
domain in various cancer types. Overexpression of AURKC is present
in many types of human cancer cells and mRNA and protein of AURKC
are also highly expressed in normal tissues, particularly in the
testis. AURKC-knockout mice are sterile due to abnormalities in the
shape and functions of sperm and AURKC is thought to have specific
actions in the formation of sperm, beyond those of AURKA and AURKB.
However, compared with the other two Aurora kinases, little is
known about the association of AURKC with carcinogenesis and only a
few AURKC inhibitors have been developed (10).
Clinical applications of Aurora kinase
inhibitors
Overexpression of Aurora kinases has been identified
in many human cancer-derived cultured cell lines and cancer
tissues. The amplification and overexpression of AURKA occurs in
breast, bladder, colon, ovarian, pancreatic and stomach cancers.
The overexpression of AURKB has been found in colon cancer, while
AURKC expression occurs in various cancer-derived cell lines,
including those from colon cancer, as well as in testis in normal
tissues. These results suggest that the overexpression of Aurora
kinases is strongly associated with tumorigenesis. For this reason,
inhibitors of Aurora kinases (Table
II) are being developed as potential anticancer drugs and many
of these agents are in phase I and II clinical trials. These drugs
only affect cells undergoing mitosis. However, many are AURKB
inhibitors that may interrupt chromosome stability by AURKB and
induce DNA damage, suggesting that examination of safety is
important for these agents.
| Table IIProperties of Aurora kinase
inhibitors. |
Table II
Properties of Aurora kinase
inhibitors.
| Drug | Chemical class | Profile | Other targets | Toxicity |
|---|
| ZM447439 | Quinazoline
derivative | B>A | MEK, SRC, LCK | |
| Hesperadin | Indolinone | B>A | Not reported | |
| VX-680/MK-0457 |
4,6-diaminopyrimidine | A=B=C | FLT3 | Grade 4:
Neutropenia
Grade 3: Herpes Zoster infection,
Others: Vomiting, diarrhea, fatigue |
| AT9283 | Pyrazole
derivative | Unknown | JAK2, 3, BCR-ABL,
V-ABL | QT prolongation
(trial halted),
Grade3: Febrile neutropenia |
| AZD1152 | Quinazoline
derivative |
B>>>A,C | Not reported | Grade3/4:
Neutropenia,
Grade1/2: Diarrhea, vomiting, nausea, lost appetite,
hypertension |
ZM447439 was reported as the first Aurora kinase
inhibitor in 2003 and is currently in phase I clinical trials.
ZM447439 inhibits the phosphorylation of histone H3 (Ser 10, 28) by
Aurora kinases. Li et al(11) showed a concentration- and
time-dependent increase in the apoptotic effect of ZM447439 in a
human colon cancer-derived cell line, HCT-116. ZM447439 targets the
ATP-binding site common to the Aurora kinase family and
theoretically should inhibit all Aurora kinases. In vitro,
ZM447439 inhibits AURKA and AURKB, however, in vivo results
show the predominant inhibition of AURKB, thus ZM447439 is now
considered to be an AURKB inhibitor. ZM447439 causes aberrant cell
division by inhibiting AURKB, inducing a p53-dependent apoptotic
pathway and apoptosis of mitochondria by caspase 3 in the presence
of Bak and Bax. Hesperadin is similar to ZM447439 in that it is a
strong inhibitor of ATP binding to AURKB, as well as an inhibitor
of ∼25 other kinases. The reason for many ATP-binding site
inhibitors exerting stronger effects on AURKB remains unclear
(12).
VX-680 is an ATP-binding site inhibitor reported by
Harrington et al(13).
VX-680 is an inhibitor of AURKA, AURKB and AURKC, but exerts the
strongest inhibition on AURKB. VX-680 has shown potential as a new
molecular-targeted drug for BCR/ABL-positive chronic myelogenous
leukemia (CML) harboring a T315I mutation resistant to the BCR/ABL
inhibitor, imatinib (13–16). However, phase II trials of VX-680
for CML harboring a T315I mutation and for Philadelphia
chromosome-positive acute lymphatic leukemia (Ph+ALL) and a phase I
trial for progressive leukemia, were discontinued in 2007 due to
prolongation of the QT interval on electrocardiograms.
