Cholangiocarcinoma (CCA), is a malignant tumor that
arises from the biliary epithelial tissue and is highly aggressive,
with no effective pharmacological treatment available. This cancer
has a poor prognosis and a high mortality rate (1). The highest worldwide incidence of CCA is
found in the North and Northeast of Thailand, at ~85 cases per
100,000 individuals per year (2). The
major predisposing factors for CCA in Asia are infection by the
liver fluke, Opisthorchis viverrini (3,4) and
exposure to groups of food-borne carcinogens, especially
N-nitrosodimethylamine compounds identified in grilled or fermented
foods (5). The only effective
treatment for the disease is surgery. For patients who are not
eligible for surgical therapy, gemcitabine- or 5-fluoro-uracil
(FU)-based treatments are given. These are largely ineffective,
since the response rate is only 20-30%.
The molecular pathology of bile duct cancer has been
a topic of intense study. The molecular pathogenesis of CCA usually
involves abnormal signal transduction and pro-inflammatory
secretion, facilitated by gene mutations and epigenetic
dysregulations (on a set of oncogenes and tumor suppressor genes)
(6). Several lines of evidence also
indicate that the abnormal expression of growth factors and
receptors, the RAS/RAF/ dual specificity mitogen-activated protein
kinase kinase 1 pathway, and the phosphatidylinositol 3 kinase
(PI3K)/protein kinase B (AKT)/mammalian target of rapamycin pathway
may be involved with CCA initiation, maintenance, and metastasis
(7). Several studies reported that
specific-target drugs or inhibitors, including epithelial growth
factor receptor (EGFR; Lapatinib or Erlotinib), fibroblast (F)GFR
and PI3K inhibitor, (8) may be
applicable to CCA. A number of novel therapeutics are under
evaluation in a phase 2 study (9).
Alternative splicing (AS) is a post-transcription
modulation process that can generate a variety of gene isoforms.
Spliced mRNA is able to be translated to differential amino acids
with various biological functions (10). Pre-mRNA is spliced through the
spliceosome; a large macromolecule comprising 5 small nuclear
ribonucleoproteins (snRNPs U1, U2, U4/U6 and U5). The AS generates
5 common splicing patterns, including alternative 5' splice site,
alternative 3' splice site, exon skipping, intron retention and
mutually exclusive exons. Previous data demonstrates that aberrant
alternative splicing also includes exonic regulatory element
mutation, splice site mutation and altered splice isoform ratios.
The differential expression of splicing factors is implicated in
various diseases and linked to hallmarks of cancer (11-15).
A number of reports demonstrated a correlation between aberrant AS
and tumor initiation/progression (16-20).
The truncated oncogenic forms of the proteins, resulted from
aberrant AS involved in cancer cell growth, apoptosis, drug
resistance and angiogenesis.
A number of articles have summarized the
interconnection between AS and cancer progression, including 17
genes in lung cancer (16), 2 reports
in breast cancer in which 7 genes (17) and 9 genes (18), respectively were demonstrated, and 9
genes in hepatocellular carcinoma (19,20). The
global cancer-specific transcript variants of five cancers
demonstrated protein metabolism and modification are the most
prevalent functional processes in cancer (24). As mentioned previously, aberrant AS
has been discovered and proven to have functional involvement in
the initiation and progression of cancer. In CCA, 623 genes
presented with alternative splicing in CCA samples when compared
with healthy bile duct tissue samples (25). In this review, atypical splicing of
nine genes, which have been investigated at the in vitro,
in vivo and clinical levels, and their relevance to CCA
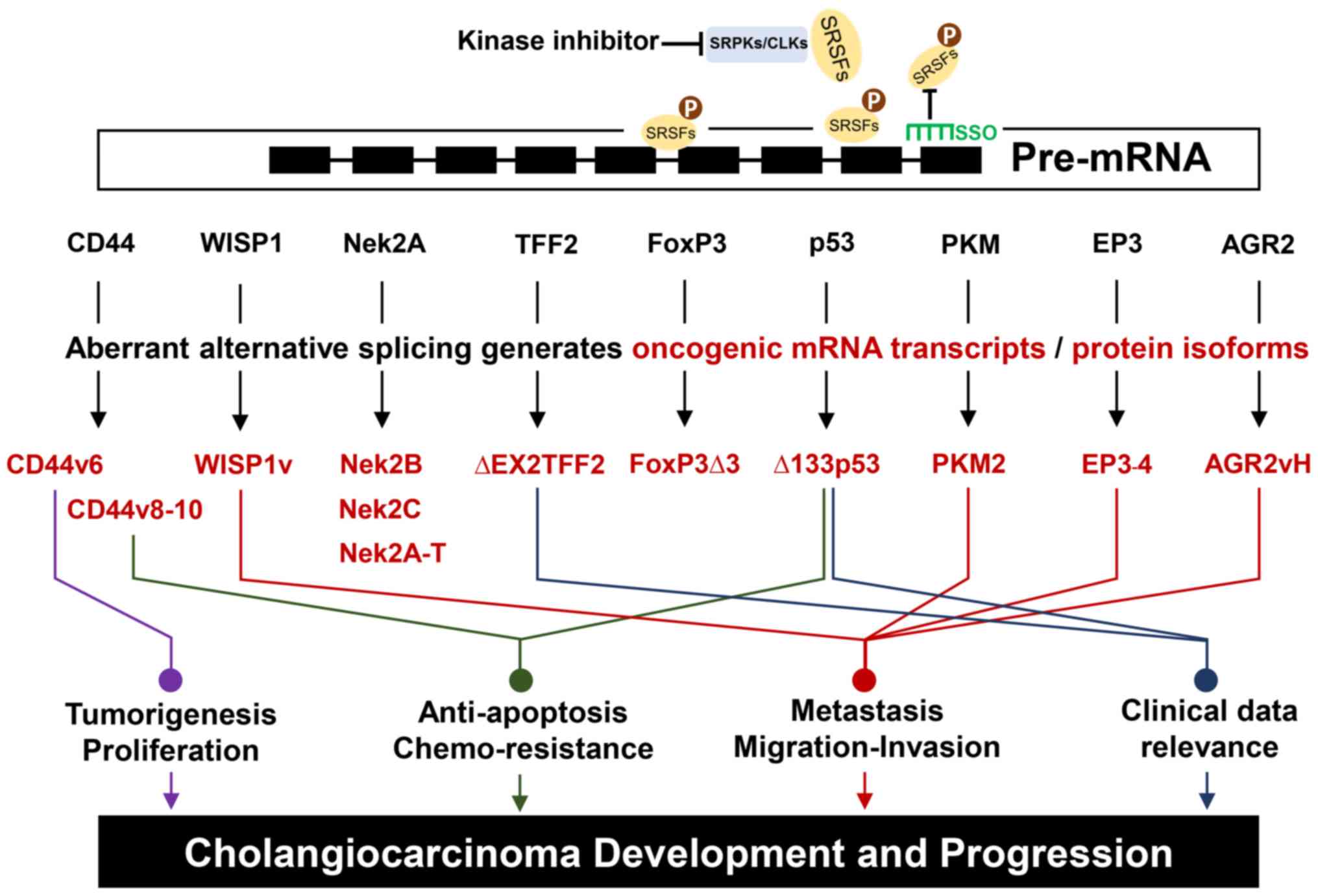
pathogenicity are summarized. The structure of nine pre-mRNAs that
undergo alternative mRNA splicing to generate wild-type mRNA or
variant transcripts are presented in Fig.
