Introduction
Platelets have an important role in primary
hemostasis, thrombus formation and the repair of vascular injury
(1). Platelets are activated by a
diverse range of stimulators, leading to alterations in shape,
adhesion, aggregation, and subsequent thrombus formation. Collagen
is well-documented as a primary stimulator of human platelets
(2,3). Platelets interact with collagen in
subendothelium at the damaged site of the vessel wall. Therefore,
once subendothelium is exposed, platelets rapidly adhere to the
exposed subendothelial collagen, which is characterized by the
interaction of glycoprotein (GP) Ib/IX/V and von Willebrand factor
(1), resulting in aggregation and
hemostasis. GPVI and integrin α2β1 are the predominant collagen
receptors located on the plasma membrane of platelets (2,3). GPVI
forms a complex with the Fc receptor γ-chain (4,5).
Activated GPVI induces the activation of various intracellular
molecules, including phospholipase Cγ2 and tyrosine kinase Syk
(6,7), resulting in the upregulation of
integrin activity (8) and the
enhancement of granule secretion (2). Platelet-derived growth factor
(PDGF)-AB, which is stored in α-granules of human platelets and is
known to exert potent proliferative effects on a variety of cells,
is released from activated platelets and has a pivotal role in
atherosclerosis via the proliferation of connective tissue,
including vascular smooth muscle cells (1).
Expression of heat shock proteins (HSPs) is induced
in response to various biological stresses, including heat,
endotoxins and reactive oxygen species (9). HSPs have been classified into seven
subtypes, including HSPA (HSP70), HSPB (low-molecular-weight HSPs)
and HSPC (HSP90) (10). It is
generally recognized that HSPBs, such as HSP27 and αB-crystallin,
possess chaperoning functions as well as HSPA (HSP70) and HSPC
(HSP90) (10). Furthermore, it has
been demonstrated that the functions of HSP27 are modulated by
post-translational modifications, such as phosphorylation (11,12).
Human HSP27 is phosphorylated at three serine residues: Ser-15,
Ser-78 and Ser-82. Although HSP27 is presented in an aggregated
form under unstimulated conditions, it is rapidly dissociated
following stimulation-responsive phosphorylation, and it has been
demonstrated that this dissociation is necessary for substrate
binding and chaperone function (13). HSP27 reportedly increases cell
viability under various unfavorable conditions, including heat and
oxidative stress (14,15). The phosphorylation of HSP27 in
platelets is known to be catalyzed by members of the
mitogen-activated protein (MAP) kinase superfamily (16). Furthermore, regarding HSP27
phosphorylation in human platelets, we have previously demonstrated
that the collagen-induced phosphorylation of HSP27 via p44/p42 MAP
kinase is sufficient for the secretion of PDGF-AB and the release
of soluble soluble cluster of differentiation 40 ligand (sCD40L)
(17).
Adenosine monophosphate (AMP)-activated protein
kinase (AMPK) has a critical role as a regulator of energy
homeostasis (18). AMPK is activated
under low energy states, including physical exercise, hypoxia and
ischemia, which lead to a decrease in the cellular ATP/AMP ratio.
It has been demonstrated that AMPK is involved in various
physiological signaling pathways associated with the metabolism of
glucose, fat and protein, and various processes, such as cell
proliferation, apoptosis and aging (19). Previous studies have reported that
AMPK is activated by the inhibition of fatty acid synthase,
resulting in the cytotoxicity observed in ovarian cancer cells
(20,21). Therefore, AMPK is considered as a
potential therapeutic target for the treatment of diabetes mellitus
(DM), cancer and obesity. Regarding the antiplatelet effect of AMPK
(22), it has previously been
reported that platelet aggregation is suppressed by
5-aminoimidazole-4-carboxamide-1-β-d-ribofuranosyl 5′-monophosphate
(AICAR), which is an activator of AMPK (23). However, the exact mechanism
underlying the effects of AMPK on human platelet functions are yet
to be clarified.
In the present study, the effects of AICAR on
collagen-induced platelet activation were examined in human
platelets.