Subsequently, VX-680 was modified to yield MK-0457, for which a
phase I trial has been completed (17). MK-0457 also binds to the ATP-binding
site and is metabolized by hepatic CYP3A4, CYP2C8 and flavin
monooxygenase. In 2011, Traynor et al(18) performed a phase I trial of MK-0457
in solid cancers and demonstrated that MK-0457 is a strong
inhibitor of AURKB and also interacts with the Flt-3 and Abl
kinases. MK-0457 produced an outcome of stable disease in
approximately half of the patients, indicating the efficacy of this
agent. However, grade 4 neutropenia, grade 3 herpes infection and
adverse reactions including nausea, vomiting, diarrhea and fatigue
occurred during the administration period. PHA-739358 (danusertib)
is also a pan-Aurora kinase inhibitor, i.e., an inhibitor of AURKA,
AURKB and AURKC, similar to VX-680 and MK-0457, as well as an
inhibitor of ABL and Ret tyrosine kinases. PHA-739358 has shown
efficacy in clinical trials in progressive solid cancers and
BCR/ABL-positive leukemia (CML, Ph+ALL) (19).
AT9283 is a synthetic heterocyclic molecule that
inhibits cancer cell proliferation in solid tumors and leukemia.
AT9283 inhibits AURKA and AURKB and also targets serine/threonine
kinases such as JAK2, JAK3 and mutant V-abl. In 2012, Arkenau et
al(20) performed a phase I
study of AT9283 in patients with progressive solid cancer, in which
Grade 3 pyrogenic neutropenia occurred, but major toxicities of
reversible, dose-dependent bone-marrow suppression,
gastrointestinal disorder, fatigue and dehairing were
tolerable.
AZD1152 (barasertib) is a pro-drug that is converted
to the therapeutically active hydroxyquinazoline pyrazole anilide
of barasertib (barasertib-hQPA) in the cytoplasm. Barasertib-hQPA
inhibits both AURKA and AURKB, although the dominant effect exerted
is on AURKB. Inhibition of AURKB leads to aberrant cell division
and generation of atypical cells with DNA tetraploidy or larger,
which then undergo apoptosis due to checkpoint regulation. In 2012,
Schwartz et al(21) reported
a phase I trial of barasertib in patients with malignant solid
cancer, in which Grade 3 or higher pyrogenic or apyrogenic
neutropenia and mild or moderate hematoxicity or gastrointestinal
toxicity emerged, but all were tolerable. In a mouse model of
hepatoma, Aihara et al(22)
reported that AZD1152 significantly decreased the number of tumor
cells and suppressed tumor proliferation.
MLN8054 and MLN8237 (alisertib) were developed as
selective AURKA inhibitors. In a phase I clinical trial of MLN8054
in progressive solid cancer, somnolence was the dose-limiting
toxicity (23). This may be because
the drug has a similar structure to that of benzodiazepine.
Clinical trials of MLN8237 in progressive solid cancer, acute
myelogenous leukemia and hematopoietic organ tumors such as
non-Hodgkin’s lymphoma are ongoing and a phase III trial in
refractory peripheral T-cell lymphoma has recently been initiated.
MLN8237 also increases cisplatin-induced apoptosis in esophageal
adenocarcinoma cells (24) and
enhances radio-sensitivity in teratoid tumor cells (25).
Gynecologic applications of Aurora kinase
inhibitors
Aurora kinases are expressed in various gynecologic
malignant tumors. Li et al(26) and Kurai et al(27) examined correlations among the
expression levels of AURKA and AURKB and prognosis in normal
endometrium, benign tumors of the corpus uteri and endometrial
cancer. The results showed significantly increased AURKA and AURKB
levels in endometrial cancer compared with those in normal
endometrium during the proliferation phase. AURKB was especially
highly expressed in poorly differentiated endometrial cancer and
AURKA and AURKB levels were correlated with worsened prognosis.
Moreno-Bueno et al(28)
found a high AURKA expression in type 2 non-endometrioid cancer
among endometrial cancers and suggested that spindle formation
checkpoint disorder in mitosis is probably involved in onset of
this type of cancer. Yang et al(29) found that AURKA may be an adverse
prognostic factor in ovarian endometrioid cancer, in addition to
BRCA2.
Chemotherapy with taxanes or platinum-containing
anticancer drugs and radiotherapy currently play key roles in the
treatment of gynecologic malignant tumors. Combination of these
regimens with Aurora kinase inhibitors is likely to produce
synergistic effects that may increase cellular sensitivity to
anticancer drugs and radiation and suppress the number of Aurora
kinase inhibitor-resistant cells. For example, taxanes, which exert
an anticancer effect by inhibition of depolymerization of
microtubules, have increased effects on cancer cells when given in
combination with Aurora kinase inhibitors. Microtubules consist of
10,000–100,000 tubulin subunits. In cells during the interphase,
hundreds of long microtubules are present in the cytoplasm and
connect different areas in the cell. In cell skeletons including
microtubules, the subunits repeatedly associate and dissociate.