1. The derived-spliced transcripts or protein isoforms are
summarized by how they can facilitate various characteristics of a
cancer cell, as presented in Fig. 2
and Table I.
CD44 is a transmembrane glycoprotein receptor that
specifically binds to extracellular hyaluronan and other
extracellular matrix (ECM) proteins to activate signal
transduction, and serves important roles in tumor proliferation,
migration, and invasion (26,27). CD44 pre-mRNA encodes transmembrane and
cytoplasmic-tail regions. The AS of CD44 can generate up to 12
isoforms of proteins with different biological functions. The CD44v
isoforms participate in cancer progression: CD44v6 promotes EMT and
activates the transforming growth factor-β pathway (28,29), and
CD44v8-10 is involved in poor cancer prognosis (30,31).
Expression of CD44v6 can be linked to CCA proliferation. CD44v6 is
a CCA-specific isoform that has never been detected in normal bile
ducts (32). Furthermore, the
CD44v8-10 transcript was overexpressed in CCA and was demonstrated
to stabilize the xCT system, a cysteine/glutamate transporter, to
elevate glutathione synthesis and inhibit reactive oxygen species
(ROS) accumulation in CCA cells. This function of CD44v8-10 was
demonstrated to facilitate cancer cell survival in cases caused by
liver fluke-induced inflammation. In addition, upregulation of
CD44v8-10 suppressed p38 mitogen-activated protein kinase 1 (MAPK),
which is a signaling protein involved in ROS suppression. Although
the mechanism by which the CD44 spliced isoform may suppress p38 is
still unclear, this observation appeared to be clinically relevant,
since patients with CD44v overexpression and
negative-phosphorylated (phospho)-p38MAPK have significantly
shorter survival times compared with low CD44v expression and
positive-phospho-p38(MAPK) (33).
Wnt-inducible secreted protein 1 [(WISP1) also known
as CNN4] is a member of the cysteine-rich CCN family of proteins,
which are highly expressed in skeletal tissues and has a role in
bone formation and maintenance. Functions of this protein involve
cell proliferation, osteoblastic differentiation and migration
(34,35). WISP1 comprises 4 domains, including
insulin-like growth factor-binding protein (IGFBP), VWC,
thrombospondin-1 (TSP-1) and CT domains and is known to have
variants with biological functions. A WISP1 variant lacking exon 3
(WISP1v) loses its VWC domain, which is thought to participate in
protein complex formation. Ectopic expression demonstrated that the
WISP1v is a secreted oncoprotein, which drives cellular
transformation and rapid cumulative growth. WISP1v overexpression
enhanced the invasive phenotype in gastric carcinoma cells, while
wild-type WISP1 exhibited no such potential. These findings
suggested that the CCN protein WISP1v was involved in the
aggressive progression of scirrhous gastric carcinoma (36). In CCA, the aberrant isoform WISP1v was
demonstrated to be overexpressed in patient CCA tissues (37). Furthermore, upregulation of WISP1v was
associated with shorter overall survival time among patients
following surgical treatment (38).
In addition, WISP1v was demonstrated to promote cell invasion in
vitro and this process was demonstrated to be mediated by p38
MAPK (37).
Nek2, or NIMA-related kinase 2, is a
serine/threonine kinase that regulates cell division through
centrosome separation (39). The
spliced isoform of Nek consists of three forms, Nek2A, Nek2B and
Nek2A-T (40). Isoforms of NEK are
demonstrated to be functionally involved with cancer formation. In
patients, overexpression of Nek2a was associated with Ki-67
expression, a cell proliferation marker (41). In addition, NEK2A cytoplasmic
expression was positively associated with cancer grade and tumor
size in breast invasive ductal carcinoma and metastatic potential
(42). Cancer cells overexpressing
the NEK2A isoform demonstrated a significant increase in colony
formation compared with control cells and small interfering
(si)RNA-based depletion of NEK2a resulted in the halting of cancer
cell proliferation (43).
Nek2A/Nek2A-T were demonstrated to be highly upregulated in CCA
cell lines, with the predominant isoform being Nek2A/Nek2A-T and
Nek2B being the lesser expressed isoform (44). Furthermore, the expression of Nek2B
was demonstrated to positively correlate with proliferation
potential in breast cancer cells (45).
Trefoil factor 2 (TFF2) is a secreted protein that
serves important roles in gastrointestinal restitution (46), chronic kidney disease and pulmonary
inflammation, through the induction of cell migration and
proliferation. Overexpression of TFF2 is commonly identified in
several types of cancer, implicating it in carcinogenesis. TFF2 was
reported to exert its pro-proliferative activity through the
EGFR-MAPK pathway in CCA (47).
Previously, ΔEX2TFF2, an exon 2- skipping isoform of TFF2 with a
stop codon (TAG) at exon 1, was uncovered as a spliced isoform of
TFF2(48). Although, the roles of
this transcript have not been clarified, the present study
demonstrated that a high expression ratio of ΔEX2TFF2/wtTFF2 in
patients was significantly associated with a longer survival time
(48). Therefore, the spliced isoform
may act as a dominant-negative form of TFF2 that counteracts the
cancer promoting wtTFF2 activity in CCA.
FOXP3 is a transcription factor in the forkhead
protein family that is involved in CD25+ regulatory T
cell (Treg) development. Not only does FOXP3 control Treg
development, it is also expressed in colorectal cancer cells, which
is associated with poor prognosis in patients (49). Exon 3 skipping of FOXP3, resulting in
an amino acid frameshift, has been reported in CCA (50). In addition, a FOXP3 splice isoform was
also observed in melanoma cells, suggesting it has a role in
suppressing immune activity (51).
