Introduction
The extracellular potassium concentration
([K+]o) is normally maintained between 3.5
and 5 mM. Hyperkalemia and hypokalemia are defined as a serum
potassium concentration above and below this range, respectively
(1). The two are important clinical
conditions (2), predisposing patients
to life-threatening ventricular arrhythmias (3,4). Of these,
hyperkalemia exerts a wide range of effects on cardiac conduction
and repolarization properties, depending on the degree of high
[K+]o. Its most common electrocardiographic
manifestations are flattened or loss of the P-wave (5), prolonged PR and QRS intervals (6), and T-wave abnormalities, particularly
peaked T-waves (7). A sine-wave
appearance can be observed at the most severely elevated levels of
[K+]o (8).
Calcium gluconate or 10% calcium chloride are used acutely to
suppress ventricular arrhythmias in hyperkalemic patients (9,10), despite
the fact that hypercalcemia alone has pro-arrhythmic effects
(11,12). There have been certain previous studies
on the electrophysiological changes during hyperkalemia (13,14), but not
on the mechanism underlying the anti-arrhythmic action of calcium
in this situation, apart from its ‘membrane-stabilizing effect’
(15). This notion has been disputed
and the protective action of high [Ca2+]o has
instead been attributed to restoration of conduction velocities
(CVs) back to normal values (16).
Mouse systems have been extensively used for the
study of arrhythmogenesis, as they permit the use of genetic and
pharmacological manipulation to produce ion channel abnormalities
with great translational potential (17–26). This
has resulted in demonstrations of the following mechanisms
(27,28). Firstly, the early-after depolarization
phenomena and triggered activity observed during hypokalemia have
been attributed to prolonged action potential durations (APDs)
(29). Secondly, several reentrant
substrates during hypokalemia have been identified: Prolonged
epicardial but unaltered endocardial APDs leading to negative
ΔAPD90 given by endocardial APD90-epicardial
APD90 (30). Reduced
ventricular effective refractory periods (VERPs) leading to
increased critical intervals given by APD90-VERP
(29). By contrast, reduced CVs were
shown to induce ventricular arrhythmias following treatment with
the gap junction and sodium channel inhibitor heptanol through a
reduction in excitation wavelengths despite unaltered APDs and even
with increased VERPs (31,32). However, to the best of our knowledge,
there have been no investigations of the arrhythmogenic effects of
hyperkalemia in the mouse system.
Therefore, in the present study, the ventricular
arrhythmogenic properties of hyperkalemia were characterized in
Langendorff-perfused mouse hearts for the first time. An increased
external calcium concentration is known to reduce membrane
excitability at the cellular level (33), but exerts pro-arrhythmic effects in the
whole heart level under normokalemic conditions (34). However, as a decrease in membrane
excitability would lead to an increase in refractoriness, it was
hypothesized that hypercalcemia would abolish arrhythmic properties
of hyperkalemia by increasing VERPs.
Materials and methods
Solutions
Krebs-Henseleit solution [119 mM NaCl, 25 mM
NaHCO3, 4 mM KCl, 1.2 mM KH2PO4, 1
mM MgCl2, 1.8 mM CaCl2, 10 mM glucose and 2
mM sodium pyruvate (pH 7.4)] that had been bicarbonate-buffered and
bubbled with 95% O2−5% CO2 (35) was used in the experiments. Hyperkalemic
solution was prepared by increasing the amount of KCl added to
produce a [K+] of 6.3 mM, whereas hypercalcemic solution
was prepared by increasing the amount of CaCl2 added to
produce a [Ca2+] of 2.2 mM.
Preparation of Langendorff-perfused
mouse hearts
Wild-type mice of the 129 genetic background between
5 and 7 months of age were used in the study. These mice were
housed in an animal facility at room temperature (21±1°C), subject
to a 12:12 h light:dark cycle and had free access to sterile rodent
chow and water. All the experiments described complied with the UK
Animals (Scientific Procedures) Act 1986. The procedures for the
preparation of Langendorff-perfused mouse hearts have been
described previously (36). Mice were
sacrificed by cervical dislocation in accordance with Sections 1(c)
and 2 of Schedule 1 of the UK Animals (Scientific Procedures) Act
1986. The hearts were quickly excised and immediately submerged in
ice-cold Krebs-Henseleit solution. The aorta was cannulated using a
tailor-made 21-gauge cannula that had been prefilled with ice-cold
buffer, secured using a micro-aneurysm clip (Harvard Apparatus,
Cambridge, UK) and attached to the perfusion system. Retrograde
perfusion was started at a rate of 2–2.5 ml min−1 using
a peristaltic pump (Watson-Marlow Bredel pumps model 505S;
Falmouth, Cornwall, UK) with the perfusate passing through 200- and
5-µm filters successively and heated to 37°C using a water jacket
and circulator prior to reaching the aorta. Approximately 90% of
the hearts that regained their pink colour and spontaneous rhythmic
activity were studied further. The remaining 10% did not and were
therefore discarded. Perfusion continued for a further 20 min to
minimise any residual effects of endogenous catecholamine release
prior to examination of the electrophysiology of the perfused
hearts.
Stimulation protocols
Electrical stimulation was achieved using paired
platinum electrodes (1 mm interpole distance) placed at the basal
right ventricular epicardium. Pacing occurred at 8 Hz, using square
wave pulses 2 msec in duration, with a stimulation voltage set to
three times the diastolic threshold (Grass S48 Stimulator;
Grass-Telefactor, Slough, UK) immediately after the start of
perfusion. This allowed direct comparisons with previous mouse
studies of arrhythmogenesis (29–32).
Programmed electrical stimulation (PES) was used to assess for
arrhythmogenicity and thereby for reentrant substrates. This
procedure consisted of a drive train of eight regularly paced S1
stimuli at a 125 msec baseline cycle length (BCL), followed by
premature S2 extra-stimuli every ninth stimulus. S1S2 intervals
first equalled the pacing interval and were successively reduced by
1 msec with each nine stimulus cycle until arrhythmic activity was
initiated or refractoriness was reached, whereupon the S2 stimulus
elicited no response.
Recording procedures
Monophasic action potentials (MAPs) recordings were
obtained from the left ventricular epicardium using an MAP
electrode (Linton Instruments, Harvard Apparatus). They were also
obtained from the left endocardium using a custom-made MAP
electrode that was made from two strands of 0.25-mm Teflon-coated
silver wire (99.99% purity; Advent Research Materials, Witney, UK).
The tips of the electrode had previously undergone galvanic
treatment with chloride to eliminate DC offset. The endocardial
electrode was introduced through a small access window made in the
inter-ventricular septum and subsequently positioned on the lateral
aspect of the left ventricular cavity. All the recordings were
performed using a BCL of 125 msec (8 Hz) to exclude rate-dependent
differences in APDs. MAPs were pre-amplified using an NL100AK head
stage, amplified with an NL104A amplifier and band-pass filtered
between 0.5 Hz and 1 kHz using an NL125/6 filter (Neurolog,
Hertfordshire, UK) and subsequently digitized (1401plus MKII;
Cambridge Electronic Design, Cambridge, UK) at 5 kHz. Following
this, they were analyzed using Spike2 software (Cambridge
Electronic Design). MAP waveforms that did not match the previously
established stringent criteria for MAP signals (37) were rejected. The MAPs must have stable
baselines, fast upstrokes, with no inflections or negative spikes,
and a rapid first phase of repolarization. The peak of the MAP was
used to measure 0% repolarization and 100% repolarization was
measured at the point of return of the potential to baseline
(37–39). The following parameters were measured:
Activation latency, defined as the time difference between the
stimulus and the peak of the MAP; APDx, defined as the
time difference between the peak of the MAP and x% repolarization;
and VERP.
Statistical analysis
All the values are expressed as mean ± standard
error of the mean. Different experimental groups were compared by
one-way analysis of variance (ANOVA) and Student's t-test was used
as appropriate. P<0.05 was considered to indicate a
statistically significant difference. Categorical data were
compared with Fisher's exact test (one-tailed).
Results
Ventricular arrhythmogenicity and
action potential characteristics
Ventricular arrhythmogenicity and its associations
to action potential characteristics were examined under
normokalemia (5.2 mM [K+]), normocalcemia (1.8 mM
[Ca2+]), hyperkalemia alone (6.3 mM [K+]) and
hyperkalemia with hypercalcemia treatment (2.2 mM
[Ca2+]).
