Contents
Introduction
Rationale and hypothesis
Test of hypothesis
Clinical relevance
Introduction
Breast cancer is the second leading cause of
cancer-related death in women (1).
The metastasis of breast cancer to bone that occurs in more than
80% of patients with advanced disease causes bone fractures,
hypercalcemia and nerve compression (2). The osteolytic breakdown of the bone
matrix by bone-resorbing osteoclasts is an important determinant of
homing and metastasis of breast cancer cells to bone (2). Interactions of breast cancer cells
with bone-forming osteoblasts and bone-resorbing osteoclasts may
possibly result in a favorable environment for the breast cancer
cells to grow rapidly (3,4). However, it is still a mystery why the
breast cancer cells metastasize to the bone. One possibility is
that altered calcium homeostasis in breast cancer cells is a
trigger for their homing to a more favorable niche, namely
calcium-rich bone. Do dynamic calcium fluxes caused by
hyperactivity of osteoclasts and simultaneous loss of osteoblasts
within bone serve as a favorable niche for the invading breast
cancer cells? What does the excessive amount of calcium released
during osteolysis have to do with the breast cancer cell that
results in further osteolysis? To address these questions at the
molecular level, one needs to consider the collective roles played
by calcium sensing receptor CaR and the prostaglandin E2 receptor,
EP2, in breast cancer cells.
Rationale and hypothesis
The breast cancer cells first detach from the
primary tumor, migrate through the systemic circulation and then
finally reach the bone (5). The
vicious cycle that operates between breast cancer cells and bone
enhances the growth of tumor upon metastasis to bone (2). The osteolytic breakdown of bone
matrix releases very high concentrations of ionized calcium and
phosphate from the dissolution of bone matrix (5–7). The
activation of CaR by extracellular Ca2+ (8) can in turn ‘trans-activate’ epidermal
growth factor receptor (EGFR) and thus initiate EGFR signaling
(Fig. 1) to activate
mitogen-activated protein kinases (MAPK) and up-regulate synthesis
and secretion of parathyroid hormone-related protein (PTHrP) from
the breast cancer cells (1,9,10).
The PTHrP produced by the breast cancer cells further stimulates
osteoclastic bone resorption.
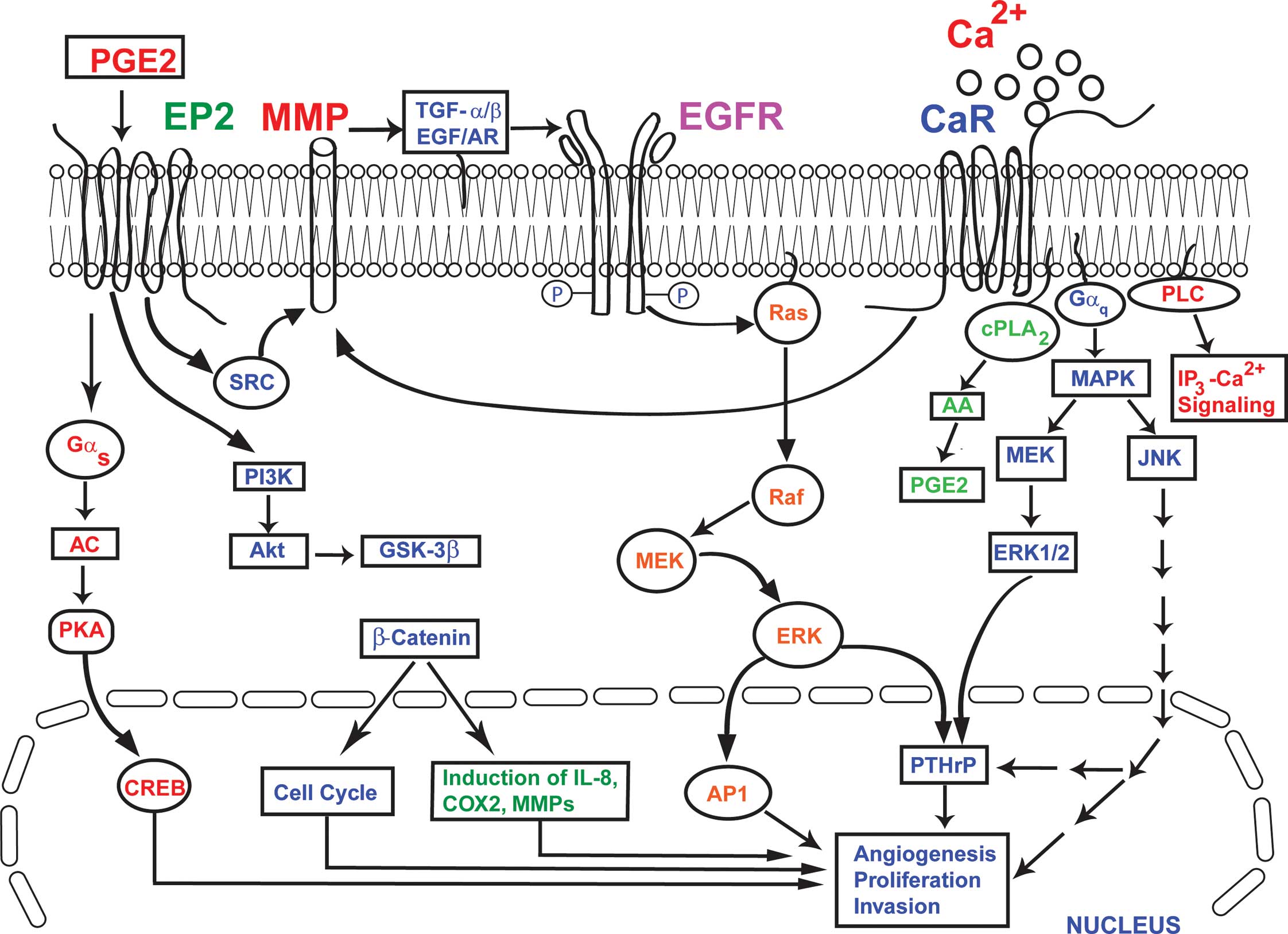 | Figure 1.The convergence of EP2 and CaR