Tumor protein 53 (TP53 or p53) is one of the most
important tumor suppressors, indicated by its high mutation rate
across all types of cancer. p53 responds to various stress signals
and orchestrates processes including cell cycle arrest, DNA repair,
cellular senescence and apoptosis in response to specific stress
signals (52). AS generates 12 p53
isoforms, including Tap53, Δ40p53, Δ133p53 and Δ160p53 among others
(53,54). The differential regulation of p53
isoforms promotes the aggressiveness of several types of cancer. A
study demonstrated that Δ133p53b enhanced breast cancer stemness
(55) and protected colorectal cells
from camptothecin-induced apoptosis (56).
p53 has been identified as a gene that frequently
mutates in a large number of CCA cases (57-59),
suggesting that a perturbed p53 pathway facilitates CCA
carcinogenesis. A study demonstrated that a high Δ133p53/p53 mRNA
expression ratio correlates with a poor overall survival (60). Notably, Δ133p53 is also associated
with resistance to certain cancer drugs; an association between
Δ133p53 and 5-FU-resistance in CCA cells was demonstrated, and
Δ133p53 was upregulated in 5-FU-resistant tumor tissues and CCA
cell lines in a dose-dependent manner (61). Given that 5-FU is a cytotoxic drug
that interferes with DNA synthesis, the Δ133p53 isoform may act as
a dominant-negative p53 that interferes with the activity of wtp53
in the ternary complex (62).
Accordingly, suppression of Δ133p53 promoted apoptosis, which
correlated with an upregulation of pro-apoptotic Bax and a
downregulation of anti-apoptotic Bcl-2(61).
PKM is a rate-limiting enzyme that catalyzes the
conversion of phosphoenolpyruvate to pyruvate during glycolysis.
PKM can be generated in 4 isoforms, which are expressed differently
in various tissues. One of the isoforms is PKM2, which lacks exon 9
and is a major isoform highly expressed in a number of types of
cancer (63). Previously data
demonstrate that overexpression of PKM2 is linked to tumor growth,
metastasis capability and a poor prognosis in hepatocellular
carcinoma, pancreatic ductal adenocarcinoma and gallbladder cancer
(64-66).
In hilar cholangiocarcinoma, immunohistochemical staining specific
to the PKM2 isoform demonstrated a great number of
positive-staining cells in the tumor tissue. Patients with
high-PKM2-expressing tumors exhibited a higher rate of tumor
recurrence and a shorter overall survival time, when compared with
patients with low PKM2 expression. However, there is still no
conclusive evidence that indicates PKM2 is a cancer driver for CCA.
In addition, PKM2 elevation was associated with CCA development and
neural invasion (67).
E prostanoid receptor 3 (EP3), or prostaglandin E2
receptor 3 (PTGER3), is a member of a G protein-coupled receptor
family, that specifically binds to prostaglandin E2 (PGE2) to
activate various responses. EP3 receptor can generate up to 11
spliced isoforms. Previous data demonstrate that EP3-5 and EP3-6
isoforms were associated with cell proliferation in the myometrium
in humans (68). Furthermore,
overexpression of the EP3-4 receptor promoted cell growth through
upregulating FUSE-binding protein 1 in liver cancer (69). In CCA, the truncated EP3-4 isoform,
which includes exon 1, 2a, 5 and 10, was detected (70). This EP3-4 isoform is activated through
the Src/EGFR/PI3K/AKT/glycogen synthase kinase-3β pathway and
promotes cell proliferation, migration, and invasion. This results
in enhanced expression of the downstream proteins c-Myc and snail.
Therefore, it is believed to serve a regulatory role in CCA cell
growth and metastasis (71).
The expression profiling of metastasis-associated
genes in CCA demonstrated that AGR2 is one of the most-upregulated
genes, specific to the metastatic CCA cell line, when compared with
the parental cell line (72). The
AGR2 gene encodes for a disulfide isomerase enzyme, which is
commonly expressed in mucus-secreting tissues. The mRNA splicing of
AGR2 was first characterized in prostate cancer (PCa). Spliced
isoforms include AGR2vC, AGR2vE, AGR2vF, AGR2vG and AGR2vH. Among
the 5 spliced isoforms and the wild-type, AGR2vG and AGR2vH were
demonstrated to be significantly upregulated in the exosome from
patient's urine sample analysis. These two exhibited high
diagnostic value, with higher sensitivity and specificity when
compared with the prostate-specific antigen used as a standard
clinical biomarker for PCa diagnosis (73). In CCA cell lines, AGR2 RNA isoforms,
namely AGR2vE, AGR2vF and AGR2vH, were recently reported that are
specific to metastatic CCA cells (74). It was demonstrated that the AGR2vH
isoform enables various metastatic-associated phenotypes in CCA
cells. Suppression of AGR2vH by the AGR2vH-specific siRNA
significantly reduced CCA cell migration and invasion.
Concordantly, AGR2vH overexpression promoted cell proliferation,
migration, invasion and adhesion potential. In addition, it was
demonstrated that the expression of AGR2vH influences
metastasis-associated phenotypes through the upregulation of
vimentin. Therefore, the results indicated that the
metastasis-specific isoform AGR2vH serves an important role in
cancer severity (74).
The prominent role of the aberrant AS in
carcinogenesis has been demonstrated, indicating that AS may be a
good target for cancer therapy. Aberrant AS can be manipulated in
several steps: For example, Pre-Trans-Splicing Molecule (PTM) is
the artificial sequence that can reprogram mRNA through replacement
of the 3'exon, 5'exon and internal exon (75,76). The
results demonstrated that the trans-splicing molecule reduced the
number of mutant p53 transcripts in the transfected cells, which
resulted in cell cycle arrest, apoptosis and tumor xenograft
suppression with colorectal cancer and hepatocellular carcinoma
cells (77,78). However, the use of PTM for targeting
oncogenic AS events is not yet well studied and the PTM
modification has limitations for cancer treatment. Therefore, this
review discussed the methodologies that may apply to cancer
therapy, including small molecule splicing modulators and SSOs,
each of which are currently under study in clinical trials.
