Introduction
Acute myocardial infarction (AMI) is among the most
severe cardiovascular diseases and the leading cause of mortality
in developed countries (1). The risk
of mortality in those who have suffered from non-ST elevation
myocardial infarctions is ~10% in the developed world (2). In addition, >3 million people suffer
from ST elevation myocardial infarctions and 4 million from non-ST
elevation myocardial infarction each year worldwide (3). An intense inflammatory response is a
crucial phenomenon in AMI. Increased numbers of inflammatory cells
are recruited to the heart following myocardial infarction, which
impacts ventricular function and remodeling.
The bromodomain and extra-terminal (BET) family is
characterized by one (in plants) or two (in animals/yeast)
N-terminal bromodomains (BRDs) and an extra-terminal domain assumed
to function as a protein-protein interaction motif. BET proteins
associate with chromatin, participating in the modulation of the
chromatin structure and influencing transcription (4,5). BET
members are implicated in a number of developmental and disease
contexts, including spermatogenesis (6), human immunodeficiency virus infection
(7), inflammatory diseases (8) and carcinogenesis (9,10).
Previous studies have indicated a central role for BET proteins in
gene control during heart failure pathogenesis (11,12). JQ1
is a potent BRD selective inhibitor that suppresses BET domain
family activity, and is widely used in laboratory research to probe
BET function in various diseases (10,13,14). BET
inhibition potently suppresses cardiomyocyte hypertrophy in
vitro and pathological cardiac remodeling in vivo
(15). However, the role of BET
proteins in AMI and the underlying mechanism of this involvement
remain unclear.
The aim of the present study was to evaluate the
potential of BET proteins as signal-responsive regulators of AMI.
Furthermore, the study investigated whether Toll-like receptor 4
(TLR4) signaling was involved in the mediation of BET protein
function.
Materials and methods
AMI rat model
A total of 30 adult Sprague-Dawley rats (age, 8–10
weeks; weight, 250–300 g) were provided by the Shanghai Laboratory
Animal Centre (Shanghai, China). This study was approved by the
Ethics Committee of Zhengzhou Central Hospital Affiliated to
Zhengzhou University (Zhengzhou, China). The investigation was
performed in accordance with the Guide for the Care and Use of
Laboratory Animals published by the National Institutes of Health
(eighth edition; 2011). The rats were randomly divided into three
groups, including the sham, AMI and AMI + JQ1 groups (n=8 per
group). The experimental AMI model was induced by the ligation of
left coronary artery in adult Sprague-Dawley rats, as previously
described (16). Briefly, the rats
were anesthetized with 80 mg/kg ketamine (Eurovet Animal Health
B.V., Bladel, Netherlands) and 5 mg/kg xylazine (Bayer AG,
Leverkusen, Germany), intraperitoneally. AMI was induced by
occlusion of the left anterior descending coronary artery using a
silk suture (Huaiyin Medical Instruments Co., Ltd., Huaian, China).
A successful intervention was demonstrated by elevated ST segments
on an electrocardiogram (MAC 1600; GE Healthcare, Pittsburgh, PA,
USA). The left ventricular end-diastolic dimension (LVEDd), left
ventricular end-systolic dimension (LVESd), ejection fraction (EF)
and fraction shortening (FS) indices of cardiac function were also
measured using an electrocardiogram. In the sham group, rats were
exposed to all surgical procedures with the exception of the
occlusion of the anterior descending coronary artery. JQ1 was
synthesized by Selleck Chemicals (Houston, TX, USA). Rats in the
AMI + JQ1 group were intraperitoneally injected with 50 mg/kg JQ1
at 12 h prior to and 12 h after AMI. At 24 h following the surgical
intervention, the rats were sacrificed by an intraperitoneal
injection of pentobarbital (200 mg/kg; Sigma-Aldrich, St. Louis,
MO, USA). Blood samples (~6 ml) were collected from the common
carotid artery and stored at −20°C, and the rat hearts were excised
and stored at −80°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the rat myocardial
tissues using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Carlsbad, CA, USA). Next, 2 µg total RNA was reverse
transcribed into cDNA using a RevertAid™ First Strand cDNA
Synthesis kit (Fermentas; Thermo Fisher Scientific, Vilnius,
Lithuania) according to the manufacturer's instructions. qPCR was
performed in an Applied Biosystems Prism 7300 Sequence Detection
system (Thermo Fisher Scientific, Inc., Foster City, CA, USA) using
a SYBR Green PCR kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Briefly, the PCR reaction mix (final volume, 25 µl)
consisted of: 1 µl cDNA, 100 nM primers and 12.5 µl 2X SYBR Green
PCR Master Mix. The PCR cycling conditions were as follows: Initial
denaturation at 94°C for 3 min, followed by 40 cycles of
denaturation at 94°C for 30 sec, annealing at 60°C for 30 sec and
elongation at 72°C for 45 sec, and a final elongation step at 72°C
for 5 min. The primers were as follows: BRD2, forward 5′-AGC ACT
GTC AAG CGG AAGAT-3′, reverse 5′-GGC AAG GCA GTA GAG ACAGG-3′;
BRD3, forward 5′-AAG ATG GTG AGG TCC CACAG-3′, reverse 5′-GGT ACT
CAC GGC TGT CCATT-3′; BDR4, forward 5′-ACA GCC CCA ACA GAA
CAAAC-3′, reverse 5′-GCT GGT TCC TTC TTG CTCAC-3′; and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward 5′-CTC
ATG ACC ACA GTC CATGC-3′ and reverse 5′-TTC AGC TCT GGG ATG
ACCTT-3′ (Sangon Biotech Co., Ltd., Shanghai, China). The relative
quantities of each mRNA were calculated using the 2-ΔΔCt
method (SDS Software version 1.4.1; Applied Biosystems; Thermo
Fisher Scientific, Inc.) with GAPDH as an internal control.