signaling in breast cancer bone metastasis. The activation of CaR
by extracellular Ca2+ leads to the activation of ERK,
MAPK, cPLA2 and PLC, which in turn causes increased synthesis and
secretion of PTHrP from breast cancer cells. Similarly, the
‘transactivation’ of EGFR by the activated CaR activates MAPK and
ERK, resulting in the up-regulation of synthesis and the secretion
of PTHrP from breast cancer cells. The PTHrP produced by these
synergistically coupled processes in the breast cancer cells
further stimulates osteoclastic bone resorption. The PGE2
activation of EP2 results in increased production of COX2, MMPs and
IL-8 via the Gαs and PI3K pathways, involving Akt, CREB,
PKA, GSK-3β and β-catenin. At the same time, the activation of EP2
by PGE2 activates MMP1 and MMP2, which in turn act to release the
EGFR ligands AR and TGF-α. These then bind to EGFR and initiate
EGFR signaling. In this model, the activation of EP2 and CaR, and
the ‘transactivation’ of EGFR by both EP2 and CaR, results in
increased production of PTHrP, COX2, MMPs and IL-8. This
exacerbates the processes of angiogenesis, cell proliferation,
invasion and the osteolytic breakdown of bone in breast cancer. |
Prostaglandin E2 (PGE2) resulting from
cyclooxygenase 2 (COX2) activity has been implicated in
stromal-cancer cell interactions that promote tumor growth,
neovascularization and metastatic spread of many highly prevalent
cancers including breast cancer (11–13).
The treatment with non-steroidal anti-inflammatory drugs (NSAIDs)
that inhibit COX2 can reduce the risk and incidence of many types
of cancer including breast cancer (13). However, in contrast to the serious
potential cardiovascular complications of COX2 inhibitors, the
direct inhibition of G-protein-linked prostaglandin E2 receptor
(EP2) could serve as a much better alternative to COX2 inhibition
as a means for breast cancer prevention and treatment (14,15).
EP2 couples to Gαs and stimulates cyclic AMP (cAMP)
accumulation, protein kinase A (PKA) activation and the
phosphorylation of cAMP response element binding protein (CREB), as
shown in Fig. 1 (16–18).
EP2 activates matrix metalloproteinases (such as MMP1 and MMP2)
that in turn act to release the EGFR ligands amphiregulin (AR) and
transforming growth factor α (TGF-α), which then bind to EGFR and
initiate EGFR-signaling (Fig. 1)
(17–19).