Splicing factors are key molecules that influence AS
regulation and are associated with cancer aggressiveness and
pathological phenotypes (79). A
previous report demonstrated that an overexpression of
serine/arginine-rich splicing factor 1 (SRSF1) can facilitate
abnormal splicing of tumor suppressors and proto-oncogenes
(80). The results demonstrated that
SRSF1 promotes 12A inclusion of an isoform of BIN1, which
interferes with the tumor-suppressing activity of this protein. In
the same study, the researchers demonstrated an increase in S6K1
isoform 2 expression resulting from SRSF1 overexpression that was
associated with colony formation activity (80). An Ov-infected hamster model was used
to identify the differentially expressed genes to study the
molecular mechanism of CCA carcinogenesis. The results demonstrated
that SRSF9 is one of the genes that are overexpressed in
Ov-infected hamsters and may be associated with CCA initiation
(81).
The data revealed that mutations in splicing factor
3B subunit 1A (SF3B1), which encodes the core component of U2
snRNP, is linked to erroneous 3' splice site selection (82-84).
The results demonstrated that the SF3B1 K700E mutation led to
differential splicing in uveal melanoma and breast cancer (85,86). In
addition, luminal B and progesterone receptor-negative breast
cancer patients with additional SF3B1 mutations have significantly
shorter survival times (87).
It is possible to modulate aberrant AS based on
small molecule inhibitors of splicing factors or mutated
spliceosomal proteins: For example, it has been demonstrated that a
natural product ‘Borrelidin’ can bind to a splicing protein, FBP21,
leading to a decrease of the vascular endothelial growth factor
(VEGF) pro-angiogenic isoform and an increase of the VEGF
anti-angiogenic isoform, in RPE cells (88). Previous studies demonstrated that a
natural product, FR901464 and its methylated derivative,
spliceostatin A, as well as E7107, specifically inhibit spliceosome
assembly through SF3B1 and lead to halted splicing reactions
(89-91).
The results demonstrated that treatment of these small molecules
inhibits cell cycle progression and inhibits the tumor angiogenesis
through decreasing the levels of VEGF transcripts (92,93).
Not only does the altered expression of splicing
regulators affect AS, but the alteration of the phosphorylation
status of the splicing factor/modulator was also implicated in
cancer progression. In head and neck squamous cell carcinoma,
hyperphosphorylation of SRPK2, a serine/arginine-rich
protein-specific kinase that phosphorylates SRSF1/2, was detected
in cancer cells; the phosphorylation promotes cell proliferation,
migration and invasion (94).
Alteration to the kinase alters the AS pattern. A previous study
demonstrated that CLKs and SRSF protein kinases (SRPKs) are targets
for kinase inhibitors to modulate AS; treatment with Cpd-1, Cpd-2,
and Cpd-3 significantly reduced the levels of phosphorylated SR
proteins, therefore affecting the splicing pattern of multiple
genes and inducing cell apoptosis (95). Furthermore, the other kinase
inhibitors, including ceramide, affect splice site selection of
Bcl-x and increases pro-apoptotic isoforms through the
dephosphorylation of the SR protein (96).
SSOs are single-stranded nucleic acids, usually
15-25 bases, that are complementary to the mRNA target transcripts
or the recognition sequence of the splice sites, that leads to
modulated splicing. A number of studies demonstrated that SSO can
inhibit aberrant RNA translation: I.e., MDM4 is the protein that
contributes to embryonic development and is undetectable in adult
tissues. An MDM4 isoform with exon 6 is frequently upregulated in
cancer cells, impairing p53 tumor-suppressor function. The
SSO-mediated skipping of exon 6 results in decreased MDM4 levels
and reduced melanoma growth (97).
Similarly, SSO targeting exon 26 of HER4 mRNA, named as SSOe26,
demonstrated its capacity on HER4 isoform switching from CYT1 to
CYP2. This treatment resulted in the depletion of the proliferation
of breast cancer cells and tumor growth in mice xenografts
(98). Furthermore, SSO targeted
B-cell lymphoma (Bcl)-x pre-mRNA, which increased the Bcl-xS
isoform, gaining pro-apoptotic activity, which was verified in the
models of murine melanoma and in human glioma cell lines (99,100).
Drug development based on targeting aberrant AS,
namely small molecule splicing modulators, is an interesting
approach for cancer treatment. Splicing regulators are upstream
molecules that control the splicing events of multiple genes.
Insight into novel target genes of the splicing regulators, can be
used to manipulate the effective inhibitor(s) of these upstream
molecules to suppress various downstream oncogenic spliced
isoforms. However, the off-target effect, toxicity (101,102)
and the effects of small splicing factors interfering with the
normal splicing patterns of global genes, should be considered. On
the other hand, the specificity of SSO technology overcomes more
than small splicing modulators by modulating AS through inhibiting
only its oncogenic target which leads to effective treatment. The
major problems of oligonucleotides include toxicity, instability
against nucleases and delivery limitations.
The present review summarized the experimental
evidence for and clinical relevance of the verification of
significant effects of aberrant mRNA splicing of well-characterized
genes with respect to CCA initiation and aggressiveness. The nine
genes discussed underwent AS and revealed an intercorrelation with
cholangiocarcinogenesis and progression. This information will
serve as an opportunity to develop novel strategies for CCA
detection and intervention. Interestingly, certain of the
cancer-specific variants may serve as potential targets for CCA
prognosis including ∆2TFF2 and ∆133p53, which demonstrate their
clinical impact on patient survival. These oncogenic isoforms may
be used as targets for cancer treatment, using specific antibodies,
or the construction of SSOs which can modulate aberrant splicing.
The regulatory machinery, including splicing factors and
regulators, represents alternative targets of precision strategies,
regarding the depletion of oncogenic isoforms. Finally, this
summarization provides new ideas for the improvement of CCA
diagnosis and treatment. Further studies should aim to investigate
the unclear linkages between AS and CCA to unlock the molecular
mechanisms governing AS regulation in CCA development and
progression.
Not applicable.
JY was supported by The Nuresuan University research
scholarship for graduate students. WK is supported by the grant
from the Thailand Research Fund and Office of the Higher Education
Commission providing to (grant no. TRF-MRG6080014). SJ is supported
by The Foundation for Cancer Care, Siriraj Hospital, the Advanced
Research on Pharmacology Fund; Siriraj Foundation (grant no.
D003421) and the Chalermphrakiat Grant, Faculty of Medicine Siriraj
Hospital, Mahidol University.
Not applicable.