Taxanes such as paclitaxel bind to microtubules and inhibit the
dissociation of tubulin subunits. Inhibition of microtubule
depolymerization by paclitaxel in tumor cells prevents the
reconstruction of micro-tubules and formation of the spindle,
generating aberrant cell division. The spindle formation checkpoint
recognizes this abnormality and triggers apoptosis of the tumor
cell, causing the tumor to decrease in size (1–4).
Overexpression of Aurora kinases induces dysfunction of checkpoints
in cell division and permits the cell to enter anaphase in an
improper state. Thus, in the presence of overexpressed Aurora
kinases, taxane-based anticancer agents are not able to induce
apoptosis of aberrant cells, resulting in reduced sensitivity
(6). Conversely, drugs that inhibit
Aurora kinases may suppress resistance to apoptosis induced by
taxanes and enhance antitumor action.
Since microtubule inhibitors such as taxanes may
also affect the microtubule network of normal nerve cells and cause
peripheral neuropathy, administration at a low dose is required to
suppress adverse reactions. Thus, inhibition of the activity of
AURKA may increase the efficacy of taxanes to the extent that the
minimum effective dose can be reduced and adverse reactions
decreased. Lin et al(30)
examined the ovarian cancer growth-inhibitory effect of MK-0457,
which inhibits AURKA, AURKB and AURKC, in mouse models. The results
showed that MK-0457 administered alone or in combination with
docetaxel significantly suppressed tumor growth by induction of
apoptosis, compared with controls and that this effect was much
greater for the combination regimen. In their study, Beussel et
al(31) demonstrated that the
response of the International Federation of Gynecology and
Obstetrics surgical stage III serous ovarian cancer to first-line
chemotherapy using taxanes following optimal debulking surgery
could be predicted based on the expression level of AURKB.
The combination of platinum-containing anticancer
agents with Aurora kinase inhibitors is also likely to increase
anti-cancer effects. Cisplatin binds to the N7 position of guanine
and adenine bases in DNA and forms crosslinks in the DNA with loss
of two chlorine atoms. Thus, replication of DNA is suppressed and
cell division is inhibited, providing an anti-tumor effect. As
described above, aberrant cell division caused by Aurora kinase
inhibitors leads to p53 activation and triggering of cell
apoptosis. However, p53 is inhibited in a number of gynecologic
tumors, including the viral protein E6 in cases with human
papillomavirus (HPV) infection. However, in the cervical cancer
cell line SiHa, Aurora kinase inhibitors have been shown to block
the expression of HPV 16E6 and BCL-2, suppressing p53 and
increasing the expression of p53, thereby increasing sensitivity to
cisplatin. Therefore, enhanced effects and reduced adverse
reactions of platinum-containing anticancer agents used for
gynecologic tumors employ combination regimens with Aurora kinase
inhibitors (32).
In cultured ovarian cancer cells, inhibition of the
Aurora kinase may increase sensitivity to anticancer drugs via
NF-κB. In human lung cancer cells and the human ovarian cancer cell
line SKOV3, which express NF-κB at high levels and are resistant to
cytotoxic anticancer agents such as adriamycin and etoposide, Sun
et al(33) showed that
Aurora kinase inhibitors caused the downregulation of NF-κB and an
increased anticancer effect. Aurora kinases inactivate IκBα by
phosphorylation, which in turn activates NF-κB and thus Aurora
kinase inhibitors suppress the inactivation of IκB and increase the
drug sensitivity of cancer cells. This mechanism was verified in
vitro(33) and suggests that
Aurora kinase inhibitors improve chemotherapy regimens by
overcoming a potential drug resistance mechanism of cancer
cells.
Combination of Aurora kinase inhibitors with
radiotherapy may also enhance treatment. For example, Tao et
al(34) found that
administration of an AURKB inhibitor, AZD1152, prior to irradiation
increased the radiosensitivity of cells in which the activity of
p53 was reduced by an inhibitor (pifithrin-α) and of p53(−/−) mice,
compared with radiation alone.
Conclusion
At present, a number of new Aurora kinase inhibitors
are being developed to target malignant tumors. However, proper
utilization of these inhibitors requires determination of the
expression levels of Aurora kinase in each type of malignant tumor.