CaR can signal through both Gαq and Gi
and activates phospholipase C (PLC), PLA2 and PLD. In breast cancer
cells MCF-7 and MDA-MB-231 signaling through the CaR increases the
secretion of PTHrP (9,20). The PKC/MEK-1/ERK1/2 pathway
representing the CaR signaling mechanism has been proposed in the
production of PTHrP from MCF-7 breast cancer cells (21). CaR activation by locally high
levels of [Ca2+]o at sites of osteolytic
metastases could exert proproliferative and anti-apoptotic effects
and/or alter the cellular phenotype, thereby rendering the cancer
cells metastasizing to the skeleton resistant to chemotherapy
and/or radiation. The development of allosteric activators
(‘calcimimetics’) and antagonists (‘calcilytics’) of the CaR has
allowed the CaR-based therapy of the disorders of the extracellular
calcium homeostasis (22).
Since EGFR can be transactivated both by CaR and
EP2, this convergence of signaling cross-talk at the level of MMPs,
EGFR and PTHrP can provide a novel opportunity to address the
issues related to breast cancer proliferation and metastasis from a
fresh perspective that has not been studied previously. Hence, we
propose a hypothesis guiding the molecular basis for the metastatic
ability of locally aggressive primary breast tumors to bone, and a
rationale for combinatorial therapeutic interventions against
metastasis. We hypothesize that ‘EP2 and CaR are collectively
involved in breast cancer cell proliferation, cell migration and
bone metastasis’. As a corollary to this hypothesis, it is
therefore suggested that combinatorial inhibition of EP2 and CaR
would attenuate proliferation, migration and bone metastasis of
breast cancer cells.
Test of hypothesis
To provide tests for this hypothesis, one may carry
out the following studies to answer four critical questions related
to the metastasis of breast cancer cells to bone and potential
pharmacological interventions to prevent it:
i) Do CaR and EP2 in breast cancer cells
collectively facilitate cell proliferation and inhibit
apoptosis? To address this question, one can use genetic,
pharmacological and immunological approaches such as a) silence the
expression of EP2 and CaR to analyze the compound knockdown by
breast cancer cells, b) pharmacological inhibition of EP2 using EP2
antagonists and CaR inhibition by ‘calcilytics’ (15,22–25),
and c) inhibition of EP2 and CaR by anti-EP2 and anti-CaR
antibodies. The expression of CaR and EP2 can be decreased
significantly by using short hairpin RNA (shRNA) in MDA-MB-231
cells. It is also possible to generate the compound knockdown cells
in which both shRNA for CaR and EP2 can be expressed in a given
cell population thereby silencing both these genes and their
encoded products CaR and EP2. The efficiency of knockdown can be
determined using quantitative RT-PCR and Western blotting. In order
to understand the relationship between biological functions and
biochemical pathways, the loss-of-function phenotype by using RNA
interference (RNAi) could be very useful. The RNAi technology has
been exploited in organisms for understanding gene function via
suppression of gene expression (26). The expression of a simple, 29-bp
hairpin from a U6 small nucleolar RNA (snRNA) promoter can induce
effective suppression of target genes when delivered either
transiently or stably from integrated constructs (27). The cell proliferation and apoptosis
of breast cancer cells can also be pharmacologically targeted using
previously characterized doses of the anti-CaR and anti-EP2
antibodies. One may use NPS 2143 which acts as an antagonist to CaR
(17) as a pharmacological agent,
and use EP2 antagonist AH6809 (25). When used as single agents, these
drugs should inhibit cell proliferation, inhibit the
trans-endothelial migration (for the migratory capacity of
MDA-MB-231 cells through an endothelial monolayer, see below), and
increase the rate of apoptosis of MDA-MB-231 cells, thereby
mimicking the effect of single genetic knockdown (i.e., either CaR
or EP2 knockdown). However, consistent with genetic studies on
combined knockdown of CaR and EP2, treatment with the anti-CaR and
anti-EP2 combinations as well as AH6809 and NPS 2143 combination
should reduce the rate of cell proliferation, reduce the
trans-endothelial migration and increase the rate of apoptosis much
more significantly. The rate of cell replication can be measured by
the incorporation of 5-bromodeoxyuridine as defined previously
(28,29). In order to study changes in the
rate of apoptosis, cleaved caspase-3 can be quantified by using
fluorescence microscopy in combination with monoclonal antibody
conjugated with fluorescein-isothiocyanate (FITC) that specifically
recognizes the active form of caspase-3 in cells.