Hyperkalemia exerts pro-arrhythmic
effects that are abolished by hypercalcemia
The initial experiments were performed on hearts
extrinsically paced at 8 Hz, which is close to the heart rate
observed in vivo under normokalemic, hyperkalemic and combined
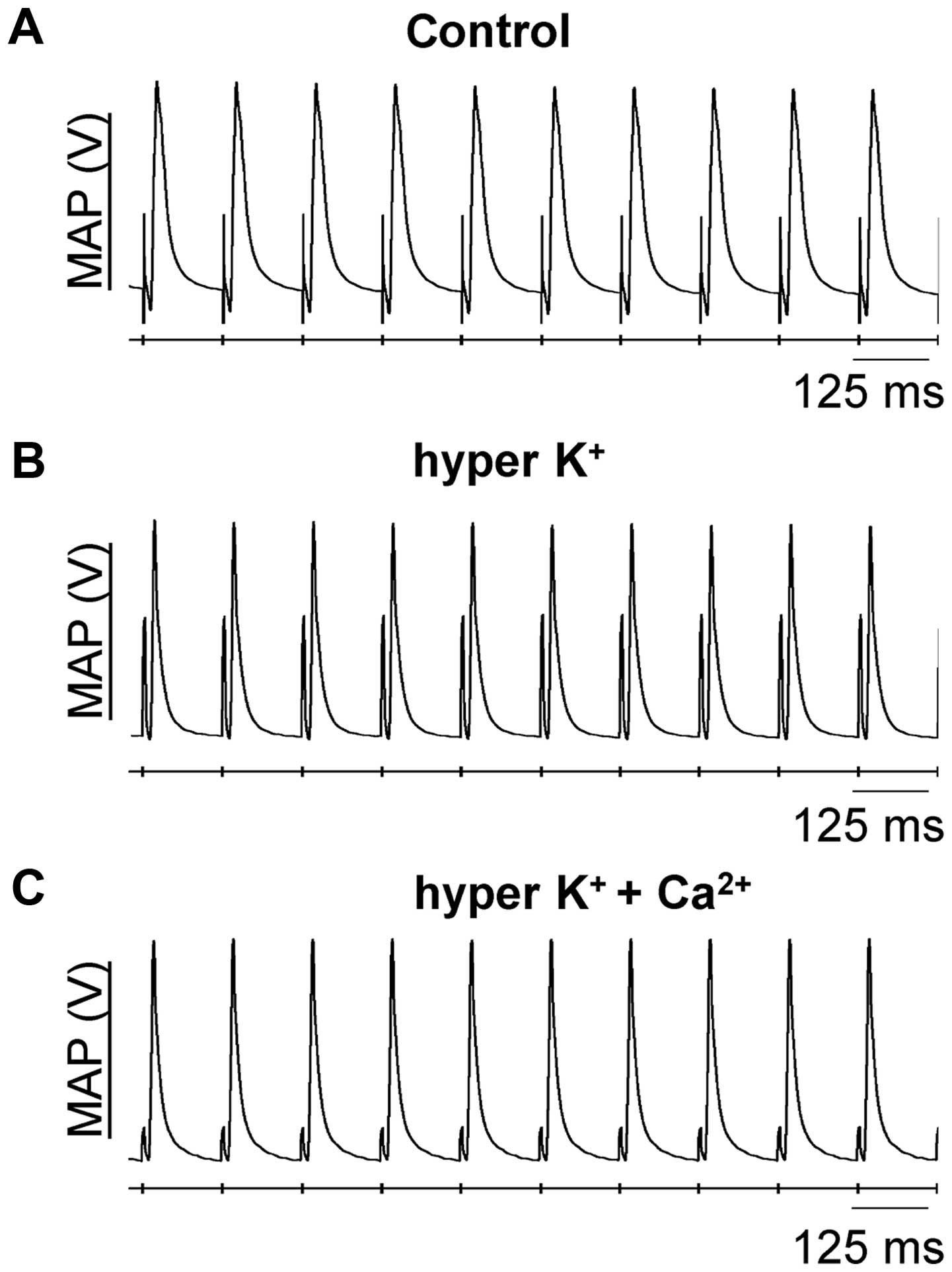
hyperkalemic and hypercalcemic conditions. Fig. 1 shows representative traces of
epicardial MAP recordings under these pharmacological conditions,
in which stable MAPs occurring directly following its preceding
stimulus, with consistent waveforms, can be observed.
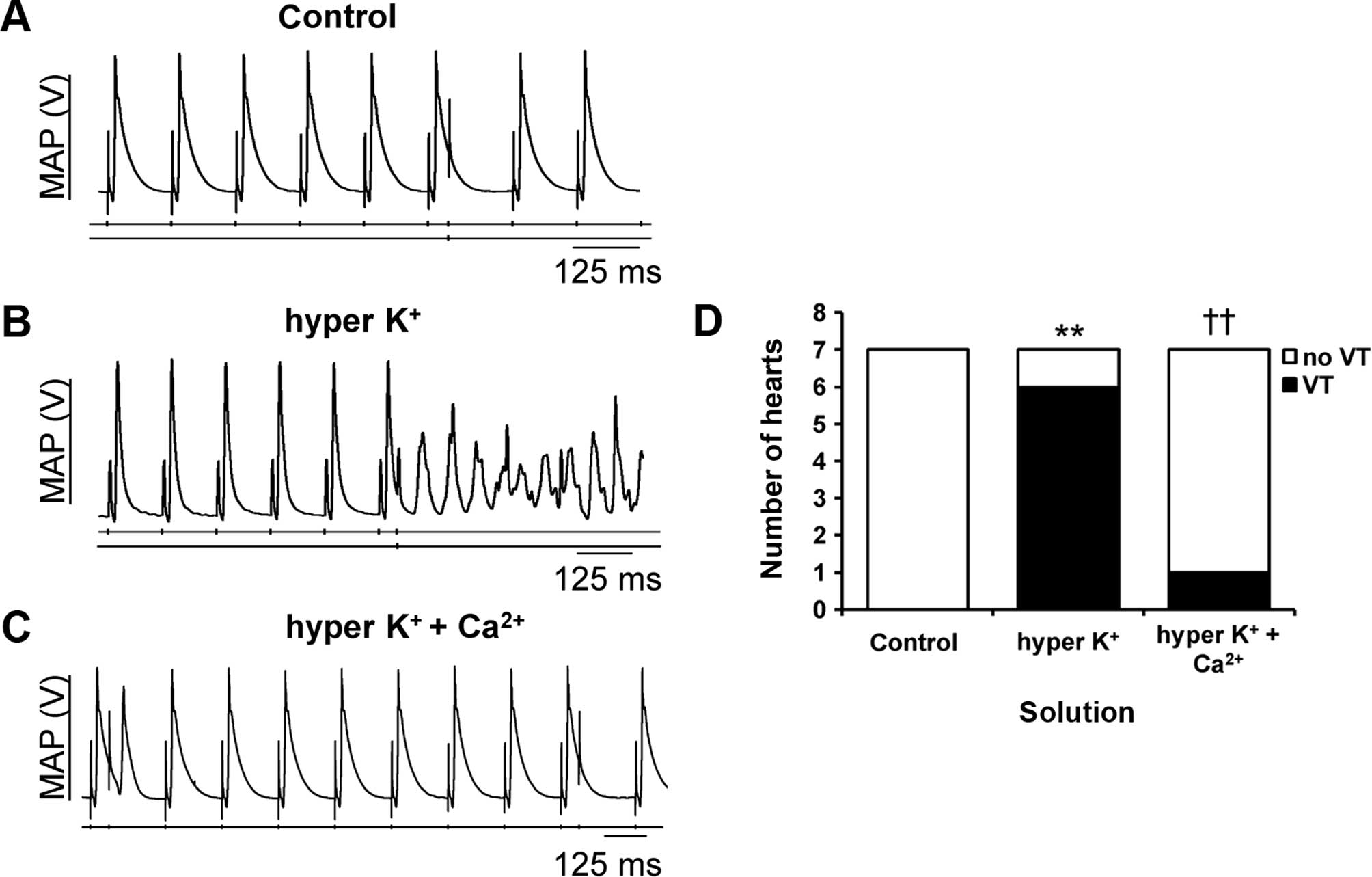
PES delivering progressively premature stimuli was
used to examine the arrhythmic tendency. It consistently failed to
provoke any arrhythmia under the control conditions (Fig. 2A; S2 extrastimulus indicated by an
arrow). By contrast, provoked ventricular tachycardia (VT) was
observed under hyperkalemic conditions alone (Fig. 2B). This was prevented by further
hypercalcemia treatment (Fig. 2C). The
incidences of provoked VT observed are summarized in Fig. 2D, demonstrating that hyperkalemia was
significantly arrhythmogenic (Fisher's exact test, P<0.01),
whereas hypercalcemia treatment was anti-arrhythmic under
hyperkalemic conditions (Fisher's exact test, P<0.01).
Shortenings in the QT interval were observed in
electrocardiograms (ECGs) obtained from patients suffering from
hyperkalemia (6). This may reflect
alterations in APD either locally or transmurally across the
myocardial wall. APDs at x=30, 50, 70 and 90% repolarization
(APDx) were therefore assessed in the epicardium and
endocardium, allowing calculation of ∆APD90 given by
endocardial APD90-epicardial APD90, thereby
providing an indication of the transmural repolarization gradient.
Epicardial APD90 was decreased from 42.2±2.6 to 24.5±1.6
msec by hyperkalemia (P<0.001; Fig.
3A), as were APD70 (P<0.001; Fig. 3B), APD50 (P<0.01;
Fig. 3C) and APD30
(P<0.05; Fig. 3D). However, the
corresponding endocardial APDx values were not altered
(P>0.05; Fig. 3E-H). These changes
corresponded to increases in ΔAPD90 (Student's t-test,
P<0.05; Fig. 4A), ΔAPD70
(P<0.01; Fig. 4B),
ΔAPD50 (P<0.01; Fig. 4C)
and ΔAPD30 (P<0.05; Fig.
4D). None of the epicardial or endocardial APDx and
ΔAPDx values were further altered upon hypercalcemia
treatment (P>0.05 in all cases).