JY and WK designed, performed and wrote the
literature review. SJ and SW revised the manuscript for
intellectual content.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Macias RI: Cholangiocarcinoma: And
pharmacological Biology, Clinical management perspectives. ISRN
Hepatol. 2014.https://doi.org/10.1155/2014/828074.
PubMed/NCBI View Article : Google Scholar
|
|
2
|
Banales JM, Cardinale V, Carpino G,
Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes
SJ and Fouassier L: et al Expert consensus document:
Cholangiocarcinoma: current knowledge and future perspectives
consensus statement from the European Network for the Study of
Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 13.
261–280. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sripa B and Pairojkul C:
Cholangiocarcinoma: Lessons from Thailand. Curr Opin Gastroenterol
24. 349–356. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Thuwajit C, Thuwajit P, Kaewkes S, Sripa
B, Uchida K, Miwa M and Wongkham S: Increased cell proliferation of
mouse fibroblast NIH-3T3 in vitro induced by excretory/secretory
product(s) from Opisthorchis viverrini. Parasitology 129. 455–464.
2004.PubMed/NCBI
|
|
5
|
Srivatanakul P, Ohshima H, Khlat M, Parkin
M, Sukaryodhin S, Brouet I and Bartsch H: Opisthorchis viverrini
infestation and endogenous nitrosamines as risk factors for
cholangiocarcinoma in Thailand. Int J Cancer 48. 821–825.
1991.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Patel T: New insights into the molecular
pathogenesis of intrahepatic cholangiocarcinoma. J Gastroenterol
49. 165–172. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Marks EI and Yee NS: Molecular genetics
and targeted therapeutics in biliary tract carcinoma. World J
Gastroenterol 22. 1335–1347. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rizvi S, Borad MJ, Patel T and Gores GJ:
Cholangiocarcinoma: Molecular pathways and therapeutic
opportunities. Semin Liver Dis 34. 456–464. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Goldstein D, Lemech C and Valle J: New
molecular and immunotherapeutic approaches in biliary cancer. ESMO
Open 2 (Suppl 1). (e000152)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Roy B, Haupt LM and Griffiths LR: Review:
Alternative splicing (AS) of genes as an approach for generating
protein complexity. Curr Genomics 14. 182–194. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Douglas AG and Wood MJ: RNA splicing:
Disease and therapy. Brief Funct Genomics 10. 151–164.
2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ghigna C, Valacca C and Biamonti G:
Alternative splicing and tumor progression. Curr Genomics 9.
556–570. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tazi J, Bakkour N and Stamm S: Alternative
splicing and disease. Biochim Biophys Acta 1792. 14–26.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Venables JP: Aberrant alternative splicing
in cancer. Cancer Res 64. 7647–7654. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ladomery M: Aberrant alternative splicing
is another hallmark of cancer. Int J Cell Biol 2013.
463786:2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pio R and Montuenga LM: Alternative
splicing in lung cancer. J Thorac Oncol 4. 674–678. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Martínez-Montiel N, Anaya-Ruiz M,
Pérez-Santos M and Martínez-Contreras RD: Alternative splicing in
breast cancer and the potential development of therapeutic tools.
Genes (Basel) 8. pii(E217)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Xiping Z, Qingshan W, Shuai Z, Hongjian Y
and Xiaowen D: A summary of relationships between alternative
splicing and breast cancer. Oncotarget 8. 51986–51993.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu L, Xie S, Zhang C and Zhu F: Aberrant
regulation of alternative pre-mRNA splicing in hepatocellular
carcinoma. Crit Rev Eukaryot Gene Expr 24. 133–149. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang L, Liu X, Zhang X and Chen R:
Identification of important long non-coding RNAs and highly
recurrent aberrant alternative splicing events in hepatocellular
carcinoma through integrative analysis of multiple RNA-Seq
datasets. Mol Genet Genomics 291. 1035–1051. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ghigna C, Giordano S, Shen H, Benvenuto F,
Castiglioni F, Comoglio PM, Green MR, Riva S and Biamonti G: Cell
motility is controlled by SF2/ASF through alternative splicing of
the Ron protooncogene. Mol Cell 20. 881–890. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Stallings-Mann ML, Waldmann J, Zhang Y,
Miller E, Gauthier ML, Visscher DW, Downey GP, Radisky ES, Fields
AP and Radisky DC: Matrix metalloproteinase induction of Rac1b, a
key effector of lung cancer progression. Sci Transl Med 4.
142ra95:2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Poulikakos PI, Persaud Y, Janakiraman M,
Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B and Gabay MT: et
al RAF inhibitor resistance is mediated by dimerization of
aberrantly spliced BRAF(V600E). Nature 480. 387–390.
2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
He C, Zhou F, Zuo Z, Cheng H and Zhou R: A
global view of cancer-specific transcript variants by subtractive
transcriptome-wide analysis. PLoS One 4. e4732:2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen Y, Liu D, Liu P, Chen Y, Yu H and
Zhang Q: Identification of biomarkers of intrahepatic
cholangiocarcinoma via integrated analysis of mRNA and miRNA
microarray data. Mol Med Rep 15. 1051–1056. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yu Q and Stamenkovic I: Cell
surface-localized matrix metalloproteinase-9 proteolytically
activates TGF-beta and promotes tumor invasion and angiogenesis.
Genes Dev 14. 163–176. 2000.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Nam K, Oh S, Lee KM, Yoo SA and Shin I:
CD44 regulates cell proliferation, migration, and invasion via
modulation of c-Src transcription in human breast cancer cells.
Cell Signal 27. 1882–1894. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Saito S, Okabe H, Watanabe M, Ishimoto T,
Iwatsuki M, Baba Y, Tanaka Y, Kurashige J, Miyamoto Y and Baba H:
CD44v6 expression is related to mesenchymal phenotype and poor
prognosis in patients with colorectal cancer. Oncol Rep 29.
1570–1578. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang J, Xiao L, Luo CH, Zhou H, Zeng L,
Zhong J, Tang Y, Zhao XH, Zhao M and Zhang Y: CD44v6 promotes
β-catenin and TGF-β expression, inducing aggression in
ovarian cancer cells. Mol Med Rep 11. 3505–3510. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yamaguchi A, Zhang M, Goi T, Fujita T,
Niimoto S, Katayama K and Hirose K: Expression of variant CD44
containing variant exon v8-10 in gallbladder cancer. Oncol Rep 7.