Western blot analysis
Total protein was lysed from tissues using ice-cold
lysis buffer containing 50 mM Tris-HCl (pH 7.5; Amresco LLC, Solon,
OH, USA), 150 mM NaCl (Dingshengxin Company, Tianjin, China), 1%
Nonidet P-40 (Amresco LLC), 1 mM sodium orthovanadate (Beyotime
Institute of Biotechnology, Shanghai, China), 1 mM dithiothreitol
(Amresco LLC), 0.2% SDS (Sigma-Aldrich) and 1 mM
phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology). Following centrifugation at 13,000 × g for 1 min,
protein concentrations were detected using a Bicinchoninic Protein
Assay kit (Beyotime Institute of Biotechnology). Subsequently, the
protein samples were separated on a 12% SDS-polyacrylamide gel via
electrophoresis and transferred to polyvinylidene difluoride
membranes (EMD Millipore, Billerica, MA, USA). The membranes were
incubated in blocking solution (Tris-buffered saline containing 5%
nonfat milk and 0.05% Tween 20) at 4°C overnight. The blot was then
probed with the following primary antibodies: Polyclonal rabbit
anti-TLR4 (1:500; cat. no. sc-30002), polyclonal goat anti-TNF
receptor-associated factor 6 (TRAF6; 1:400; cat. no. sc-33897),
monoclonal mouse anti-nuclear factor (NF)-κB (1:800; cat. no.
sc-8008) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
polyclonal goat anti-BRD2 (1:400; cat. no. ab3718), polyclonal
rabbit anti-BRD3 (1:200; cat. no. ab83478) and polyclonal rabbit
anti-BRD4 (1:400; cat. no. ab75898) (Abcam, Cambridge, MA, USA) at
37°C for 2 h. Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies: Goat
anti-rabbit immunoglobulin (Ig)G (1:10,000; cat. no. sc-2004),
rabbit anti-goat IgG (1:10,000; cat. no. sc-2768) and goat
anti-mouse IgG (1:10,000; cat. no. sc-2031) (Santa Cruz
Biotechnology, Inc.) at 37°C for 1 h. The signal was detected using
an enhanced chemiluminescence western blotting kit (Pierce
Biotechnology, Inc., Rockford, IL, USA). The intensity of the bands
was quantified using a ScanJet 4C Flatbed Scanner (Hewlett-Packard,
Palo Alto, CA, USA) with National Institutes of Health Image v1.52
software (http://rsb.info.nih.gov/nih-image).
Lactate dehydrogenase (LDH) and
creatine kinase-MB (CK-MB) isozyme measurement
Blood samples were centrifuged at 1,500 × g for 15
min at 4°C, and then the serum was separated and stored at −80°C
until analyzed. An LDH assay kit was purchased from Jiancheng
Bioengineering Institute (Nanjing, China) and a CK-MB isozyme assay
kit was obtained from Ningbo Medical System Biotechnology Co., Ltd.
(Ningbo, China). LDH and CK-MB activities in the serum were
determined according to the manufacturer's instructions.
High-sensitivity C-reactive protein
(hs-CRP) and interleukin (IL)-6 measurement
An hs-CRP assay kit was purchased from Shanghai
ExCell Biology, Inc. (Shanghai, China) and the serum hs-CRP
concentration was quantified using a Cobas Integra 400 Plus®
Automatic Chemistry analyzer (Roche Diagnostics, Basel,
Switzerland) using an immunoturbidimetric method. The IL-6 levels
in the myocardial tissue were determined using enzyme-linked
immunosorbent assay kits obtained from eBioscience, Inc. (San
Diego, CA, USA), following the manufacturer's instructions.