Materials and methods
Reagents and materials
Collagen was purchased from Takeda Austria GmbH
(Linz, Austria). AICAR was purchased from Sigma-Aldrich (St. Louis,
MO, USA). PDGF-AB ELISA kit and sCD40L ELISA kit were purchased
from R&D Systems, Inc., (Minneapolis, MN, USA). Phosphorylated
(p)-p44/p42 MAP kinase rabbit anti-human polyclonal antibody (cat.
no. 9101), p44/p42 MAP kinase rabbit anti-human polyclonal antibody
(cat. no. 9102), p-HSP27 (Ser-15) rabbit anti-human polyclonal
antibody (cat. no. 2404), p-HSP27 (Ser-78) rabbit anti-human
polyclonal antibody (cat. no. 2405) and p-HSP27 (Ser-82) rabbit
anti-human polyclonal antibody (cat. no. 2401) were purchased from
Cell Signaling Technology, Inc., (Beverly, MA, USA). HSP27 goat
polyclonal antibodies (cat. no. sc-1049) and GAPDH rabbit
polyclonal antibodies (cat. no. sc-25778) were purchased from Santa
Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). Enhanced
chemiluminescence (ECL) reagent was purchased from GE Healthcare
(Chalfont, UK).
Preparation of platelets
Human blood (10 ml) was donated by healthy
volunteers (mean age, 39.2±9.0 years; mean±standard deviation) and
supplemented with 3.8% sodium citrate (1:10). Platelet-rich plasma
(PRP) was obtained from the blood samples by centrifugation at 155
× g for 12 min at room temperature. Platelet-poor plasma (PPP) was
prepared from residual blood by centrifugation at 1,400 × g for 5
min at room temperature. Written informed consent was obtained from
all participants signed an informed consent following a detailed
explanation of the study. The protocol of the present study was
approved by the Committee of Ethics at Gifu University Graduate
School of Medicine (Gifu, Japan).
Measurement of platelet aggregation
induced by collagen
Platelet aggregation analysis of citrated PRP was
performed using an aggregometer (PA-200; Kowa Co. Ltd., Tokyo,
Japan), which determines the size of platelet aggregates based on a
particle counting laser light scattering system at 37°C with a
stirring speed of 800 rpm. Aggregate sizes were determined as
follows: Small, 9–25 µm; medium, 25–50 µm; and large, 50–70 µm.
Platelets were preincubated for 1 min prior to platelet aggregation
monitoring for 4 min. Percentage transmittance of the isolated
platelets was recorded as 0%, and the appropriate PPP blank was
recorded as 100%. When indicated, PRP was pretreated with AICAR for
15 min.
Measurement of PDGF-AB and sCD40L
levels
Following stimulation by collagen, platelet
aggregation was terminated by the addition of 10 mM ice-cold EDTA
solution and the mixture was subsequently centrifuged at 10,000 × g
for 2 min at 4°C. In order to measure PDGF-AB and sCD40L levels,
the supernatant was isolated and stored at −20°C. PDGF-AB and
sCD40L levels were determined using respective ELISA kits,
according to the manufacturer's protocol.
Western blot analysis
As described, following stimulation by collagen,
platelet aggregation was terminated and the mixture was
subsequently centrifuged at 10,000 × g for 2 min at 4°C. For
western blot analysis, the pellet was washed twice with
phosphate-buffered saline, lysed and immediately boiled in a lysis
buffer containing 62.5 mM Tris/Cl (pH 6.8), 2% sodium dodecyl
sulfate (SDS), 50 mM dithiothreitol and 10% glycerol. Western blot
analysis was performed as previously described (24). Briefly, proteins were separated by
SDS-polyacrylamide gel electrophoresis PAGE according to the
Laemmli method (25) with 10% or 12%
polyacrylamide gel, and were subsequently transferred onto a
polyvinylidine fluoride (PVDF) membranes. Following blocking with
5% fat-free dry milk in Tris-buffered saline with Tween 20 [TBS-T;
20 mM Tris (pH 7.6), 137 mM NaCl, 0.1% Tween 20] for 2 h at room
temperature, the membranes were incubated with the primary
antibodies p-p44/p42 MAP kinase, p44/p42 MAP kinase, p-HSP27
(Ser-15, Ser-78 and Ser-82) and GAPDH at a dilution of 1:1,000 in
5% milk in TBS-T overnight at 4°C. Following washing, the membranes
were incubated with anti-rabbit (cat. no. 7074S; Cell Signaling
Technology, Inc.) or anti-goat (cat. no. sc-2020; Santa Cruz
Biotechnology, Inc.) IgG secondary antibodies at a dilution of
1:1,000 in 5% milk in TBS-T for 1 h at room temperature. Using an
enhanced chemiluminescence western blotting detection system,
peroxidase activity on PVDF membranes was visualized on X-ray film,
according to the manufacturer's protocol. The densitometry of bands
were analyzed by Image J software program. The quantitative data of
each bands were measured as the counts of pixels.