In addition, since cancer cells often acquire resistance to
low-molecular-weight agents, as seen in the emergence of
imatinib-resistant CML, it is important to examine mechanisms of
drug resistance to Aurora kinase inhibitors and to develop methods
to overcome this resistance. Emerging short- and long-term
toxicities also require evaluation as Aurora kinases play important
roles in normal cells. In particular, since Aurora kinase
inhibitors target the cell division system, study of the risk of
secondary carcinogenesis is imperative prior to the long-term use
of these inhibitors. Development of numerous Aurora kinase
inhibitors is likely to increase the number of selectable drugs
during treatment and contribute to the growth of tailor-made
chemotherapy in which drugs are selected based on the individual
patient and characteristics of the cancer during progression. Thus,
the introduction of new molecular-targeted agents is likely to
diversify the treatment options for intractable gynecologic
cancers.
Acknowledgements
The authors gratefully acknowledge
grant support from the Japan Society for the Promotion of Science
(JSPS) through a Grant-in-Aid for Scientific Research (KAKENHI), a
Grant-in-Aid for Scientific Research (B) (22390313), a Grant-in-Aid
for Scientific Research (C) (22591866) and a Grant-in-Aid for Young
Scientists (B) (24791718); the Ichiro Kanehara Foundation;
Kobayashi Foundation for Cancer Research; Keio University
Grant-in-Aid for Encouragement of Young Medical Scientists and the
Keio University Medical Science Fund through a Research Grant for
Life Sciences and Medicine.
References
|
1
|
Katayama H, Brinkly WR and Sen S: The
Aurora kinases: role in cell transformation and tumorigenesis.
Cancer Metastasis Rev. 22:451–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Agnese V, Bazan V, Fiorentino FP, et al:
The role of Aurora-A inhibitors in cancer therapy. Ann Oncol.
6:vi47–vi52. 2007.PubMed/NCBI
|
|
3
|
Carmena M and Earnshaw WC: The cellular
geography of aurora kinases. Nat Rev Mol Cell Biol. 4:842–854.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glover DM, Leibowitz MH, McLean DA and
Parry H: Mutations in aurora prevent centrosome separation leading
to the formation of monopolar spindles. Cell. 81:95–105. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chung CM, Man C, Jin Y, et al:
Amplification and overexpression of aurora kinase A (AURKA) in
immortalized human ovarian epithelial (HOSE) cells. Mol Carcinog.
43:165–174. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Anand S, Penrhyn-Lowe S and Venkitaraman
AR: AURORA-A amplification overrides the mitotic spindle assembly
checkpoint, inducing resistance to Taxol. Cancer cell. 3:51–62.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang D, Shimizu T, Araki N, et al: Aurora
A overexpression induces cellular senescence in mammary gland
hyperplastic tumors developed in p53-deficient mice. Oncogene.
27:4305–4314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsueh KW, Fu SL, Huang CY and Lin CH:
Aurora-A phosphorylates hnRNPK and disrupts its interaction with
p53. FEBS Lett. 585:2671–2675. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santaguida S, Vernieri C, Villa F,
Ciliberto A and Musacchio A: Evidence that Aurora B is implicated
in spindle checkpoint signalling independently of error correction.
EMBO J. 30:1508–1519. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kimmins S, Crosio C, Kotaja N, et al:
Differential functions of the Aurora B and Aurora C kinases in
mammalian spermatogenesis. Mol Endocrinol. 21:726–739. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li M, Jung A, Ganswindt U, Marini P, et
al: Aurora kinase inhibitor ZM447439 induces apoptosis via
mitochondrial pathways. Biochem Pharmacol. 79:122–129. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hauf S, Cole RW, LaTerra S, et al: The
small molecule Hesperadin reveals a role for Aurora B in correcting
kinetochore-microtubule attachment and in maintaining the spindle
assembly checkpoint. J Cell Biol. 161:281–294. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harrington EA, Bebbington D, Moore J, et
al: VX-680, a potent and selective small-molecule inhibitor of the
Aurora kinases, suppresses tumor growth in vivo. Nat Med.
10:262–267. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gora-Tybor J and Robak T: Targeted drugs
in chronic myeloid leukemia. Current Med Chem. 15:3036–3051. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Young MA, Shah NP, Chao LH, et al:
Structure of the kinase domain of an imatinib-resistant Abl mutant
in complex with the Aurora kinase inhibitor VX-680. Cancer Res.