Statistical analysis
The results were analyzed using SPSS statistical
software, version 19.0 (IBM SPSS, Armonk, NY, USA) and all the data
are expressed as the mean ± standard deviation. Student's t-test
was used for comparison of differences between the groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of BET domain family inhibition
on cardiac function in the cardiomyocytes
Following the establishment of the AMI model, it was
observed that the LVEDd and LVESd were significantly increased,
while the EF and FS were significantly decreased. Subsequently,
JQ1, a well-known selective BRD inhibitor, was used to investigate
the effect of BET inhibition on cardiac function in the established
AMI model. The results indicated that LVEDd and LVESd were
significantly decreased, while EF and FS were significantly
increased, in the AMI + JQ1 group compared with the AMI group
(P<0.05; Table I).
 | Table I.Indices of cardiac function in the
three groups. |
Table I.
Indices of cardiac function in the
three groups.
| Parameter | Sham | AMI | AMI + JQ1 |
|---|
| LVEDd (mm) |
5.62±0.09 |
9.38±0.14a |
6.22±0.11b |
| LVESd (mm) |
2.21±0.04 |
7.61±0.11a |
4.15±0.06b |
| EF (%) | 75.1±6.2 | 34.2±2.1a | 48.3±4.9b |
| FS (%) | 45.2±3.6 | 14.2±1.1a | 36.3±2.8b |
Expression of BET domain family in the
cardiomyocytes
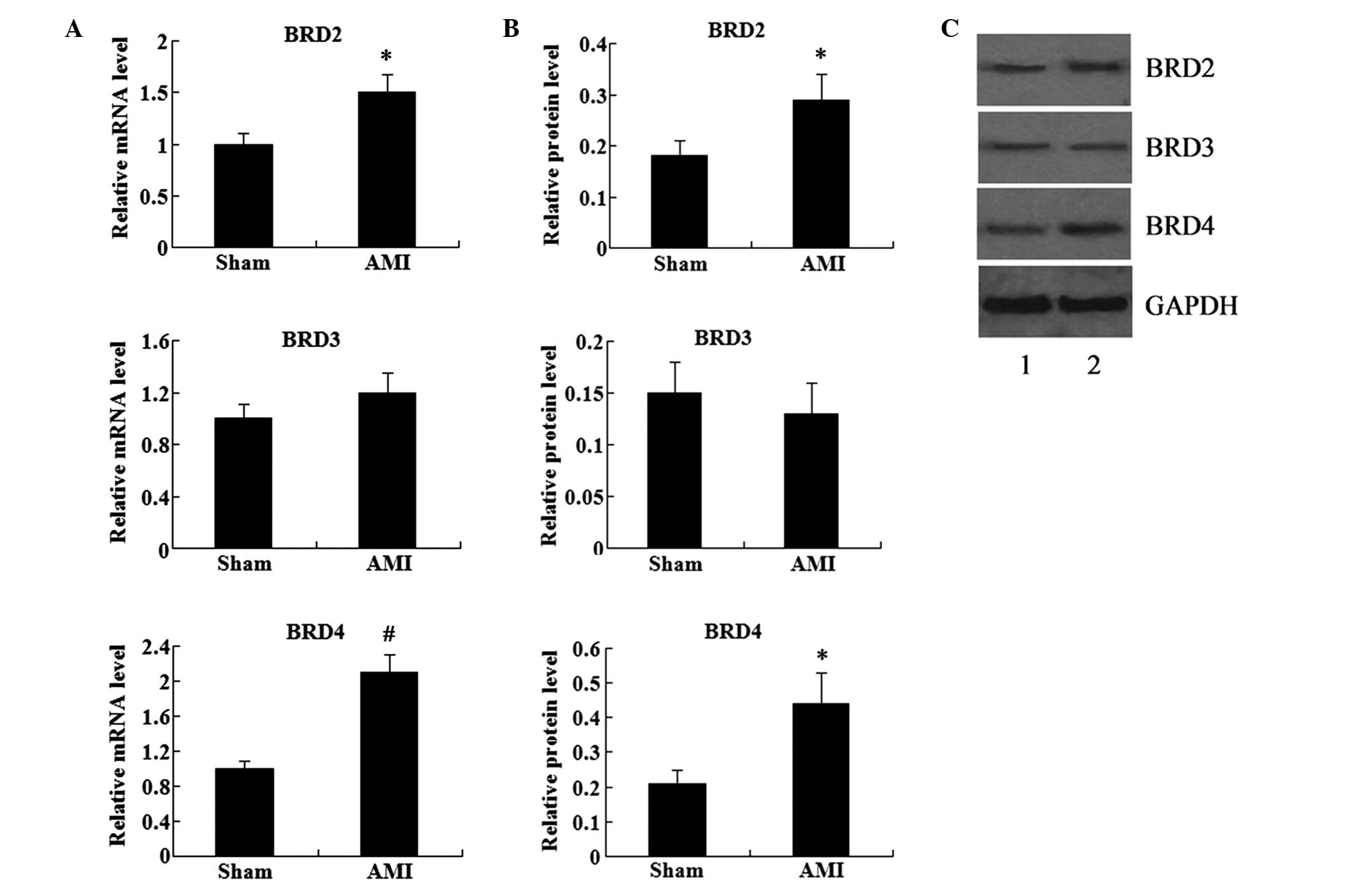
To detect the expression of BETs in the myocardial
tissue, RT-qPCR and western blot analysis were used to determine
the mRNA and protein expression levels, respectively, of BRD2, BRD3
and BRD4. The results showed that the mRNA and protein expression
levels of BRD2, BRD3 and BRD4 were detectable in the myocardial
tissue (Fig. 1). Compared with the
sham group, the mRNA and protein expression levels of BRD2 and BRD4
were significantly increased in the AMI group. However, BRD3
expression was not found to be significantly different in the AMI
group compared with that in the sham group (P>0.05; Fig. 1).