541–544. 2000.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sosulski A, Horn H, Zhang L, Coletti C,
Vathipadiekal V, Castro CM, Birrer MJ, Nagano O, Saya H and Lage K:
et al CD44 splice variant v8-10 as a marker of serous ovarian
cancer prognosis. PLoS One 11. e0156595:2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yun KJ, Yoon KH and Han WC:
Immunohistochemical study for CD44v6 in hepatocellular carcinoma
and cholangiocarcinoma. Cancer Res Treat 34. 170–174.
2002.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Thanee M, Loilome W, Techasen A, Sugihara
E, Okazaki S, Abe S, Ueda S, Masuko T, Namwat N and Khuntikeo N: et
al CD44 variant-dependent redox status regulation in liver
fluke-associated cholangiocarcinoma: A target for
cholangiocarcinoma treatment. Cancer Sci 107. 991–1000.
2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu H, Dong W, Lin Z, Lu J, Wan H, Zhou Z
and Liu Z: CCN4 regulates vascular smooth muscle cell migration and
proliferation. Mol Cells 36. 112–118. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ono M, Inkson CA, Kilts TM and Young MF:
WISP-1/CCN4 regulates osteogenesis by enhancing BMP-2 activity. J
Bone Miner Res 26. 193–208. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tanaka S and Sugimachi K, Saeki H,
Kinoshita J, Ohga T, Shimada M, Maehara Y and Sugimachi K: A novel
variant of WISP1 lacking a Von Willebrand type C module
overexpressed in scirrhous gastric carcinoma. Oncogene 20.
5525–5532. 2001.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Tanaka S, Sugimachi K, Kameyama T, Maehara
S, Shirabe K, Shimada M, Wands JR and Maehara Y: Human WISP1v, a
member of the CCN family, is associated with invasive
cholangiocarcinoma. Hepatology 37. 1122–1129. 2003.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wu Q, Jorgensen M, Song J, Zhou J, Liu C
and Pi L: Members of the Cyr61/CTGF/NOV protein family: Emerging
players in hepatic progenitor cell activation and intrahepatic
cholangiocarcinoma. Gastroenterol Res Pract 2016.
2313850:2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Helps NR, Luo X, Barker HM and Cohen PT:
NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase
localized to centrosomes, is complexed to protein phosphatase 1.
Biochem J 349. 509–518. 2000.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fardilha M, Wu W, Sá R, Fidalgo S, Sousa
C, Mota C, da Cruz e Silva OA and da Cruz e Silva EF: Alternatively
spliced protein variants as potential therapeutic targets for male
infertility and contraception. Ann N Y Acad Sci 1030. 468–478.
2004.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhong X, Guan X, Dong Q, Yang S, Liu W and
Zhang L: Examining Nek2 as a better proliferation marker in
non-small cell lung cancer prognosis. Tumour Biol 35. 7155–7162.
2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang S, Li W, Liu N, Zhang F, Liu H, Liu
F, Liu J, Zhang T and Niu Y: Nek2A contributes to tumorigenic
growth and possibly functions as potential therapeutic target for
human breast cancer. J Cell Biochem 113. 1904–1914. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lai XB, Nie YQ, Huang HL, Li YF, Cao CY,
Yang H, Shen B and Feng ZQ: NIMA-related kinase 2 regulates
hepatocellular carcinoma cell growth and proliferation. Oncol Lett
13. 1587–1594. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kokuryo T, Senga T, Yokoyama Y, Nagino M,
Nimura Y and Hamaguchi M: Nek2 as an effective target for
inhibition of tumorigenic growth and peritoneal dissemination of
cholangiocarcinoma. Cancer Res 67. 9637–9642. 2007.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wang Y, Shen H, Yin Q, Zhang T, Liu Z,
Zhang W and Niu Y: Effect of NIMA-related kinase 2B on the
sensitivity of breast cancer to paclitaxel in vitro and vivo.
Tumour Biol 39. 1010428317699754:2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Xue L, Aihara E, Podolsky DK, Wang TC and
Montrose MH: In vivo action of trefoil factor 2 (TFF2) to speed
gastric repair is independent of cyclooxygenase. Gut 59. 1184–1191.
2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kosriwong K, Menheniott TR, Giraud AS,
Jearanaikoon P, Sripa B and Limpaiboon T: Trefoil factors: Tumor
progression markers and mitogens via EGFR/MAPK activation in
cholangiocarcinoma. World J Gastroenterol 17. 1631–1641.
2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kamlua S, Patrakitkomjorn S, Jearanaikoon
P, Menheniott TR, Giraud AS and Limpaiboon T: A novel TFF2 splice
variant (∆EX2TFF2) correlates with longer overall survival time in
cholangiocarcinoma. Oncol Rep 27. 1207–1212. 2012.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Kim M, Grimmig T, Grimm M, Lazariotou M,
Meier E, Rosenwald A, Tsaur I, Blaheta R, Heemann U and Germer CT:
et al Expression of Foxp3 in colorectal cancer but not in Treg
cells correlates with disease progression in patients with
colorectal cancer. PLoS One 8. e53630:2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Harada K, Shimoda S, Kimura Y, Sato Y,
Ikeda H, Igarashi S, Ren XS, Sato H and Nakanuma Y: Significance of
immunoglobulin G4 (IgG4)-positive cells in extrahepatic
cholangiocarcinoma: Molecular mechanism of IgG4 reaction in cancer
tissue. Hepatology 56. 157–164. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Ebert LM, Tan BS Browning J, Svobodova S,
Russell SE, Kirkpatrick N, Gedye C, Moss D, Ng SP and MacGregor D:
et al The regulatory T cell-associated transcription factor FoxP3
is expressed by tumor cells. Cancer Res 68. 3001–3009.