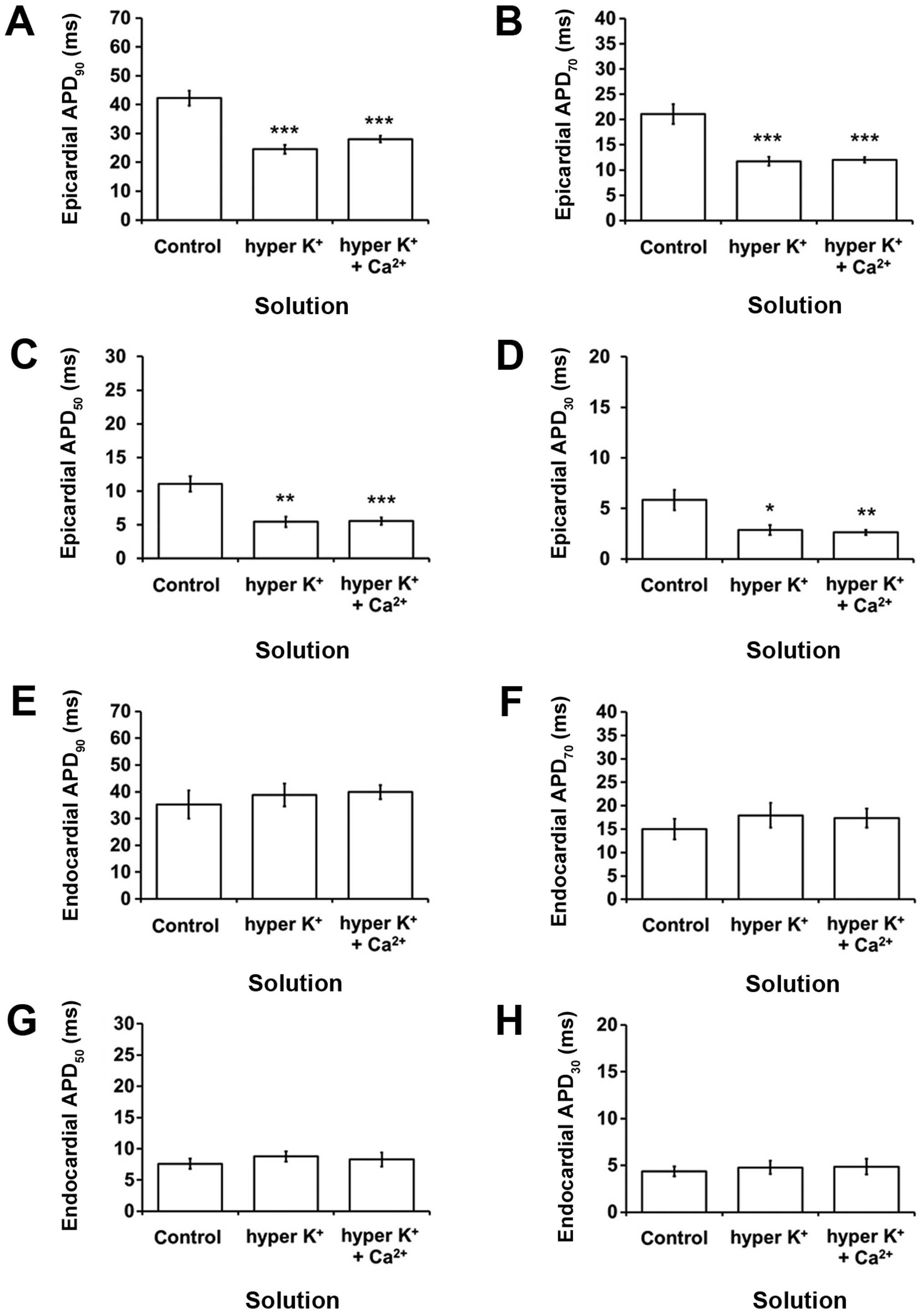 | Figure 3.Epicardial action potential durations
(APDx) at x=(A) 90, (B) 70, (C) 50 and (D) 30%
repolarization (msec) (mean ± SEM) (C) under control conditions,
hyperkalemia alone or following hypercalcemia treatment during 8 Hz
pacing (n=7). All APDx values were shortened by
hyperkalemia (ANOVA, ***P<0.001, ***P<0.001, **P<0.01,
*P<0.05, respectively), which were not further altered by
hypercalcemia treatment (ANOVA, P>0.05). Endocardial
APDx at x=(E) 90, (F) 70, (G) 50 and (H) 30%
repolarization (msec) (mean ± SEM) obtained under the same
experimental conditions. None of these values was altered by
hyperkalemia alone or following hypercalcemia treatment (ANOVA,
P>0.05). APD, action potential duration; SEM, standard error of
the mean; ANOVA, analysis of variance. |
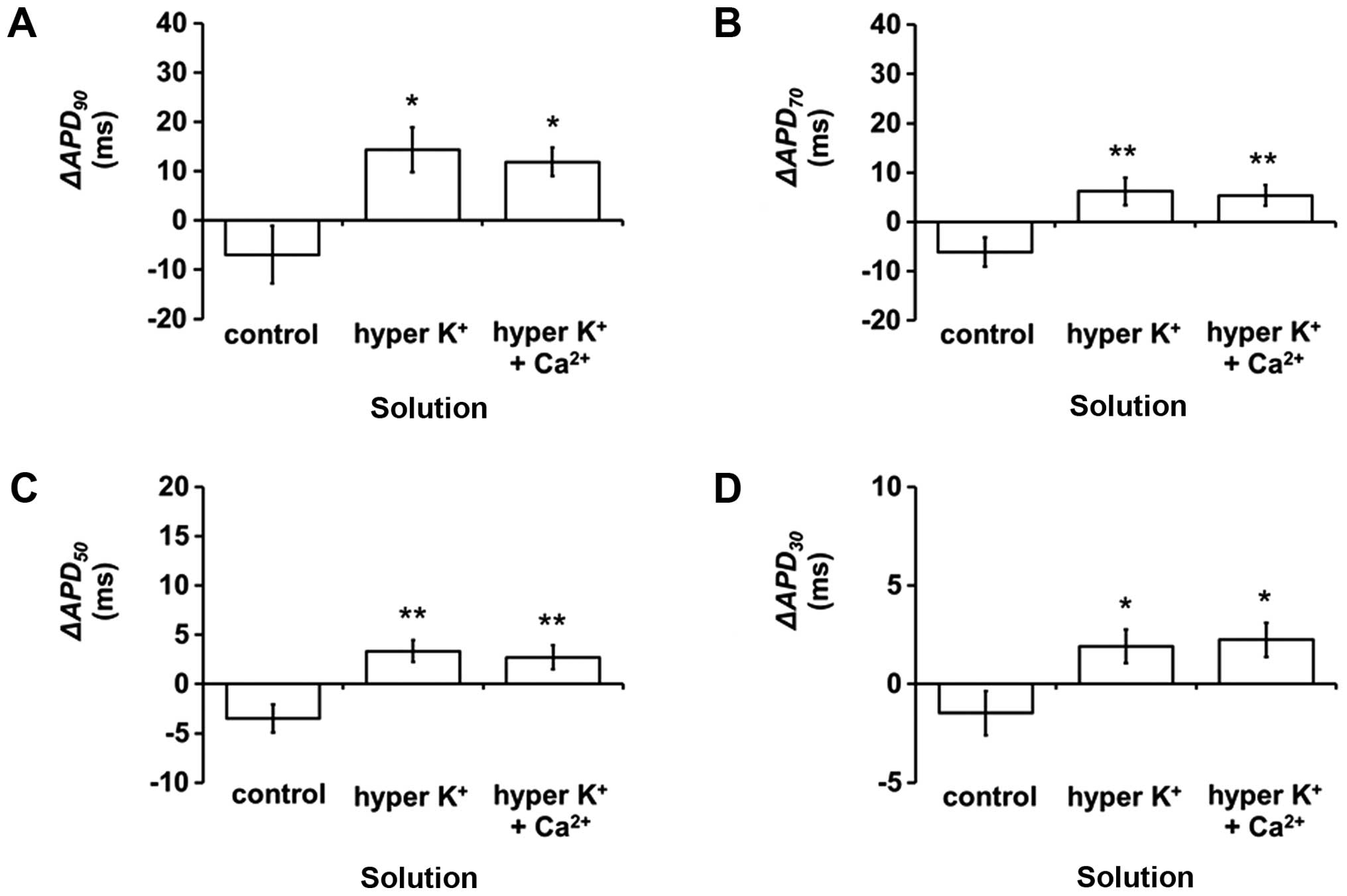 | Figure 4.ΔAPDx (endocardial
APDx-epicardial APDx) at x=(A) 90, (B) 70,
(C) 50 and (D) 30% repolarization (msec) (mean ± standard error of
the mean) under control conditions, hyperkalemia alone or after
hypercalcemia treatment during 8 Hz pacing (n=7).