Statistical analysis
Data were analyzed using a paired t-test.
P<0.05 was considered to indicate a statistically significant
difference. All data are presented as the mean ± standard error of
the mean. All statistical analyses were performed using PASW
Statistics software, version 18 (SPSS Japan, Tokyo, Japan).
Results
Effects of AICAR on collagen induced
platelet aggregation
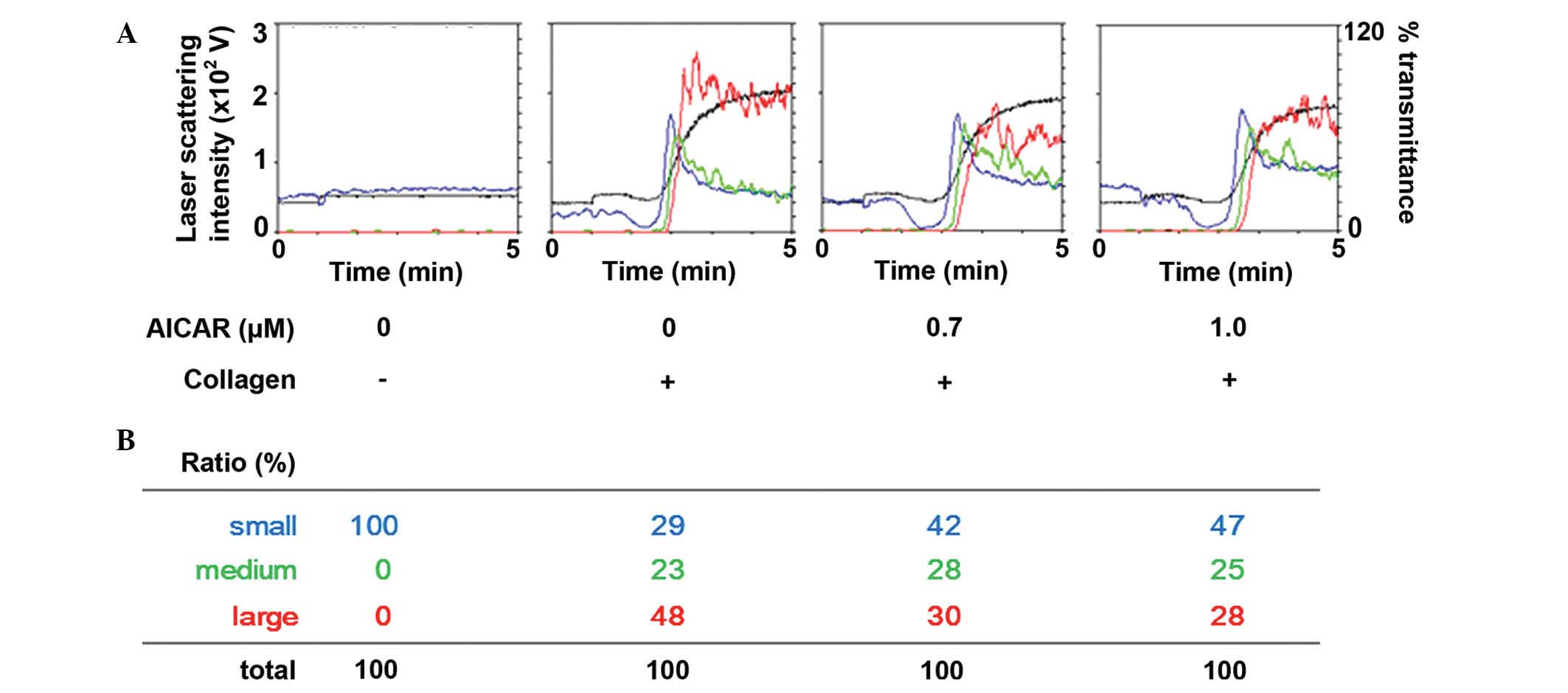
The effect of AICAR on collagen-stimulated platelet
aggregation was determined in human platelets. AICAR administration
markedly reduced the platelet aggregation induced by collagen in a
dose-dependent manner up to 1.0 µM (Fig.
1A). Furthermore, analysis of the size of platelet aggregates
demonstrated that treatment with 1 µm AICAR decreased the ratio of
large aggregates (50–70 µm) from 48 to 28%; whereas the ratio of
small aggregates (9–25 µm) increased from 29 to 47% (Fig. 1B), as compared with the control.