Effect of BET domain family inhibition
on LDH and CK-MB activity in the cardiomyocytes
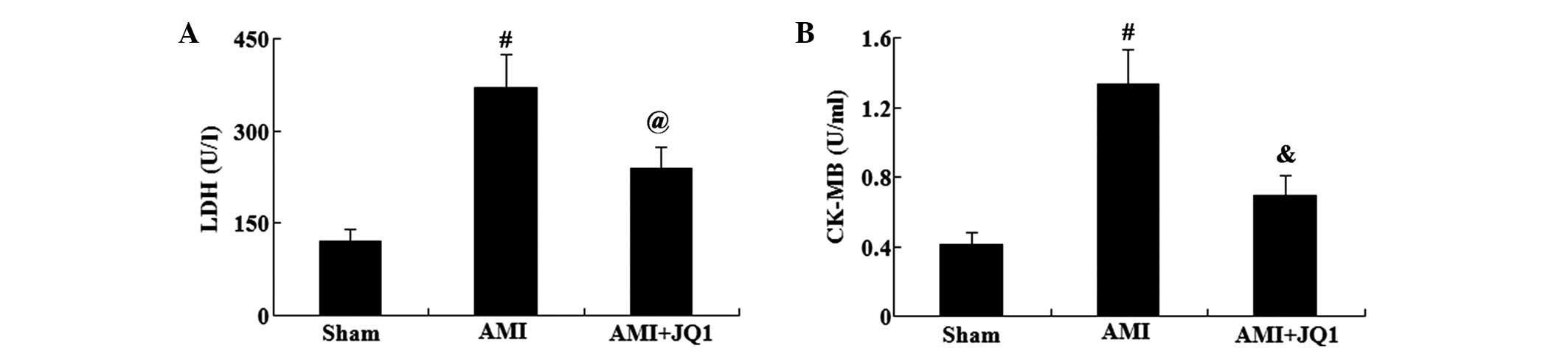
The myocardial enzymes, LDH and CK-MB, are
indicators of myocardial damage following AMI. Serum activities of
LDH and CK-MB were evaluated using commercially available kits
(Fig. 2). Compared with the sham
group, the serum activities of LDH and CK-MB were significantly
elevated in the AMI group (P<0.01). However, JQ1 treatment was
found to significantly reduce the serum activities of LDH
(P<0.05) and CK-MB (P<0.01) in rats with AMI.
Effect of BET domain family inhibition
on inflammatory reaction in the cardiomyocytes
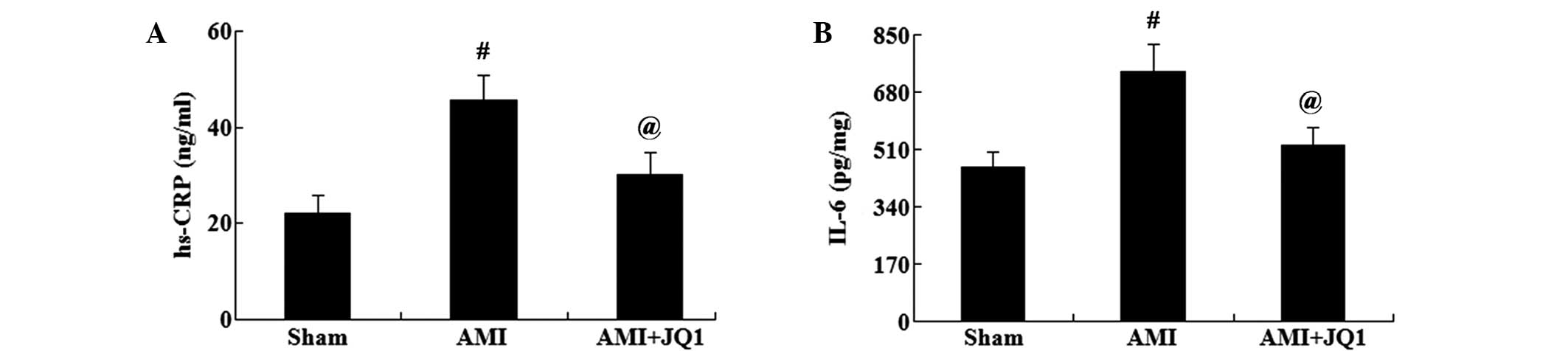
To assess the effect of BETs inhibition on the
inflammatory response, the levels of hs-CRP and IL-6 were
determined using commercially available kits. As shown in Fig. 3, the levels of hs-CRP in the serum
and of IL-6 in the myocardial tissue were significantly increased
in the AMI group compared with those in the sham group (P<0.01).