ΔAPD90, ΔAPD70, ΔAPD50 and
ΔAPD30 were increased by hyperkalemia (Student's t-test,
*P<0.05, **P<0.01, **P<0.01 and *P<0.05, respectively)
and were not further altered by hypercalcemia treatment
(P>0.05). APD, action potential duration. |
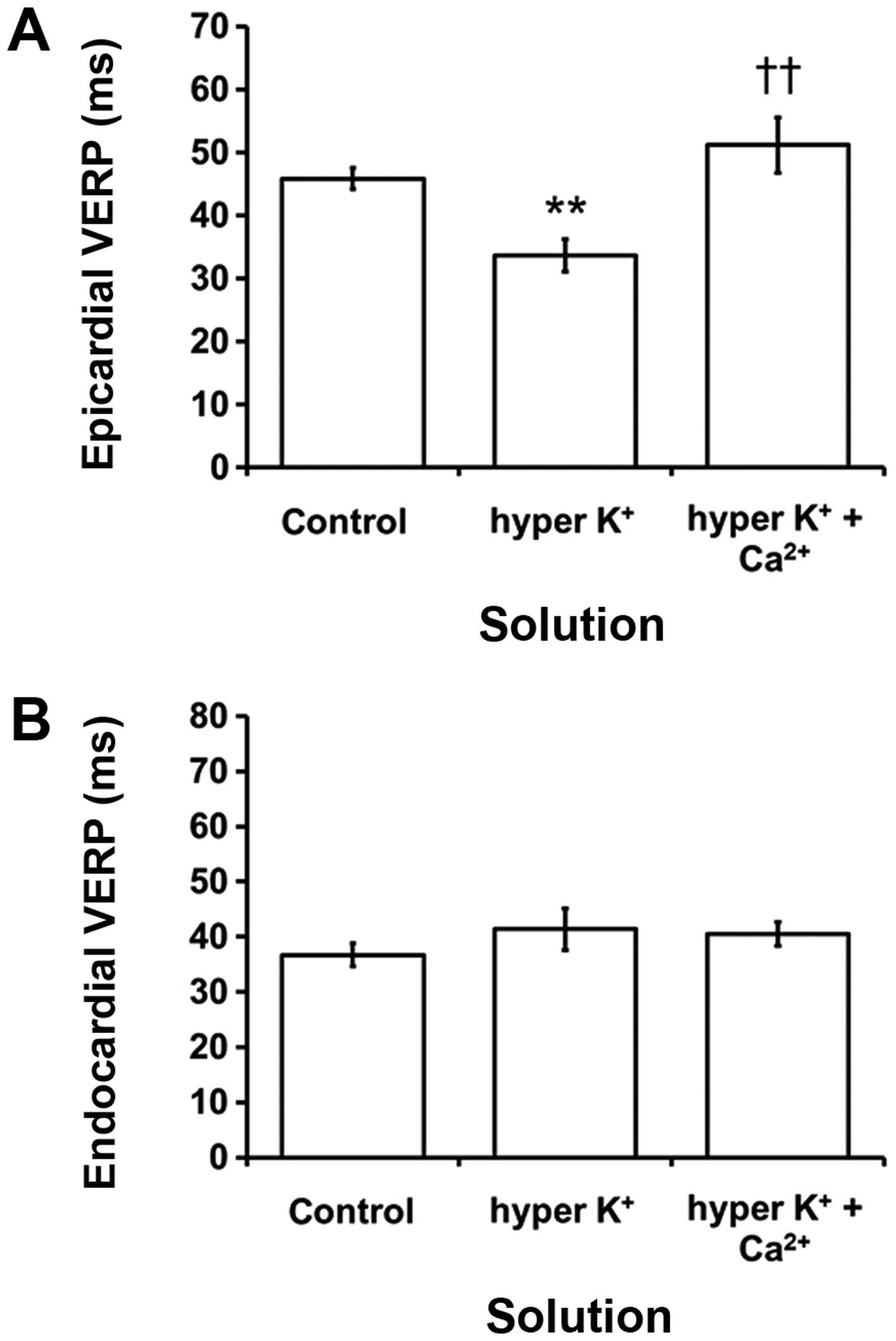
Epicardial VERPs were decreased from 45.9±1.7 to
33.7±2.6 msec during hyperkalemia (ANOVA, P<0.001; Fig. 5A) and reversed by hypercalcemia
treatment (P>0.05). By contrast, endocardial VERPs had a mean
value of 36.7±2.1 msec under control conditions and this was not
altered by hyperkalemia alone or following hypercalcemia treatment
(P>0.05; Fig. 5B). Epicardial VERPs
were significantly shorter compared to the corresponding
endocardial VERPs under hyperkalemic conditions alone (P<0.05)
but not under control conditions or hyperkalemic conditions
following hypercalcemia treatment (P>0.05).
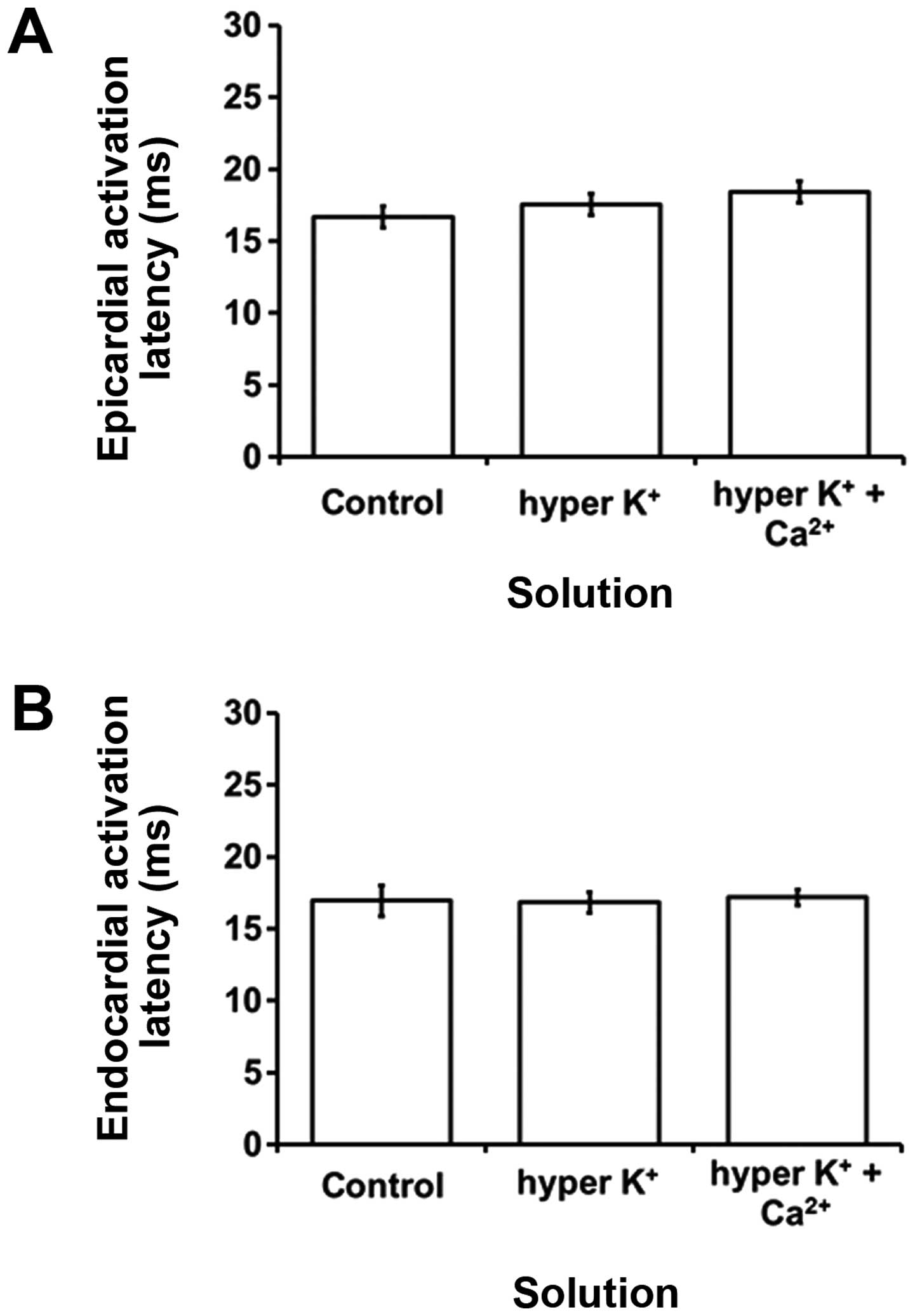
Hyperkalemia is known to cause prolongations in QRS
durations, reflecting slowed ventricular conduction in humans
(6). Reduced CVs have been shown to be
an important factor in producing ventricular arrhythmogenesis
following heptanol treatment (31).
Therefore, in the studythe activation latencies, which provide an
indication of the CVs, were quantified to determine whether changes
in these values contribute to the arrhythmogenic substrate.
Epicardial and endocardial activation latencies had values of
16.7±0.8 (Fig. 6A) and 17.0±1.1 msec
(Fig. 6B), respectively, under
normokalemic conditions. These values were not altered by
hyperkalemia alone or following hypercalcemia treatment (ANOVA,
P>0.05). Epicardial activation latencies were not significantly
different from their corresponding endocardial activation latencies
under any of the aforementioned pharmacological conditions studied
(P>0.05).