66:1007–1014. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mancini M, Aluigi M, Leo E, et al: Histone
H3 covalent modifications driving response of
BCR-ABL1+cells sensitive and resistant to imatinib to
Aurora kinase inhibitor MK-0457. Br J Haematol. 156:265–268.
2012.PubMed/NCBI
|
|
17
|
Tyler RK, Shpiro N, Marquez R and Eyers
PA: VX-680 inhibits Aurora A and Aurora B kinase activity in human
cells. Cell Cycle. 6:2846–2854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Traynor AM, Hewitt M, Liu G, et al: Phase
I dose escalation study of MK-0457, a novel Aurora kinase
inhibitor, in adult patients with advanced solid tumors. Cancer
Chemother Pharmacol. 67:305–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gontarewicz A, Balabanov S, Keller G, et
al: Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by
the small molecule inhibitor PHA-739358 is effective against
imatinib-resistant BCR-ABL mutations including T315I. Blood.
111:4355–4364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arkenau HT, Plummer R, Molife LR, et al: A
phase I dose escalation study of AT9283, a small molecule inhibitor
of aurora kinases, in patients with advanced solid malignancies.
Ann Oncol. 23:1307–1313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schwartz GK, Carvajal RD, Midgley R, et
al: Phase I study of barasertib (AZD1152), a selective inhibitor of
Aurora B kinase, in patients with advanced solid tumors. Invest New
Drugs. Jun 2–2012.(E-pub ahead of print).
|
|
22
|
Aihara A, Tanaka S, Yasen M, et al: The
selective Aurora B kinase inhibitor AZD1152 as a novel treatment
for hepatocellular carcinoma. J Hepatol. 52:63–71. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Macarulla T, Cervantes A, Elez E, et al:
Phase I study of the selective Aurora A kinase inhibitor MLN8054 in
patients with advanced solid tumors: safety, pharmacokinetics, and
pharmacodynamics. Mol Cancer Ther. 9:2844–2852. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sehdev V, Peng D, Soutto M, et al: The
aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell
death in esophageal adenocarcinoma cells. Mol Cancer Ther.
11:763–774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Venkataraman S, Alimova I, Tello T, et al:
Targeting Aurora Kinase A enhances radiation sensitivity of
atypical teratoid rhabdoid tumor cells. J Neurooncol. 107:517–526.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li P, Zhou Q, Ren L and Xiao L: Clinical
implication of the expression of Aurora B in normal endometrium and
endometrial carcinoma. J Huazhong Univ Sci Technolog Med Sci.
337–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kurai M, Shiozawa T, Shih HC, et al:
Expression of Aurora kinases A and B in normal, hyperplastic, and
malignant human endometrium: Aurora B as a predictor for poor
prognosis in endometrial carcinoma. Hum Pathol. 36:1281–1288. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moreno-Bueno G, Sánchez-Estévez C, Cassia
R, et al: Differential gene expression profile in endometrioid and
nonendometrioid endometrial carcinoma: STK15 is frequently
overexpressed and amplified in nonendometrioid carcinomas. Cancer
Res. 63:5697–5702. 2003.
|
|
29
|
Yang F, Guo X, Yang G, Rosen DG and Liu J:
AURKA and BRCA2 expression highly correlate with prognosis of
endometrioid ovarian carcinoma. Mod Pathol. 24:836–845. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin YG, Immaneni A, Merritt WM, et al:
Targeting aurora kinase with MK-0457 inhibits ovarian cancer
growth. Clin Cancer Res. 14:5437–5446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Beussel S, Hasenburg A, Bogatyreva L,
Hauschke D, Werner M and Lassmann S: Aurora-B protein expression is
linked to initial response to taxane-based first-line chemotherapy
in stage III ovarian carcinoma. J Clin Pathol. 65:29–35. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang L and Zhang S: ZM447439, the Aurora
kinase B inhibitor, suppresses the growth of cervical cancer SiHa
cells and enhances the chemosensitivity to cisplatin. J Obstet
Gynaecol Res. 37:591–600. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun C, Chan F, Briassouli P and
Linardopoulos S: Aurora kinase inhibition downregulates NF-kappaB
and sensitises tumour cells to chemotherapeutic agents. Biochem
Biophys Res Commun. 352:220–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tao Y, Zhang P, Girdler F, et al:
Enhancement of radiation response in p53-deficient cancer cells by
the Aurora-B kinase inhibitor AZD1152. Oncogene. 27:3244–3255.
2008. View Article : Google Scholar : PubMed/NCBI
|