However, the levels of hs-CRP and IL-6 were significantly decreased
in the AMI + JQ1 group compared with those in the AMI group
(P<0.05).
Effect of BET domain family inhibition
on the protein expression levels of TLR4/TRAF6/NF-κB in rats with
AMI
Western blot analysis was used to determine the
association between the BET domain family and the expression of
TLR4/TRAF6/NF-κB in the cardiomyocytes of rats with AMI. As shown
in Fig. 4, the protein expression
levels of TLR4, TRAF6 and NF-κB (P<0.01, P<0.01 and
P<0.05, respectively) were significantly increased in the AMI
group compared with those in the sham group. By contrast, the
administration of JQ1 significantly reduced the protein expression
levels of TLR4, TRAF6 and NF-κB (P<0.01, P<0.05 and
P<0.05, respectively) in rats with AMI.
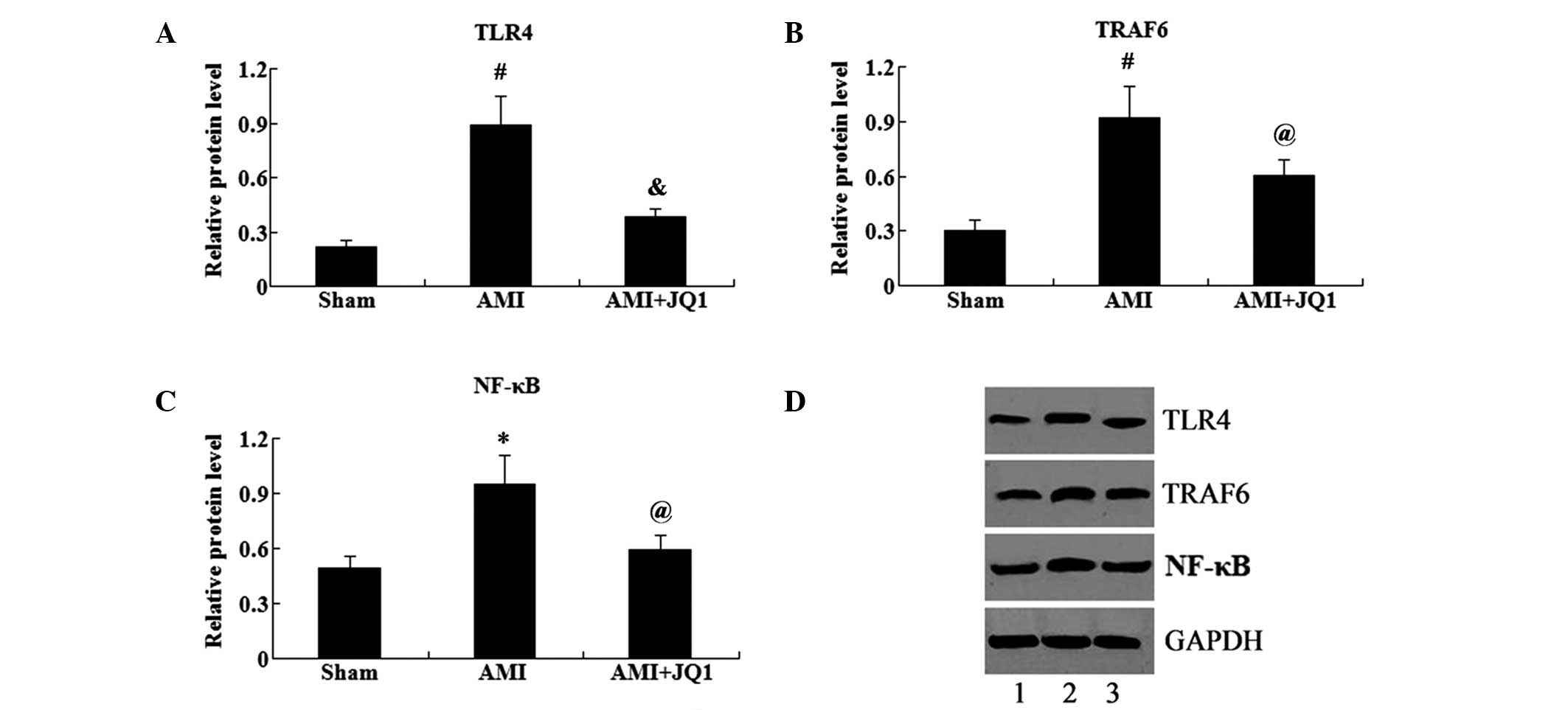 | Figure 4.Effect of BET domain family inhibition
on the expression of TLR4/TRAF6/NF-κB in cardiomyocytes. Relative
protein expression levels of (A) TLR4, (B) TRAF6 and (C) NF-κB in
cardiomyocytes in the sham, AMI and AMI + JQ1 groups are shown. (D)
Western blot, showing the protein expression in the sham (lane 1),
AMI (lane 2) and AMI + JQ1 groups. *P<0.05 and
#P<0.01 vs. sham group; @P<0.05 and
&P<0.01 vs. AMI group. BET, bromodomain and
extra-terminal; AMI, acute myocardial infarction; TLR4, Toll-like
receptor 4; TRAF6, TNF receptor-associated factor 6; NF-κB, nuclear
factor-κB; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
Discussion
The acetylation of N-terminal histone tails serves a
crucial function in gene regulation (17). Members of the BET family of
bromodomain (BRD)-containing reader proteins, including BRD2, BRD3,
BRD4 and testis-specific BRDT, bind to acetylated lysine residues
on histone and nonhistone proteins, and recruit transcriptional
regulators, leading to the activation or repression of gene
transcription (18–20). In the present study, RT-qPCR and
western blot analysis were performed to determine the mRNA and
protein expression levels, respectively, of the BET family members
BRD2, BRD3 and BRD4 in rat myocardial tissue. The results
demonstrated that the mRNA and protein expression levels of BRD2,