Increased critical intervals for reexcitation have
previously been associated with increased arrhythmogenicity in
hypokalemic mouse hearts (40). To
determine their possible roles in hyperkalemia-induced
arrhythmogenesis, these values were accordingly calculated for all
the pharmacological conditions studied. The local critical interval
for the epicardium was −7.0±4.1 msec under control conditions (n=7;
Fig. 7A). The interval was not altered
by hyperkalemia alone but was reduced by hypercalcemia treatment to
−23.1±4.5 msec (ANOVA, P<0.05). By contrast, the local critical
interval for the endocardium had a value of −1.4±3.5 msec (n=7;
Fig. 7B) but this was not altered by
either hyperkalemia alone or following further hypercalcemia
treatment (P>0.05). The transmural critical interval for
reexcitation of the endocardium by the epicardium had a value of
5.3±3.5 msec (n=7; Fig. 7C). This was
reduced by hyperkalemia to −16.2±4.2 msec (Student's t-test,
P<0.01) and not further altered by hypercalcemia treatment
(P>0.05). By contrast, the critical interval for reexcitation of
the epicardium by the endocardium had a value of −10.3±5.7 msec,
and was not altered by either hyperkalemia alone or following
hypercalcemia treatment (Fig. 7D;
P>0.05).
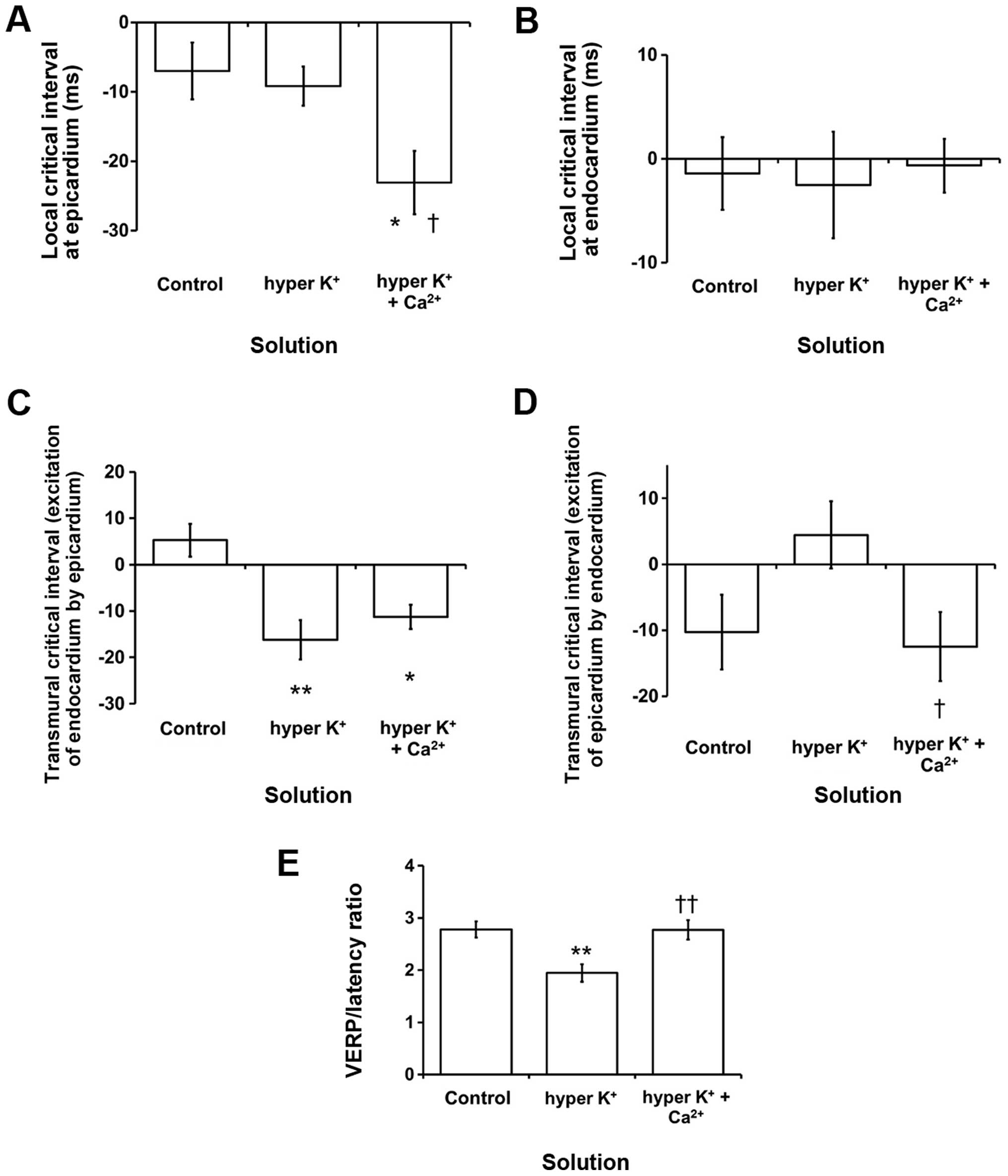 | Figure 7.(A-D) Critical intervals
(APD90-VERP) and (E) VERP/latency ratio. * and **
indicate significant differences from control values, and †
indicates significant differences from values obtained during
hyperkalemia alone. Local critical intervals obtained from the (A)
epicardium were not significantly affected by hyperkalemia alone
but were reduced by hypercalcemia treatment (ANOVA,
*,†P<0.05). The local critical interval obtained from
the (B) endocardium was not altered by either hyperkalemia alone or
following hypercalcemia treatment (P>0.05). The transmural
critical interval for reexcitation of the endocardium by the (C)
epicardium was reduced (Student's t-test, *P<0.05 and
**P<0.01) and not further altered by hypercalcemia treatment
(P>0.05). (D) The transmural critical interval for reexcitation
of the epicardium by the endocardium was not altered by either
hyperkalemia alone or following hypercalcemia treatment (P>0.05;
but there was a difference between K+ and K+
+ Ca2+, †P<0.05). (E) VERP/latency was
decreased by hyperkalemia from 2.8±0.2 to 1.9±0.2 mm (n=8; ANOVA,
**P<0.01; K+ vs. K+ + Ca2+,
††P<0.01) and subsequently restored to 2.8±0.2 mm by
hypercalcemia treatment, a value that was not statistically
different when compared to the control value (ANOVA, P>0.05).
APD, action potential duration; VERP, ventricular effective
refractory periods; ANOVA, analysis of variance. |
Reductions in the wavelength of excitation, defined
as the product of VERP and CV, increase the likelihood of
arrhythmogenesis (41). The
VERP/latency can be used as a surrogate marker of wavelength
(42). VERP/latency was decreased by
hyperkalemia from 2.8±0.2 to 1.9±0.2 mm (n=8; ANOVA, P<0.01;
Fig. 7E) and subsequently restored to
2.8±0.2 mm by hypercalcemia treatment, a value that was not
statistically different from the control value (ANOVA,
P>0.05).
Such reexcitation criteria employing the concept of
the critical interval therefore correlated poorly with
arrhythmogenicity in this hyperkalemia model, unlike the case of
hypokalemia described previously. This would suggest
arrhythmogenesis may not be due to APD exceeding VERP, but may
arise from reductions in VERP/latency ratios.
Discussion
Hyperkalemia is one of the most common electrolyte
abnormalities observed in hospitalized patients, predisposing them
to life-threatening ventricular arrhythmias (43). The mechanisms of arrhythmogenesis have
been studied using animal models as they permit the use of genetic
or pharmacological manipulation to study the consequences of ion
channel abnormalities (19,20,22,23,44–46). In the present study, arrhythmogenic
effects of hyperkalemia were examined in Langendorff-perfused mouse
hearts. The potential anti-arrhythmic effects of hypercalcemia were
also examined under this condition, mimicking 10% calcium chloride
administration used clinically to suppress ventricular arrhythmias
in patients suffering from hyperkalemia (10). In the present experiments, epicardial
and endocardial MAPs were recorded from the left ventricle during
electrical stimulation at the right ventricular epicardium. This
led to several new conclusions.
Stable epicardial and endocardial MAP recordings
were demonstrated under control conditions, hyperkalemia alone and
following hypercalcemia treatment during regular pacing. There was
no evidence of spontaneous arrhythmias under these conditions. This
subsequently permitted the use of PES to assess arrhythmogenicity
and detect the presence of reentrant substrates. No inducible
arrhythmias were observed under the control conditions. By
contrast, episodes of provoked VT was observed during hyperkalemia,
recapitulating clinical findings of increased arrhythmogenicity in
humans (9). These arrhythmogenic
effects were associated with reductions in the epicardial
APD90 and VERP in an absence of alterations in
activation latencies, which is inversely proportional to CV.
Endocardial APD90 and VERP were not altered. These
findings are consistent with the shortened QT intervals observed in
ECGs of patients suffering from hyperkalemia (6). The QT interval is a reflection of
ventricular repolarization time that is determined by the balance
between influx and efflux of ions across the cell membrane
(47). Initially, increased
[K+]o would produce hyperpolarization of the
myocardial membrane, but upon reaching a steady state, there is a
depolarizing shift in the resting membrane potential (RMP), as
described by the Goldman field relationship (48). This has been shown to increase the
conductance of the IKr channel (49,50), which
would accelerate repolarization durations. As well as affecting
action potential repolarization, it also influences its initiation
and subsequent propagation through the myocardium. Thus,
hyperkalemia produces a positive shift in the threshold potential
(TP) to a smaller extent than the depolarizing shift in RMP,
thereby increasing myocardial excitability given by 1/(TP-RMP)
(48,51). This could explain why mild hyperkalemia
increases CV. However, hyperkalemia also increases the proportion
of inactivated sodium channels, reducing dV/dtmax, and
therefore the CV, of the propagating cardiac excitation (51,52).