Reduction of CaR or EP2 expression individually is
expected to have statistically significant, yet limited, effects on
cell proliferation and apoptosis. Nevertheless, silencing of CaR
and EP2 genes is expected to increase cell death and decrease the
rate of cell proliferation. By using genetic, pharmacological and
immunological approaches, it is possible to determine if CaR and
EP2 in breast cancer collectively facilitate cell proliferation and
inhibit apoptosis. The pharmacological inhibition of EP2 using EP2
antagonists and CaR inhibition by ‘calcilytics’ can be carried out
in MDA-MB-231 breast cancer cells.
It has been previously reported that when any of
four lung-metastatic signature (LMS) genes is inactivated by
genetic manipulation in breast cancer cells that are metastasizing
to the lung, there is only a moderate inhibition of primary-tumor
growth and lung metastasis (30).
However, when combinations of these genes are inactivated, additive
or synergistic effects are apparent, with an almost complete
abrogation of both primary-tumor growth and lung metastasis when
all four genes are inactivated. Gupta et al (30) also used pharmacological agents to
specifically inhibit the products of the four genes. Combinatorial
treatment of mice transplanted with the lung metastatic variant of
the MDA-MB-231 cell line recapitulated the results of the genetic
knockdown studies.
ii) Do CaR and EP2 in breast cancer collectively
facilitate cell migration? The in vitro assay of
trans-endothelial migration of MDA-MB-231 cells can be carried out
by seeding human-umbilical vascular endothelial cells (HUVECs) into
collagen-coated trans-well inserts and allowing them to grow to
confluence. Breast cancer cells can then be loaded with a
fluorescent cell tracker green dye before being conditioned
overnight in cell culture media without growth factors. The next
day, breast cancer cells can be seeded into trans-well inserts with
or without a confluent HUVEC endothelial monolayer, and the wells
can be fixed in 4% paraformaldehyde after 10 h. Cells on the apical
side of each insert can then be removed and the trans-well membrane
mounted onto slides. The migration of MDA-MB-231 to the basolateral
side of the membrane can be visualized with a fluorescence
microscope. The images of 6–10 random fields across three replicate
wells can be acquired for quantification, and migration of the
cells is plotted as a percentage of migrating MDA-MB-231 control
cells.
It is expected that trans-endothelial migration, the
migratory capacity of MDA-MB-231 cells through an endothelial
monolayer, will be inhibited by the a) combined knockdown of CaR
and EP2, b) anti-CaR and anti-EP2 antibodies, and c)
pharmacological agents NPS 2143 and AH6809. This result should
provide evidence that the expression of CaR and EP2 by cancer cells
can collectively promote trans-endothelial migration, a step
involved in metastatic intravasation and extravasation
processes.
iii) Do CaR and EP2 in breast cancer cells
collectively promote bone colonization? One can utilize 2- and
3-D cultures of MC3T3-E1, a murine preosteoblast cell line as an
in vitro model of bone to study the collective roles of CaR
and EP2 in breast cancer cell colonization of the bone (6,7,31).
It is feasible to use both 2- and 3-D cultures (using a specially
designed bioreactor) of MC3T3-E1, a murine preosteoblast cell line
as an in vitro model of bone to study breast cancer
colonization of bone. By adding MDA-MB-231 breast cancer cells to
these 2- and 3-D cultures, one can show by using Z-stack sectioning
and analysis of expression of differentiation proteins type 1
collagen, osteocalcin and osteonectin in the culture media that the
combinatorial inhibition of EP2 and CaR by a) combined knockdown of
CaR and EP2, b) antibodies against CaR and EP2, and c)
pharmacological agents NPS 2143 a ‘calcilytics’ and AH6809 a EP2
antagonist, in breast cancer cells indeed inhibits bone
colonization (7).
iv) Signaling cross-talk between EP2 and CaR at
the downstream targets, MMPs and EGFR. An emerging body of
evidence indicates that GPCRs are able to transactivate receptor
tyrosine kinases (RTKs) including EGFR (Fig. 1) (10,32,33).