2008.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Hu W, Feng Z and Levine AJ: The regulation
of multiple p53 stress responses is mediated through MDM2. Genes
Cancer 3. 199–208. 2012.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Khoury MP and Bourdon JC: The isoforms of
the p53 protein. Cold Spring Harb Perspect Biol 2.
a000927:2010.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Surget S, Khoury MP and Bourdon JC:
Uncovering the role of p53 splice variants in human malignancy: A
clinical perspective. Onco Targets Ther 7. 57–68. 2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Arsic N, Gadea G, Lagerqvist EL, Busson M,
Cahuzac N, Brock C, Hollande F, Gire V, Pannequin J and Roux P: The
p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell
Reports 4. 531–540. 2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Arsic N, Ho-Pun-Cheung A, Evelyne C,
Assenat E, Jarlier M, Anguille C, Colard M, Pezet M, Roux P and
Gadea G: The p53 isoform delta133p53ß regulates cancer cell
apoptosis in a RhoB-dependent manner. PLoS One 12.
e0172125:2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Della Torre G, Pasquini G, Pilotti S,
Alasio L, Civelli E, Cozzi G, Milella M, Salvetti M, Pierotti MA
and Severini A: TP53 mutations and mdm2 protein overexpression in
cholangiocarcinomas. Diagn Mol Pathol 9. 41–46. 2000.PubMed/NCBI
|
|
58
|
Tullo A, D'Erchia AM, Honda K, Kelly MD,
Habib NA, Saccone C and Sbisà E: New p53 mutations in hilar
cholangiocarcinoma. Eur J Clin Invest 30. 798–803. 2000.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liu XF, Zhang H, Zhu SG, Zhou XT, Su HL,
Xu Z and Li SJ: Correlation of p53 gene mutation and expression of
P53 protein in cholangiocarcinoma. World J Gastroenterol 12.
4706–4709. 2006.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Nutthasirikul N, Limpaiboon T, Leelayuwat
C, Patrakitkomjorn S and Jearanaikoon P: Ratio disruption of the
∆133p53 and TAp53 isoform equilibrium correlates with poor clinical
outcome in intrahepatic cholangiocarcinoma. Int J Oncol 42.
1181–1188. 2013.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Nutthasirikul N, Hahnvajanawong C,
Techasen A, Limpaiboon T, Leelayuwat C, Chau-In S and Jearanaikoon
P: Targeting the ∆133p53 isoform can restore chemosensitivity in
5-fluorouracil-resistant cholangiocarcinoma cells. Int J Oncol 47.
2153–2164. 2015.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Liu K, Zang Y, Guo X, Wei F, Yin J, Pang L
and Chen D: The Δ133p53 isoform reduces wtp53-induced stimulation
of DNA Pol γ activity in the presence and absence of D4T. Aging Dis
8. 228–239. 2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
David CJ, Chen M, Assanah M, Canoll P and
Manley JL: HnRNP proteins controlled by c-Myc deregulate pyruvate
kinase mRNA splicing in cancer. Nature 463. 364–368.
2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Liu WR, Tian MX, Yang LX, Lin YL, Jin L,
Ding ZB, Shen YH, Peng YF, Gao DM and Zhou J: et al PKM2 promotes
metastasis by recruiting myeloid-derived suppressor cells and
indicates poor prognosis for hepatocellular carcinoma. Oncotarget
6. 846–861. 2015.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Li C, Zhao Z, Zhou Z and Liu R: PKM2
promotes cell survival and invasion under metabolic stress by
enhancing Warburg effect in pancreatic ductal adenocarcinoma. Dig
Dis Sci 61. 767–773. 2016.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lu W, Cao Y, Zhang Y, Li S, Gao J, Wang
XA, Mu J, Hu YP, Jiang L and Dong P: et al Up-regulation of PKM2
promote malignancy and related to adverse prognostic risk factor in
human gallbladder cancer. Sci Rep 6. 26351:2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Yu G, Yu W, Jin G, Xu D, Chen Y, Xia T, Yu
A, Fang W, Zhang X and Li Z: et al PKM2 regulates neural invasion
of and predicts poor prognosis for human hilar cholangiocarcinoma.
Mol Cancer 14. 193:2015.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Kotani M, Tanaka I, Ogawa Y, Suganami T,
Matsumoto T, Muro S, Yamamoto Y, Sugawara A, Yoshimasa Y and Sagawa
N: et al Multiple signal transduction pathways through two
prostaglandin E receptor EP3 subtype isoforms expressed in human
uterus. J Clin Endocrinol Metab 85. 4315–4322. 2000.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Ma J, Chen M, Xia SK, Shu W, Guo Y, Wang
YH, Xu Y, Bai XM, Zhang L and Zhang H: et al Prostaglandin E2
promotes liver cancer cell growth by the upregulation of
FUSE-binding protein 1 expression. Int J Oncol 42. 1093–1104.
2013.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Kotelevets L, Foudi N, Louedec L,
Couvelard A, Chastre E and Norel X: A new mRNA splice variant
coding for the human EP3-I receptor isoform. Prostaglandins Leukot
Essent Fatty Acids 77. 195–201. 2007.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Du M, Shi F, Zhang H, Xia S, Zhang M, Ma
J, Bai X, Zhang L, Wang Y and Cheng S: et al Prostaglandin E2
promotes human cholangiocarcinoma cell proliferation, migration and
invasion through the upregulation of β-catenin expression via EP3-4
receptor. Oncol Rep 34. 715–726. 2015.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Uthaisar K, Vaeteewoottacharn K, Seubwai
W, Talabnin C, Sawanyawisuth K, Obchoei S, Kraiklang R, Okada S and
Wongkham S: Establishment and characterization of a novel human
cholangiocarcinoma cell line with high metastatic activity. Oncol
Rep 36. 1435–1446. 2016.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Neeb A, Hefele S, Bormann S, Parson W,
Adams F, Wolf P, Miernik A, Schoenthaler M, Kroenig M and Wilhelm
K: et al Splice variant transcripts of the anterior gradient 2 gene
as a marker of prostate cancer. Oncotarget 5. 8681–8689.
2014.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Yosudjai J, Inpad C, Chomwong S, Dana P,
Sawanyawisuth K, Phimsen S, Wongkham S, Jirawatnotai S and Kaewkong
W: An aberrantly spliced isoform of anterior gradient-2, AGR2vH
promotes migration and invasion of cholangiocarcinoma cell. Biomed
Pharmacother 107. 109–116. 2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Yang Y and Walsh CE: Spliceosome-mediated
RNA trans-splicing. Mol Ther 12. 1006–1012. 2005.PubMed/NCBI
|
|
76
|
Mansfield SG, Chao H and Walsh CE: RNA
repair using spliceosome-mediated RNA trans-splicing. Trends Mol
Med 10. 263–268. 2004.PubMed/NCBI View Article : Google Scholar
|
|
77
|
He X, Liao J, Liu F, Yan J, Yan J, Shang
H, Dou Q, Chang Y, Lin J and Song Y: Functional repair of p53
mutation in colorectal cancer cells using trans-splicing.