Activation latency was not altered by the hyperkalemia in the
present study ([K+]o of 6.3 mM), suggestive
of little conduction abnormalities. This may be due to a balance
between increased myocardial excitability and reduced proportion of
sodium channels available for activation. This is consistent with
previous findings that QRS duration was increased or CV was reduced
when [K+]o was >6.5–7.5 mM (53).
The differing effects of hyperkalemia upon
endocardial and epicardial APD90 led to increased
∆APD90 given by their difference, which is a measure of
the transmural repolarization gradient (54). Under normokalemic conditions, the time
courses of repolarization are longer in the endocardium compared to
the epicardium, giving rise to a positive ∆APD90. This
ensures a normal unidirectional spread of the excitation through
the heart (28,55), preventing the epicardium from
reexciting the endocardium by phase 2 reentry. It is also
responsible for the upright electrocardiographic T-waves in the
right precordial leads (56,57). ∆APD90 remained positive
during exposure to hyperkalemia, suggesting the increased
arrhythmogenicity here was not due to reversal of such gradients,
as was the case in hypokalemia (29).
Instead, the arrhythmogenesis observed can be explained by
decreases in the VERP through shortening in the action potential,
which would reduce VERP/latency ratios and therefore predispose to
reentry (58). However, the critical
intervals for reexcitation given by the difference between
APD90 and VERP (40) were
either unchanged or decreased, which would be expected to have no
effect on or decrease, rather than increase, arrhythmogenicity.
Hypercalcemia treatment exerted anti-arrhythmic
effects during experimental hyperkalemia, complementing clinical
findings that calcium chloride administration is effective in
suppressing arrhythmia episodes in hyperkalemic patients (10). It reversed VERP changes without
correcting for the shortenings in APDs, and left activation
latencies unaltered. Consequently, the VERP/latency ratio returned
to control values and the critical intervals were either unchanged
or decreased. High [Ca2+]o causes a positive
shift in the TP without significant effects on the RMP (59). This effect can be explained by
adsorption of calcium ions to the outer surface of the cell
membrane, generating an electric field that shifts the threshold of
INa activation to more depolarized potentials
(60). It also has a positively
inotropic effect in the context of hyperkalemia in a rabbit model
(15). Although ventricular
tachyarrhythmias attributable to hypercalcemia has been reported in
humans (61) and mouse studies
(34) under normokalemic conditions,
they are nevertheless rare occurrences (62). Due to these protective effects of
calcium on the heart, it has been used clinically to treat patients
with hyperkalemia acutely prior to correcting for the plasma levels
of potassium through the use of insulin and glucose infusion with
nebulized salbutamol (63). Notably,
no further APD shortening was found in the presence of
hypercalcemia. Although hypercalcemia has been shown to cause QT
shortening, in certain instances it may be associated with a normal
QT interval (64,65). The QT interval may therefore be an
unreliable indicator of the level of hypercalcemia (66). In addition to the effects of
[Ca2+]o on the RMP and TP, it is possible
that the calcium-dependent potassium currents or calcium currents
are also altered, and this remains to be studied in the future.
There are several limitations of the present study.
First, whilst the mouse is a common animal model for studying
cardiac arrhythmias, certain caution must be taken when attempting
to extrapolate the results to human findings. In mouse hearts,
cardiac action potentials are triangular with the transient outward
current (Ito) being the major repolarizing
current (67). By contrast, the human
action potential shows a characteristic plateau phase (68). Repolarization is initially mediated by
Ito, followed by a delayed plateau phase mediated
by a balance between the inward calcium current
(ICa) and outward delayed rectifier potassium
currents (IKr and IKs). Guinea
pig and rabbit hearts have the same action potential morphology
with similar ionic contributions, and may therefore provide better
translational results when used as model systems for studying human
arrhythmic syndromes (69–80).
Secondly, the MAP technique was chosen for studying
electrophysiology in the present study, which allows electrical
recordings to be made from intact, isolated, perfused hearts. This
has the advantage of preserving intercellular coupling, meaning
that the experimental system would be more physiological. MAP
recordings close recapitulate the intracellular action potential
obtained from single cells (37,81,82). Previous studies have shown that the MAP
technique is sufficiently sensitive for detecting alterations in
activation latencies, APD and VERP from atrial and ventricular
tissue under a variety of experimental conditions (29–32).
Furthermore, parameters derived from MAP recordings only show small
variations on repeated measurements and between different hearts,
suggesting that this technique is sufficiently reliable for
studying cardiac electrophysiology. However, certain limitations of
the MAP technique must be noted, as recently reviewed (83). One major disadvantage is that MAPs,
unlike microelectrode recordings, do not provide information on the
upstroke velocity of the cellular action potential. Although the CV
of the propagating excitation wave could not be determined,
activation latency could be measured instead. This also permitted
the calculation of VERP/latency ratios that are used clinically for
approximating the excitation wavelength. Future studies can
investigate further by using optical mapping, which would allow
simultaneous measurements of cellular activation from numerous
recording sites, and CVs as well as excitation wavelength to be
calculated. Measurement of magnetic signals, such as cardiac
magnetic resonance imaging, has been used for the characterization
of structural properties (84–86), and future studies could utilize
magnetocardiography to detect electrical abnormalities and predict
arrhythmic risk (87–92).
Finally, why epicardial APDs are altered by
hyperkalemia whereas endocardial APDs are not remains to be
elucidated. Such differences were also observed in experimental
hypokalemia, where it was noted that epicardial APDs were prolonged
but endocardial APDs remained unaltered (40). This may be due to differences in ion
channel types or their levels of expression between these regions,
but these issues remain to be clarified in future studies.
Taken together, the present study produced an
arrhythmia model of hyperkalemia for the first time in the mouse,
in which ventricular tachyarrhythmias were associated with
shortenings in APD and VERP. Hypercalcemia treatment was able to
prevent this arrhythmogenesis through correction of VERP alone
without influencing APD, thereby scaling the VERP/latency ratio.
Therefore, excitation wavelength appears to be a central
determinant of arrhythmogenesis in this system, as has been
demonstrated for other models (93).
Acknowledgements
GT received a Biotechnology and Biological Sciences
Research Council (BBSRC) Doctoral Training Award supplemented by
Xention Discovery. GT was supported by Xention Discovery. The
company did not influence the writing of this manuscript in any
manner.
References
|
1
|
No authors listed, . Guidelines 2000 for
Cardiopulmonary Resuscitation and Emergency Cardiovascular Care.
Part 8: Advanced challenges in resuscitation: section 1:
Life-threatening electrolyte abnormalities. The American Heart
Association in collaboration with the International Liaison
Committee on Resuscitation. Circulation. 102(Suppl 8): I217–I222.
2000.PubMed/NCBI
|
|
2
|
Gettes LS: Electrolyte abnormalities
underlying lethal and ventricular arrhythmias. Circulation.
85(Suppl 1): I70–I76. 1992.PubMed/NCBI
|
|
3
|
Friedensohn A, Faibel HE, Bairey O,
Goldbourt U and Schlesinger Z: Malignant arrhythmias in relation to
values of serum potassium in patients with acute myocardial
infarction. Int J Cardiol. 32:331–338. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ahmed MI, Ekundayo OJ, Mujib M, Campbell
RC, Sanders PW, Pitt B, Perry GJ, Bakris G, Aban I, Love TE, et al:
Mild hyperkalemia and outcomes in chronic heart failure: A
propensity matched study. Int J Cardiol. 144:383–388. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bashour TT and Cheng TO: Evidence for
specialized atrioventricular conduction in hyperkalemia. J
Electrocardiol. 8:65–68. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dittrich KL and Walls RM: Hyperkalemia:
ECG manifestations and clinical considerations. J Emerg Med.
4:449–455. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Freeman K, Feldman JA, Mitchell P, Donovan
J, Dyer KS, Eliseo L, White LF and Temin ES: Effects of
presentation and electrocardiogram on time to treatment of
hyperkalemia. Acad Emerg Med. 15:239–249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cooper WD, Kuan P, Reuben SR and
VandenBurg MJ: Cardiac arrhythmias following acute myocardial
infarction: Associations with the serum potassium level and prior
diuretic therapy. Eur Heart J. 5:464–469. 1984.PubMed/NCBI
|
|
9
|
Weiner ID and Wingo CS: Hyperkalemia: A
potential silent killer. J Am Soc Nephrol. 9:1535–1543.