The initial transactivation process involves stimulation of MMPs,
which results in the extracellular release of a latent
membrane-spanning precursor of a member of the family of ligands
known to activate these groups of receptors (19,33).
These ligands, either heparin-bound (HB)-EGF or TGF-α, then
secondarily activate the EGFR to phosphorylate specific tyrosine
residues residing in intracellular domains of EGFR, thereby
activating downstream proteins such as MAPKs (10,34).
Inhibiting MMPs by a broadly selective MMP inhibitor, GM-6001 and
neutralizing heparin-bound EGF with a neutralizing antibody has
been shown to prevent the CaR-mediated increases in phospho-ERK and
PTHrP release, consistent with a triple-membrane-spanning signaling
requirement for transactivation of the EGFR by the CaR (Fig. 1) (35). Similarly, EP2 can also promote the
transactivation of EGFR expressed in cancer cells and thus initiate
the EGFR-signaling network (Fig.
1) (17,18). It is possible to study convergence
of signaling cross-talk both in control and in MDA-MD-231 cells a)
silenced genetically by shRNA against EP2 and CaR, b)
combinatorially inhibited by pharmacological agents of CaR
‘calcilytics’ and EP2 antagonist, and c) inhibited by antibodies
directed against EP2 and CaR.
The downstream targets signaling cross-talks between
EP2 and CaR can be determined at the level of MMPs and EGFR
stimulation by both EP2 and CaR. In order to determine EGFR
stimulation by EP2 and CaR, EGFR can be immunoprecipitated and
probed for tyrosine phosphorylation by using polyclonal antisera
against phosphotyrosine and the EGFR. The silencing of CaR or EP2
gene expression individually is expected to cause a statistically
significant decrease in transactivation of EGFR. Nevertheless, the
combinatorial silencing of CaR and EP2 genes is expected to
significantly decrease transactivation of EGFR further. Similarly,
when used as single agents, the drugs anti-CaR or anti-EP2 or
AH6809 or NPS 2143 alone should inhibit transactivation of EPGR
thereby mimicking the effect of single genetic knockdown (i.e.,
either CaR or EP2 knockdown). However, consistent with genetic
studies on combined knockdown of CaR and EP2, treatment with the
anti-CaR and anti-EP2 combinations as well as AH6809 and NPS 2143
combination should lead to extensive decrease in the
transactivation of EGFR.
Determination of MMP1 and MMP
For analysis of MMP1 and MMP2 secreted protein,
MDA-MB-231 cells can be plated in cell culture medium. The
experimental conditioned media can then be collected at defined
intervals and MMP1 and MMP2 concentrations can be analyzed in
conditioned media using ELISA kits. The reduction of CaR or EP2
expression individually is expected to cause a statistically
significant decrease in the release of MMP1 and MMP2. Nevertheless,
the combinatorial silencing of CaR and EP2 genes is expected to
decrease the release of MMP1 and MMP2 further. Similarly, when used
as single agents, the drugs anti-CaR or anti-EP2 or AH6809 or NPS
2143 alone should decrease the release of MMP1 and MMP2 thereby
mimicking the effect of single genetic knockdown (i.e., either CaR
or EP2 knockdown). However, consistent with genetic studies on
combined knockdown of CaR and EP2, treatment with the anti-CaR and
anti-EP2 combinations as well as AH6809 and NPS 2143 combination
should decrease the release of MMP1 and MMP2 very
significantly.
Clinical relevance
The present hypothesis provides a molecular basis
for the metastatic ability of aggressive primary breast tumors, and
a rationale for a therapeutic approach against metastasis via
combinatorial interventions involving EP2 and CaR. As we progress
towards the molecular understanding of the biological functions
required for metastasis, it may become possible to develop
antimetastatic strategies that target metastatic cells and their
interactions with newly acquired microenvironments. The inhibition
of EP2 and CaR via combinatorial therapies can abate metastatic
progression in a clinically relevant model of breast cancer. The
inhibition of EP2 and CaR activities would benefit breast cancer
patients in a number of novel ways such as a) blocking the breast
cancer cell proliferation at the primary site, b) blocking the
trans-endothelial migration of breast cancer cells in the
vasculature, and finally c) blocking the bone breakdown. These
studies can provide novel insights as to how breast cancer cells
become metastatic, specifically homing to and targeting bone. This
knowledge will pave the way to develop clinical strategies to
prevent breast cancer metastasis to bone.