Oncotarget 6. 2034–2045. 2015.PubMed/NCBI View Article : Google Scholar
|
|
78
|
He X, Liu F, Yan J, Zhang Y, Yan J, Shang
H, Dou Q, Zhao Q and Song Y: Trans-splicing repair of mutant p53
suppresses the growth of hepatocellular carcinoma cells in vitro
and in vivo. Sci Rep 5. 8705:2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Gout S, Brambilla E, Boudria A, Drissi R,
Lantuejoul S, Gazzeri S and Eymin B: Abnormal expression of the
pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non
small cell lung carcinoma. PLoS One 7. e46539:2012.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Karni R, de Stanchina E, Lowe SW, Sinha R,
Mu D and Krainer AR: The gene encoding the splicing factor SF2/ASF
is a proto-oncogene. Nat Struct Mol Biol 14. 185–193.
2007.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Loilome W, Yongvanit P, Wongkham C,
Tepsiri N, Sripa B, Sithithaworn P, Hanai S and Miwa M: Altered
gene expression in Opisthorchis viverrini-associated
cholangiocarcinoma in hamster model. Mol Carcinog 45. 279–287.
2006.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Cretu C, Schmitzová J, Ponce-Salvatierra
A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H,
Lührmann R and Pena V: Molecular Architecture of SF3b and
Structural Consequences of Its Cancer-Related Mutations. Mol Cell
64. 307–319. 2016.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Darman RB, Seiler M, Agrawal AA, Lim KH,
Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B and Colla S: et al
Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3' Splice
Site Selection through Use of a Different Branch Point. Cell
Reports 13. 1033–1045. 2015.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Alsafadi S, Houy A, Battistella A, Popova
T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S
and Roman-Roman S: et al Cancer-associated SF3B1 mutations affect
alternative splicing by promoting alternative branchpoint usage.
Nat Commun 7. 10615:2016.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Furney SJ, Pedersen M, Gentien D, Dumont
AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la
Grange P and Roman-Roman S: et al SF3B1 mutations are associated
with alternative splicing in uveal melanoma. Cancer Discov 3.
1122–1129. 2013.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Maguire SL, Leonidou A, Wai P, Marchiò C,
Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B and Natrajan
RC: SF3B1 mutations constitute a novel therapeutic target in breast
cancer. J Pathol 235. 571–580. 2015.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Fu X, Tian M, Gu J, Cheng T, Ma D, Feng L
and Xin X: SF3B1 mutation is a poor prognostic indicator in luminal
B and progesterone receptor-negative breast cancer patients.
Oncotarget 8. 115018–115027. 2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Woolard J, Vousden W, Moss SJ,
Krishnakumar A, Gammons MV, Nowak DG, Dixon N, Micklefield J,
Spannhoff A and Bedford MT: et al Borrelidin modulates the
alternative splicing of VEGF in favour of anti-angiogenic isoforms.
Chem Sci (Camb) 2011. 273–278. 2011.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kaida D, Motoyoshi H, Tashiro E, Nojima T,
Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T and
Nakajima H: et al Spliceostatin A targets SF3b and inhibits both
splicing and nuclear retention of pre-mRNA. Nat Chem Biol 3.
576–583. 2007.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Folco EG, Coil KE and Reed R: The
anti-tumor drug E7107 reveals an essential role for SF3b in
remodeling U2 snRNP to expose the branch point-binding region.
Genes Dev 25. 440–444. 2011.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Roybal GA and Jurica MS: Spliceostatin A
inhibits spliceosome assembly subsequent to prespliceosome
formation. Nucleic Acids Res 38. 6664–6672. 2010.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Satoh T and Kaida D: Upregulation of p27
cyclin-dependent kinase inhibitor and a C-terminus truncated form
of p27 contributes to G1 phase arrest. Sci Rep 6.
27829:2016.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Furumai R, Uchida K, Komi Y, Yoneyama M,
Ishigami K, Watanabe H, Kojima S and Yoshida M: Spliceostatin A
blocks angiogenesis by inhibiting global gene expression including
VEGF. Cancer Sci 101. 2483–2489. 2010.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Radhakrishnan A, Nanjappa V, Raja R, Sathe
G, Chavan S, Nirujogi RS, Patil AH, Solanki H, Renuse S and
Sahasrabuddhe NA: et al Dysregulation of splicing proteins in head
and neck squamous cell carcinoma. Cancer Biol Ther 17. 219–229.
2016.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Araki S, Dairiki R, Nakayama Y, Murai A,
Miyashita R, Iwatani M, Nomura T and Nakanishi O: Inhibitors of CLK
protein kinases suppress cell growth and induce apoptosis by
modulating pre-mRNA splicing. PLoS One 10. e0116929:2015.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Massiello A, Salas A, Pinkerman RL, Roddy
P, Roesser JR and Chalfant CE: Identification of two RNA
cis-elements that function to regulate the 5' splice site selection
of Bcl-x pre-mRNA in response to ceramide. J Biol Chem 279.
15799–15804. 2004.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Dewaele M, Tabaglio T, Willekens K, Bezzi
M, Teo SX, Low DH, Koh CM, Rambow F, Fiers M and Rogiers A: et al
Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs
tumor growth. J Clin Invest 126. 68–84. 2016.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Nielsen TO, Sorensen S, Dagnæs-Hansen F,
Kjems J and Sorensen BS: Directing HER4 mRNA expression towards the
CYT2 isoform by antisense oligonucleotide decreases growth of
breast cancer cells in vitro and in vivo. Br J Cancer 108.
2291–2298. 2013.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Bauman JA, Li SD, Yang A, Huang L and Kole
R: Anti-tumor activity of splice-switching oligonucleotides.
Nucleic Acids Res 38. 8348–8356. 2010.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Li Z, Li Q, Han L, Tian N, Liang Q, Li Y,
Zhao X, Du C and Tian Y: Pro-apoptotic effects of splice-switching
oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell
lines. Oncol Rep 35. 1013–1019. 2016.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Eskens FA, Ramos FJ, Burger H, O'Brien JP,
Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ and Baselga
J: et al Phase I pharmacokinetic and pharmacodynamic study of the
first-in-class spliceosome inhibitor E7107 in patients with
advanced solid tumors. Clin Cancer Res 19. 6296–6304.
2013.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Hong DS, Kurzrock R, Naing A, Wheler JJ,
Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O'Brien J and
LoRusso P: A phase I, open-label, single-arm, dose-escalation study
of E7107, a precursor messenger ribonucleic acid (pre-mRNA)
splicesome inhibitor administered intravenously on days 1 and 8
every 21 days to patients with solid tumors. Invest New Drugs 32.
436–444. 2014.PubMed/NCBI View Article : Google Scholar
|