1998.PubMed/NCBI
|
|
10
|
Sood MM and Pauly RP: A case of severe
hyperkalemia: Fast, safe and effective treatment is required. J
Crit Care. 23:431–433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kiewiet RM, Ponssen HH, Janssens EN and
Fels PW: Ventricular fibrillation in hypercalcaemic crisis due to
primary hyperparathyroidism. Neth J Med. 62:94–96. 2004.PubMed/NCBI
|
|
12
|
Curione M, Letizia C, Amato S, Di Bona S,
Di Fazio F, Minisola S, Mazzuoli G and D'Erasmo E: Increased risk
of cardiac death in primary hyperparathyroidism: What is a role of
electrical instability? Int J Cardiol. 121:200–202. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
George SA, Sciuto KJ, Lin J, Salama ME,
Keener JP, Gourdie RG and Poelzing S: Extracellular sodium and
potassium levels modulate cardiac conduction in mice heterozygous
null for the Connexin43 gene. Pflugers Arch. 467:2287–2297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kodama I, Wilde A, Janse MJ, Durrer D and
Yamada K: Combined effects of hypoxia, hyperkalemia and acidosis on
membrane action potential and excitability of guinea-pig
ventricular muscle. J Mol Cell Cardiol. 16:247–259. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leitch SP and Paterson DJ: Role of Ca2+ in
protecting the heart from hyperkalemia and acidosis in the rabbit:
Implications for exercise. J Appl Physiol (1985). 77:2391–2399.
1994.PubMed/NCBI
|
|
16
|
Piktel JS, Wan X, Infeld M, Rosenbaum D
and Wilson LD: Beneficial effect of calcium treatment for
hyperkalemia is mediated by calcium-dependent conduction, not
‘membrane stabilization’. Ann Emerg Med. 56:S92010. View Article : Google Scholar
|
|
17
|
Tse G and Yeo JM: Conduction abnormalities
and ventricular arrhythmogenesis: The roles of sodium channels and
gap junctions. Int J Cardiol Heart Vasc. 9:75–82. 2015.PubMed/NCBI
|
|
18
|
Choy L, Yeo JM, Tse V, Chan SP and Tse G:
Cardiac disease and arrhythmogenesis: Mechanistic insights from
mouse models. Int J Cardiol Heart Vasc. 12:1–10. 2016.
|
|
19
|
Tse G: Both transmural dispersion of
repolarization and transmural dispersion of refractoriness are poor
predictors of arrhythmogenicity: A role for the index of Cardiac
Electrophysiological Balance (QT/QRS)? J Geriatr Cardiol. (In
press). PubMed/NCBI
|
|
20
|
Tse G, Lai ET, Yeo JM and Yan BP:
Electrophysiological mechanisms of Bayés syndrome: Insights from
clinical and mouse studies. Front Physiol. May 31–2016.(Epub ahead
of print). View Article : Google Scholar
|
|
21
|
Tse G, Lai ET, Lee AP, Yan BP and Wong SH:
Electrophysiological mechanisms of gastrointestinal
arrhythmogenesis: Lessons from the heart. Front Physiol. (In
press).
|
|
22
|
Tse G, Lai TH, Yeo JM, Tse V and Wong SH:
Mechanisms of electrical activation and conduction in the
gastrointestinal system: Lessons from cardiac electrophysiology.
Front Physiol. May 31–2016.(Epub ahead of print). View Article : Google Scholar
|
|
23
|
Tse G, Lai ET, Tse V and Yeo JM: Molecular
and electrophysiological mechanisms underlying cardiac
arrhythmogenesis in diabetes mellitus. J Diabetes Res. (In
press).
|
|
24
|
Tse G and Yan BP: Novel arrhythmic risk
markers incorporating QRS dispersion: QRSd x (Tpeak-Tend)/QRS and
QRSd x (Tpeak-Tend)/(QT x QRS). Ann Noninvasive Electrocardiol. (In
press).
|
|
25
|
Tse G: Novel conduction-repolarization
indices for the stratification of arrhythmic risk. J Geriatr
Cardiol. (In press). PubMed/NCBI
|
|
26
|
Tse G: (Tpeak-Tend)/QRS and
(Tpeak-Tend)/(QT x QRS): Novel markers for predicting arrhythmic
risk in Brugada syndrome. Europace. (In press).
|
|
27
|
Tse G, Wong ST, Tse V, Lee YT, Lin HY and
Yeo JM: Cardiac dynamics: alternans and arrhythmogenesis. J
Arrhythm. Mar 28–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tse G: Mechanisms of cardiac arrhythmias.
J Arrhythm. 32:75–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tse G, Tse V and Yeo JM: Ventricular
anti-arrhythmic effects of heptanol in hypokalaemic,
Langendorff-perfused mouse hearts. Biomed Rep. 4:313–324.
2016.PubMed/NCBI
|
|
30
|
Tse G, Wong ST, Tse V and Yeo JM:
Restitution analysis of alternans using dynamic pacing and its
comparison with S1S2 restitution in heptanol-treated, hypokalaemic
Langendorff-perfused mouse hearts. Biomed Rep. 4:673–680.
2016.PubMed/NCBI
|
|
31
|
Tse G, Hothi SS, Grace AA and Huang CL:
Ventricular arrhythmogenesis following slowed conduction in
heptanol-treated, Langendorff-perfused mouse hearts. J Physiol Sci.
62:79–92. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tse G, Tse V, Yeo JM and Sun B: Atrial
anti-arrhythmic effects of heptanol in Langendorff-perfused mouse
hearts. PLoS One. 11:e01488582016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hogan PM and Spitzer KW: Manganese amd
electrogenic phenomena in canine Purkinje fibers. Circ Res.
36:377–391. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Balasubramaniam R, Chawla S, Mackenzie L,
Schwiening CJ, Grace AA and Huang CL: Nifedipine and diltiazem
suppress ventricular arrhythmogenesis and calcium release in mouse
hearts. Pflugers Arch. 449:150–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Balasubramaniam R, Grace AA, Saumarez RC,
Vandenberg JI and Huang CL: Electrogram prolongation and
nifedipine-suppressible ventricular arrhythmias in mice following
targeted disruption of KCNE1. J Physiol. 552:535–546. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Head CE, Balasubramaniam R, Thomas G,
Goddard CA, Lei M, Colledge WH, Grace AA and Huang CL: Paced
electrogram fractionation analysis of arrhythmogenic tendency in
DeltaKPQ Scn5a mice. J Cardiovasc Electrophysiol. 16:1329–1340.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Knollmann BC, Katchman AN and Franz MR:
Monophasic action potential recordings from intact mouse heart:
Validation, regional heterogeneity, and relation to refractoriness.
J Cardiovasc Electrophysiol. 12:1286–1294. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gussak I, Chaitman BR, Kopecky SL and
Nerbonne JM: Rapid ventricular repolarization in rodents:
Electrocardiographic manifestations, molecular mechanisms, and
clinical insights. J Electrocardiol. 33:159–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fabritz L, Kirchhof P, Franz MR, Eckardt
L, Mönnig G, Milberg P, Breithardt G and Haverkamp W: Prolonged
action potential durations, increased dispersion of repolarization,
and polymorphic ventricular tachycardia in a mouse model of
proarrhythmia. Basic Res Cardiol. 98:25–32. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sabir IN, Fraser JA, Killeen MJ, Grace AA
and Huang CL: The contribution of refractoriness to arrhythmic
substrate in hypokalemic Langendorff-perfused murine hearts.
Pflugers Arch. 454:209–222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kléber AG and Rudy Y: Basic mechanisms of
cardiac impulse propagation and associated arrhythmias. Physiol
Rev. 84:431–488. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thomas GP, Howlett SE and Ferrier GR:
Saralasin suppresses arrhythmias in an isolated guinea pig
ventricular free wall model of simulated ischemia and reperfusion.
J Pharmacol Exp Ther. 274:1379–1386. 1995.PubMed/NCBI
|
|
43
|
Nolan JP, Soar J, Zideman DA, Biarent D,
Bossaert LL, Deakin C, Koster RW, Wyllie J and Böttiger B: ERC
Guidelines Writing Group: European Resuscitation Council Guidelines
for Resuscitation 2010 Section 1. Executive summary. Resuscitation.
81:1219–1276. 2010.
|
|
44
|
Tse G, Wong ST, Tse V and Yeo JM:
Depolarization vs. repolarization: What is the mechanism of
ventricular arrhythmogenesis underlying sodium channel
haploinsufficiency in mouse hearts? Acta Physiol (Oxf). Apr
30–2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen Z, Sun B, Tse G, Jiang J and Xu W:
Reversibility of both sinus node dysfunction and reduced HCN4 mRNA
expression level in an atrial tachycardia pacing model of
tachycardia-bradycardia syndrome in rabbit hearts. Int J Clin Exp
Pathol. (In press).
|
|
46
|
Tse G, Wong ST, Tse V and Yeo JM:
Determination of action potential wavelength restitution in Scn5a
+/− mouse hearts modelling human Brugada syndrome. J Physiol. (In
press).
|
|
47
|
Antzelevitch C: Cellular basis for the
repolarization waves of the ECG. Ann N Y Acad Sci. 1080:268–281.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fisch C: Relation of electrolyte
disturbances to cardiac arrhythmias. Circulation. 47:408–419. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sanguinetti MC and Jurkiewicz NK: Role of
external Ca2+ and K+ in gating of cardiac delayed rectifier K+
currents. Pflugers Arch. 420:180–186. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sanguinetti MC, Jiang C, Curran ME and
Keating MT: A mechanistic link between an inherited and an acquired
cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell.