BRD3 and BRD4 were detectable in the myocardial tissue.
Furthermore, a rat model of AMI was established, and the results
showed that BRD2 and BRD4 expression was significantly increased in
the AMI group compared with the sham group. However, BRD3
expression was not altered between the AMI and sham groups. These
results indicated that BRD2 and BRD4 proteins were associated with
AMI, whereas BRD3 may not be associated with AMI.
JQ1 is a well-known selective BRD inhibitor that
competes with members of the BET family of BRDs by displacing BET
BRDs from chromatin and resulting in the suppression of downstream
signaling events to RNA polymerase II (Pol II) (9,10,21).
Hence, JQ1 was utilized to probe BET function in a rat
cardiomyocyte model of AMI.
Myocardial infarction is the primary cause of the
development of congestive heart failure in the left ventricle.
CK-MB and LDH are sensitive biochemical indicators of myocardial
injury. AMI leads to cardiomyocyte death, which in turn results in
the coordinated activation of a cytokine cascade that initiates an
acute inflammatory reaction (22).
hs-CRP is considered to be a sensitive indicator of inflammation
that is associated with AMI, as increased serum hs-CRP levels have
been detected in post-infarction patients (23). Increased levels of hs-CRP indicate a
poor prognosis in patients following an AMI (24). A previous study indicated that the
serum IL-6 concentrations of the AMI group were significantly
higher compared with the control group (25). JQ1 is a selective BRD inhibitor. In
the present study, a model of AMI was established in rats, and it
was observed that treatment with JQ1 reversed cardiac function
injury, decreased serum LDH and CK-MB activities, and decreased the
expression levels of hs-CRP and IL-6. These findings suggested that
BET inhibition may potently suppress myocardial infarction in
vivo.
Toll-like receptors (TLRs) are germline-encoded
pattern recognition receptors that are involved in the initiation
of innate immune responses (26–28).
TLR4 is a key member of the TLR family that may be activated by
lipopolysaccharide and nonbacterial agonists (29,30).