Acknowledgements
The authors acknowledge the Florida
International University Foundation’s Faculty Research Award to
J.P., and the Research Conference Awards from the National
Institutes of Health, Flight Attendant Medical Research Institute,
Society for Free Radical Research International and the Oxygen Club
of California to K.A.
References
|
1.
|
Kingsley LA, Fournier PGJ, Chirgwin JM and
Guise TA: Molecular biology of bone metastasis. Mol Cancer Ther.
6:2609–2617. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Kozlow W and Guise TA: Breast cancer
metastasis to bone: mechanisms of osteolysis and implications for
therapy. J Mammary Gland Biol Neoplasia. 10:169–180. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Fidler IJ: The pathogenesis of cancer
metastasis. The ‘seed and soil’ hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003.
|
|
4.
|
Paget S: The distribution of secondary
growths in cancers of breast. Lancet. 1:571–573. 1889. View Article : Google Scholar
|
|
5.
|
Kakonen SM and Mundy GR: Mechanisms of
osteolytic bone metastases in breast carcinoma. Cancer. 97:834–839.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Doherty TM, Uzui H, Fitzpatrick LA,
Tripathi PV, Dunstan CR, Asotra K and Rajavashisth TB: Mechanism of
arterial calcification in the context of atherosclerosis. FASEB J.
16:577–582. 2002.
|
|
7.
|
Doherty TM, Asotra K, Fitzpatrick LA,
Dunstan CR, Qiao J-H, Wilkin DJ, Detrano RC, Shah PK and
Rajavashisth TB: Calcification in atherosclerosis: bone biology and
chronic inflammation at the arterial crossroads. Proc Natl Acad Sci
USA. 100:11201–11206. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Parkash J, Chaudhry MA and Rhoten WB:
Calbindin-D28k and calcium sensing receptor cooperate in MCF-7
human breast cancer cells. Int J Oncol. 24:1111–1119.
2004.PubMed/NCBI
|
|
9.
|
Sanders JL, Chattopadhyay N, Kifor O,
Yamaguchi T, Butters RR and Brown EM: Extracellular calcium-sensing
receptor expression and its potential role in regulating
parathyroid hormone-related peptide secretion in human breast
cancer cell lines. Endocrinology. 141:4357–4364. 2000.
|
|
10.
|
Schafer B, Gschwind A and Ullrich A:
Multiple G-protein receptor signals converge on the epidermal
growth factor receptor to promote migration and invasion. Oncogene.
23:991–999. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chang SH, Ai Y, Breyer RM, Lane TF and Hla
T: The prostaglandin E2 receptor EP2 is required for cyclooxygenase
2-mediated mammary hyperplasia. Cancer Res. 65:4496–4499. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hiraga T, Myoui A, Choi ME, Yoshikawa H
and Yoneda T: Stimulation of cyclooxygenase-2 expression by
bone-derived transforming growth factor-β enhances bone metastases
in breast cancer. Cancer Res. 66:2067–2073. 2006.
|
|
13.
|
Mazhar D, Ang R and Waxman J: COX
inhibitors and breast cancer. Br J Cancer. 94:346–350. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Bresalier RS, Sandler RS, Quan H, et al:
Cardiovascular events associated with rofecoxib in a colorectal
adenoma chemoprevention trial. N Engl J Med. 352:1092–1102. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fulton AM, Ma X and Kundu N: Targeting
prostaglandin E EP receptors to inhibit metastasis. Cancer Res.
66:9794–9797. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Dorsam RT and Gutkind JS: G-protein
coupled receptors and cancer. Nat Rev. 7:79–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Pai R, Soreghan B, Szabo IL, Pavelka M,
Baatar D and Tarnawski AS: Prostaglandin E2 transactivates EGF
receptor: a novel mechanism for promoting colon cancer growth and
gastrointestinal hypertrophy. Nat Med. 8:289–293. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Willmarth NE, Baillo A, Dziubinski ML,
Wilson K, Riese DJ II and Ethier SP: Altered EGFR localization and
degradation in human breast cancer cells with an amphiregulin/EGFR
autocrine loop. Cell Signal. 21:212–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Doherty TM, Asotra K, Pei D, Uzui H,
Wilkin DJ, Shah PK and Rajavashisth TB: Therapeutic developments in
matrix metal-loproteinase inhibition. Expert Opinion Therapeutic
Patents. 12:665–707. 2002. View Article : Google Scholar
|
|
20.
|
Brown EM and MacLeod RJ: Extracellular
calcium sensing and extracellular calcium signaling. Physiol Rev.