81:299–307. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kishida H, Surawicz B and Fu LT: Effects
of K+ and K+-induced polarization on (dV/dt)max, threshold
potential, and membrane input resistance in guinea pig and cat
ventricular myocardium. Circ Res. 44:800–814. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dominguez G and Fozzard HA: Influence of
extracellular K+ concentration on cable properties and excitability
of sheep cardiac Purkinje fibers. Circ Res. 26:565–574. 1970.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ettinger PO, Regan TJ and Oldewurtel HA:
Hyperkalemia, cardiac conduction, and the electrocardiogram: A
review. Am Heart J. 88:360–371. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Killeen MJ, Thomas G, Gurung IS, Goddard
CA, Fraser JA, MahautSmith MP, Colledge WH, Grace AA and Huang CL:
Arrhythmogenic mechanisms in the isolated perfused hypokalaemic
murine heart. Acta Physiol (Oxf). 189:33–46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nerbonne JM and Guo W: Heterogeneous
expression of voltage-gated potassium channels in the heart: Roles
in normal excitation and arrhythmias. J Cardiovasc Electrophysiol.
13:406–409. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Antzelevitch C: Transmural dispersion of
repolarization and the T wave. Cardiovasc Res. 50:426–431. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yan GX and Martin J: Electrocardiographic
T wave: A symbol of transmural dispersion of repolarization in the
ventricles. J Cardiovasc Electrophysiol. 14:639–640. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mines GR: On dynamic equilibrium in the
heart. J Physiol. 46:349–383. 1913. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Weidmann S: Effects of calcium ions and
local anesthetics on electrical properties of Purkinje fibres. J
Physiol. 129:568–582. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hélie F, Cossette J, Vermeulen M and
Cardinal R: Differential effects of lignocaine and hypercalcaemia
on anisotropic conduction and reentry in the ischaemically damaged
canine ventricle. Cardiovasc Res. 29:359–372. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Corlew DS, Bryda SL, Bradley EL III and
DiGirolamo M: Observations on the course of untreated primary
hyperparathyroidism. Surgery. 98:1064–1071. 1985.PubMed/NCBI
|
|
62
|
Rosenqvist M, Nordenström J, Andersson M
and Edhag OK: Cardiac conduction in patients with hypercalcaemia
due to primary hyperparathyroidism. Clin Endocrinol (Oxf).
37:29–33. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Parham WA, Mehdirad AA, Biermann KM and
Fredman CS: Hyperkalemia revisited. Tex Heart Inst J. 33:40–47.
2006.PubMed/NCBI
|
|
64
|
Rumancik WM, Denlinger JK, Nahrwold ML and
Falk RB Jr: The QT interval and serum ionized calcium. JAMA.
240:366–368. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Scheidegger D and Drop LJ: The
relationship between duration of Q-T interval and plasma ionized
calcium concentration: Experiments with acute, steady-state [Ca++]
changes in the dog. Anesthesiology. 51:143–148. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wortsman J and Frank S: The QT interval in
clinical hypercalcemia. Clin Cardiol. 4:87–90. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Killeen MJ, Thomas G, Sabir IN, Grace AA
and Huang CL: Mouse models of ventricular arrhythmias. Acta Physiol
(Oxf). 192:455–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Nerbonne JM and Kass RS: Molecular
physiology of cardiac repolarization. Physiol Rev. 85:1205–1253.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hsieh YC, Lin JC, Hung CY, Li CH, Lin SF,
Yeh HI, Huang JL, Lo CP, Haugan K, Larsen BD, et al: Gap junction
modifier rotigaptide decreases the susceptibility to ventricular
arrhythmia by enhancing conduction velocity and suppressing
discordant alternans during therapeutic hypothermia in isolated
rabbit hearts. Heart Rhythm. 13:251–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wu TJ, Lin SF, Weiss JN, Ting CT and Chen
PS: Two types of ventricular fibrillation in isolated rabbit
hearts: Importance of excitability and action potential duration
restitution. Circulation. 106:1859–1866. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Osadchii OE: Effects of ventricular pacing
protocol on electrical restitution assessments in guinea-pig heart.
Exp Physiol. 97:807–821. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Osadchii OE, Larsen AP and Olesen SP:
Predictive value of electrical restitution in hypokalemia-induced
ventricular arrhythmogenicity. Am J Physiol Heart Circ Physiol.
298:H210–H220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Osadchii OE: Mechanisms of
hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin
Pharmacol. 24:547–559. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Osadchii OE, Bentzen BH and Olesen SP:
Chamber-specific effects of hypokalaemia on ventricular
arrhythmogenicity in isolated, perfused guinea-pig heart. Exp
Physiol. 94:434–446. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Osadchii OE and Olesen SP:
Electrophysiological determinants of hypokalaemia-induced
arrhythmogenicity in the guinea-pig heart. Acta Physiol (Oxf).
197:273–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Osadchii OE: Flecainide attenuates rate
adaptation of ventricular repolarization in guinea-pig heart. Scand
Cardiovasc J. 50:28–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Osadchii OE: Impact of hypokalemia on
electromechanical window, excitation wavelength and repolarization
gradients in guinea-pig and rabbit hearts. PLoS One. 9:e1055992014.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Osadchii OE: Impaired epicardial
activation-repolarization coupling contributes to the proarrhythmic
effects of hypokalaemia and dofetilide in guinea pig ventricles.
Acta Physiol (Oxf). 211:48–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Osadchii OE: Flecainide-induced
proarrhythmia is attributed to abnormal changes in repolarization
and refractoriness in perfused guinea-pig heart. J Cardiovasc
Pharmacol. 60:456–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hsieh YC, Lin SF, Lin TC, Ting CT and Wu
TJ: Therapeutic hypothermia (30 degrees C) enhances arrhythmogenic
substrates, including spatially discordant alternans, and
facilitates pacing-induced ventricular fibrillation in isolated
rabbit hearts. Circ J. 73:2214–2222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Franz MR, Burkhoff D, Spurgeon H,
Weisfeldt ML and Lakatta EG: In vitro validation of a new cardiac
catheter technique for recording monophasic action potentials. Eur
Heart J. 7:34–41. 1986.PubMed/NCBI
|
|
82
|
Hoffman BF, Cranefield PF, Lepeschkin E,
Surawicz B and Herrlich HC: Comparison of cardiac monophasic action
potentials recorded by intracellular and suction electrodes. Am J
Physiol. 196:1297–1301. 1959.PubMed/NCBI
|
|
83
|
Tse G, Wong ST, Tse V and Yeo JM, Lin HY
and Yeo JM: Monophasic action potential recordings: Which is the
recording electrode? J Basic Clin Physiol Pharmacol. Apr
30–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tse G, Ali A, Alpendurada F, Prasad S,
Raphael CE and Vassiliou V: Tuberculous Constrictive Pericarditis.
Res Cardiovasc Med. 4:e296142015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tse G, Ali A, Prasad SK, Vassiliou V and
Raphael CE: Atypical case of post-partum cardiomyopathy: an overlap
syndrome with arrhythmogenic right ventricular cardiomyopathy?
BJR|case reports. 1:201501822015. View Article : Google Scholar
|
|
86
|
Vassiliou V, Chin C, Perperoglou A, Tse G,
Ali A, Raphael C, Jabbour A, Newby D, Pennell D, Dweck M and Prasad
S: 93 Ejection Fraction by Cardiovascular Magnetic Resonance
Predicts Adverse Outcomes Post Aortic Valve Replacement. Heart.
100(Suppl 3): A53–A54. 2014. View Article : Google Scholar
|
|
87
|
Kwong JS, Leithäuser B, Park JW and Yu CM:
Diagnostic value of magnetocardiography in coronary artery disease
and cardiac arrhythmias: A review of clinical data. Int J Cardiol.
167:1835–1842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Steinhoff U, KnappeGrueneberg S, Schnabel
A, Trahms L, Smith F, Langley P, Murray A and Koch H:
Magnetocardiography for pharmacology safety studies requiring high
patient throughput and reliability. J Electrocardiol. 37(Suppl):
187–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yoshida K, Ogata K, Inaba T, Nakazawa Y,
Ito Y, Yamaguchi I, Kandori A and Aonuma K: Ability of
magnetocardiography to detect regional dominant frequencies of
atrial fibrillation. J Arrhythm. 31:345–351. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ito Y, Shiga K, Yoshida K, Ogata K,
Kandori A, Inaba T, Nakazawa Y, Sekiguchi Y, Tada H, Sekihara K, et
al: Development of a magnetocardiography-based algorithm for
discrimination between ventricular arrhythmias originating from the
right ventricular outflow tract and those originating from the
aortic sinus cusp: A pilot study. Heart Rhythm. 11:1605–1612. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sato Y, Yoshida K, Ogata K, Inaba T, Tada
H, Sekiguchi Y, Ito Y, Ishizu T, Seo Y, Yamaguchi I, et al: An
increase in right atrial magnetic strength is a novel predictor of
recurrence of atrial fibrillation after radiofrequency catheter
ablation. Circ J. 76:1601–1608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tse G, Wong ST, Tse V and Yeo JM:
Variability in local action potential durations, dispersion of
repolarization and wavelength restitution in aged wild-type and
Scn5a+/− mouse hearts modelling human Brugada syndrome. Journal of
Geriatric Cardiology. (In press). PubMed/NCBI
|
|
93
|
Tse G, Yeo JM, Tse V and Sun B: Gap
junction inhibition by heptanol increases ventricular
arrhythmogenicity by decreasing conduction velocity without
affecting repolarization properties or myocardial refractoriness in
Langendorff-perfused mouse hearts. MMR. (In Press).
|