Furthermore, TLR4 is localized on the cell surface and mediates
transmembrane signaling transduction. Previous studies demonstrated
that AMI is accompanied by the activation of TLR4 in circulating
blood cells (31,32). In animal models, TLR4 deficiency or
signaling inhibition have been shown to reduce inflammatory cell
influx into the infarcted area, ameliorate cardiac remodeling and
decrease infarct size (33–35). In addition, TLR pathway activation
was indicated by increased expression of TLR4 in patients with
acute ST elevation myocardial infarction (36). The activation of TLR4 signaling leads
to the synthesis and release of cytokines and inflammatory
mediators. For instance, the nuclear factor (NF)-κB pathway has
been suggested to serve a crucial function in TLR4-mediated
inflammatory regulation (37). In
myocardial infarction models, NF-κB activation has been associated
with the remodeling and dysfunction of the myocardium (38). TNF receptor-associated factor 6
(TRAF6) may be activated by TLR4, which in turn activates the
inhibitor of κB kinase, leading to the activation of NF-κB
(39). In the present study, the
involvement of the TLR4/TRAF6/NF-κB pathway in the effect of BET
proteins on AMI was investigated. The results suggested that TLR4
signaling was activated by the increased expression of TLR4, TRAF6
and NF-κB in the cardiomyocytes of rats that underwent AMI.
However, JQ1 treatment suppressed this TLR4 signaling activation.
These results indicate that the regulation of TLR4/TRAF6/NF-κB
signaling by BET family members may underlie the effect of BET
proteins on gene expression following AMI.
In conclusion, the present study demonstrated that
the inhibition of BET family proteins suppresses certain
manifestations of AMI, and that this effect was partially mediated
by the inhibition of the TLR4/TRAF6/NF-κB pathway. These results
suggest that BET proteins may be associated with the pathogenesis
of AMI, and may be a novel target for the treatment of AMI.
References
|
1
|
Ozaki K, Sato H, Inoue K, Tsunoda T,
Sakata Y, Mizuno H, Lin TH, Miyamoto Y, Aoki A, Onouchi Y, et al:
SNPs in BRAP associated with risk of myocardial infarction in Asian
populations. Nat Genet. 41:329–333. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Task Force on the management of ST-segment
elevation acute myocardial infarction of the European Society of
Cardiology (ESC). Steg PG, James SK, Atar D, Badano LP,
Blömstrom-Lundqvist C, Borger MA, Di Mario C, Dickstein K, Ducrocq
G, Fernandez-Aviles F, et al: ESC Guidelines for the management of
acute myocardial infarction in patients presenting with ST-segment
elevation. Eur Heart J. 33:2569–2619. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White HD and Chew DP: Acute myocardial
infarction. Lancet. 372:570–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Florence B and Faller DV: You bet-cha: A
novel family of transcriptional regulators. Front Biosci.
6:D1008–D1018. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Florence BL and Faller DV: Drosophila
female sterile (1) homeotic is a multifunctional transcriptional
regulator that is modulated by Ras signaling. Dev Dyn. 237:554–564.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matzuk MM, McKeown MR, Filippakopoulos P,
Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, et al:
Small-molecule inhibition of BRDT for male contraception. Cell.
150:673–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Banerjee C, Archin N, Michaels D, Belkina
AC, Denis GV, Bradner J, Sebastiani P, Margolis DM and Montano M:
BET bromodomain inhibition as a novel strategy for reactivation of
HIV-1. J Leukoc Biol. 92:1147–1154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nicodeme E, Jeffrey KL, Schaefer U, Beinke
S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H,
et al: Suppression of inflammation by a synthetic histone mimic.
Nature. 468:1119–1123. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB,
Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et
al: BET bromodomain inhibition as a therapeutic strategy to target
c-Myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Filippakopoulos P, Qi J, Picaud S, Shen Y,
Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et
al: Selective inhibition of BET bromodomains. Nature.
468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anand P, Brown JD, Lin CY, Qi J, Zhang R,
Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, et al: BET
bromodomains mediate transcriptional pause release in heart
failure. Cell. 154:569–582. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haldar SM and McKinsey TA: BET-ting on
chromatin-based therapeutics for heart failure. J Mol Cell Cardiol.
74:98–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB,
Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et
al: BET bromodomain inhibition as a therapeutic strategy to target
c-Myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang X, Peng R, Ren Y, Apparsundaram S,
Deguzman J, Bauer CM, Hoffman AE, Hamilton S, Liang Z, Zeng H, et
al: BET bromodomain proteins mediate downstream signaling events
following growth factor stimulation in human lung fibroblasts and
are involved in bleomycin-induced pulmonary fibrosis. Mol
Pharmacol. 83:283–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spiltoir J, Stratton MS, Cavasin MA,
Demos-Davies K, Reid BG, Qi J, Bradner JE and McKinsey TA: BET
acetyl-lysine binding proteins control pathological cardiac
hypertrophy. J Mol Cell Cardiol. 63:175–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eckle T, Grenz A, Köhler D, Redel A, Falk
M, Rolauffs B, Osswald H, Kehl F and Eltzschig HK: Systematic
evaluation of a novel model for cardiac ischemic preconditioning in
mice. Am J Physiol Heart Circ Physiol. 291:H2533–H2540. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marushige K: Activation of chromatin by
acetylation of histone side chains. Proc Natl Acad Sci USA.