81:239–297. 2001.PubMed/NCBI
|
|
21.
|
Lindemann RK, Braig M, Hauser CA, Nordheim
A and Dittmer J: Ets2 and protein kinase C epsilon are important
regulators of parathyroid hormone-related protein expression in
MCF-7 breast cancer cells. Biochem J. 372:787–797. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Nemeth EF, Delmar EG, Heaton WL, et al:
Calcilytic compounds: potent and selective Ca2+ –
receptor antagonists that stimulate secretion of parathyroid
hormone. J Pharmacol Exp Ther. 299:323–331. 2001.PubMed/NCBI
|
|
23.
|
Brown EM: Clinical lessons from the
calcium-sensing receptor. Nat Clin Pract Endocrinol Metab.
3:122–133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Mihai R, Stevens J, McKinney C and Ibrahim
NB: Expression of the calcium receptor in human breast cancer – a
potential new marker predicting the risk of bone metastases. Eur J
Surg Oncol. 32:511–515. 2006.
|
|
25.
|
Woodward DF, Pepperl DJ, Burkey TH and
Regan JW: 6-Isopropoxy-9-oxoanthene-2-carboxylic acid (AH6809), a
human EP2 receptor antagonist. Biochem Pharmacol. 50:1731–1733.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Silva J, Chang K, Hannon GJ and Rivas FV:
RNA-interference-based functional genomics in mammalian cells:
reverse genetics coming of age. Oncogene. 23:8401–8409. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Paddison PJ, Caudy AA, Bernstein E, Hannon
GJ and Conklin DS: Short hairpin RNAs (shRNAs) induce
sequence-specific silencing in mammalian cells. Genes Dev.
16:948–958. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Parkash J, Chaudhry MA and Rhoten WB:
Tumor necrosis factor-α induced changes in insulin-producing
β-cells. Anatom Rec A Discov Mol Cell Evol Biol. 286A:982–993.
2005.
|
|
29.
|
Parkash J, Felty Q and Roy D: Estrogen
exerts a spatial and temporal influence on reactive oxygen species
generation that precedes calcium uptake in high capacity
mitochondria: implications for rapid nongenomic signaling of cell
growth. Biochemistry. 45:2872–2881. 2006. View Article : Google Scholar
|
|
30.
|
Gupta GP, Nguyen DX, Chiang AC, et al:
Mediators of vascular remodelling co-opted for sequential steps in
lung metastasis. Nature. 446:765–770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Dhurjati R, Krishnan V, Shuman LA, Mastro
AM and Vogler EA: Metastatic breast cancer cells colonize and
degrade three-dimensional osteoblastic tissue in vitro. Clin Exp
Metastasis. 25:741–752. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Fisher OM, Hart S, Gshwind A and Ullrich
A: EGFR signal trans-activation in cancer cells. Biochem Soc Trans.
31:1203–1208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Prenzel N, Zwick E, Daub H, Leserer M,
Abraham R, Wallasch C and Ullrich A: EGF receptor transactivation
by G-protein-coupled receptors requires metalloproteinase cleavage
of proHB-EGF. Nature. 402:884–888. 1999.PubMed/NCBI
|
|
34.
|
Gilmore JL, Scott JA, Bouizar Z, Robling
A, Pitfield SE, Riese DJ II and Foley J: Amphiregulin-EGFR
signaling regulates PTHrP gene expression in breast cancer cells.
Breast Cancer Res Treat. 110:493–505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
MacLeod RJ, Yano S, Chattopadhyay N and
Brown EM: Extracellular calcium-sensing receptor transactivates the
epidermal growth factor receptor by a triple-membrane-spanning
signaling mechanism. Biochem Biophys Res Commun. 320:455–460. 2004.
View Article : Google Scholar
|