73:3937–3941. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dhalluin C, Carlson JE, Zeng L, He C,
Aggarwal AK and Zhou MM: Structure and ligand of a histone
acetyltransferase bromodomain. Nature. 399:491–496. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jacobson RH, Ladurner AG, King DS and
Tjian R: Structure and function of a human TAFII250 double
bromodomain module. Science. 288:1422–1425. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans
PR, Ballario P, Neuhaus D, Filetici P and Travers AA: The
structural basis for the recognition of acetylated histone H4 by
the bromodomain of histone acetyltransferase gcn5p. EMBO J.
19:6141–6149. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chung CW, Coste H, White JH, Mirguet O,
Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, et
al: Discovery and characterization of small molecule inhibitors of
the BET family bromodomains. J Med Chem. 54:3827–3838. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frangogiannis NG, Smith CW and Entman ML:
The inflammatory response in myocardial infarction. Cardiovasc Res.
53:31–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Freitas F, Brucker N, Durgante J, Bubols
G, Bulcão R, Moro A, Charão M, Baierle M, Nascimento S, Gauer B, et
al: Urinary 1-hydroxypyrene is associated with oxidative stress and
inflammatory biomarkers in acute myocardial infarction. Int J
Environ Res Public Health. 11:9024–9037. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang YU, Kim MJ, Choi JS, Kim CS, Bae EH,
Ma SK, Ahn YK, Jeong MH, Kim YJ, Cho MC, et al: Other Korea Acute
Myocardial Infarction Registry Investigators: Concomitant impact of
high-sensitivity C-reactive protein and renal dysfunction in
patients with acute myocardial infarction. Yonsei Med J.
55:132–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang XH, Liu SQ, Wang YL and Jin Y:
Correlation of serum high-sensitivity C-reactive protein and
interleukin-6 in patients with acute coronary syndrome. Genet Mol
Res. 13:4260–4266. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beutler B: Inferences, questions and
possibilities in Toll-like receptor signalling. Nature.
430:257–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Poltorak A, He X, Smirnova I, Liu MY, Van
Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al:
Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations
in Tlr4 gene. Science. 282:2085–2088. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee JY, Ye J, Gao Z, Liu MY, Van Huffel C,
Du X, Birdwell D, Alejos E, Silva M and Galanos C: Reciprocal
modulation of Toll-like receptor-4 signaling pathways involving
MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and
polyunsaturated fatty acids. J Biol Chem. 278:37041–37051. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arslan F, de Kleijn DP and Pasterkamp G:
Innate immune signaling in cardiac ischemia. Nat Rev Cardiol.
8:292–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Timmers L, Pasterkamp G, de Hoog VC,
Arslan F, Appelman Y and de Kleijn DP: The innate immune response
in reperfused myocardium. Cardiovasc Res. 94:276–283. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chong AJ, Shimamoto A, Hampton CR,
Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH and Verrier
ED: Toll-like receptor 4 mediates ischemia/reperfusion injury of
the heart. J Thorac Cardiovasc Surg. 128:170–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Timmers L, Sluijter JP, van Keulen JK,
Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ,
Joles JA, Quax PH, et al: Toll-like receptor 4 mediates maladaptive
left ventricular remodeling and impairs cardiac function after
myocardial infarction. Circ Res. 102:257–264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shimamoto A, Chong AJ, Yada M, Shomura S,
Takayama H, Fleisig AJ, Agnew ML, Hampton CR, Rothnie CL, Spring
DJ, et al: Inhibition of toll-like receptor 4 with eritoran
attenuates myocardial ischemia-reperfusion injury. Circulation.
114(Suppl 1): S1270–S1274. 2006.
|
|
36
|
van der Pouw Kraan TC, Bernink FJ,
Yildirim C, Koolwijk P, Baggen JM, Timmers L, Beek AM, Diamant M,
Chen WJ, van Rossum AC, et al: Systemic toll-like receptor and
interleukin-18 pathway activation in patients with acute ST
elevation myocardial infarction. J Mol Cell Cardiol. 67:94–102.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Akira S and Takeda K: Toll-like receptor
signaling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Timmers L, van Keulen JK, Hoefer IE, Meijs
MF, van Middelaar B, den Ouden K, van Echteld CJ, Pasterkamp G and
de Kleijn DP: Targeted deletion of nuclear factor kappaB p50
enhances cardiac remodeling and dysfunction following myocardial
infarction. Circ Res. 104:699–706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yin Q, Lin SC, Lamothe B, Lu M, Lo YC,
Hura G, Zheng L, Rich RL, Campos AD, Myszka DG, et al: E2
interaction and dimerization in the crystal structure of TRAF6. Nat
Struct Mol Biol. 16:658–666. 2009. View Article : Google Scholar : PubMed/NCBI
|