These results suggest that AICAR inhibits collagen-stimulated human
platelet aggregation.
Effects of AICAR on the
phosphorylation of p44/p42 MAP kinase and HSP27 induced by collagen
in human platelets
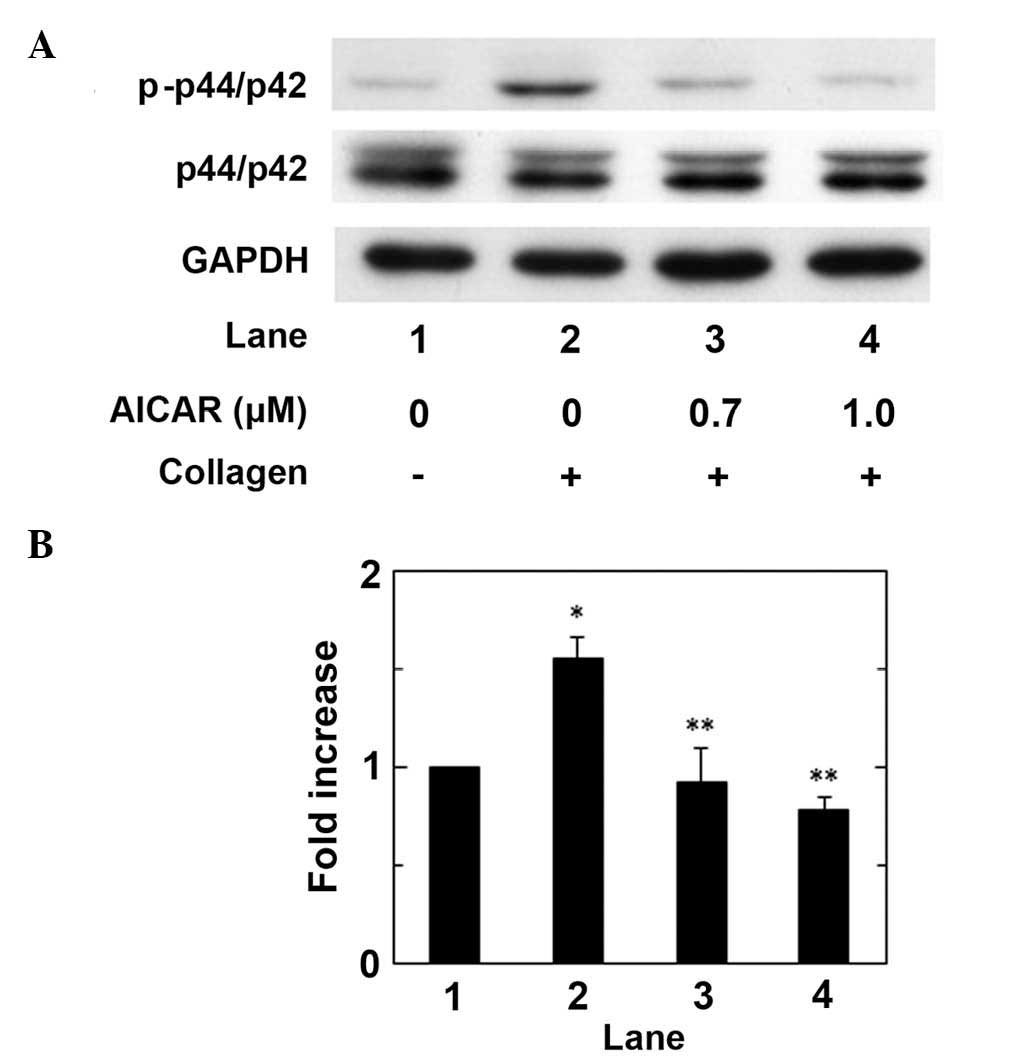
We have previously reported that the
collagen-induced phosphorylation of HSP27 is positively regulated
by the activation of p44/p42 MAP kinase in human platelets
(17). In order to clarify whether
AICAR administration affects the collagen-induced activation of
p44/p42 MAPK in human platelets, the effect of AICAR on the
collagen-induced phosphorylation of p44/p42 MAP kinase was examined
using western blot analysis. AICAR administration significantly
suppressed collagen-stimulated p44/p42 MAP kinase phosphorylation
in a dose-dependent manner up to 1.0 µM, as compared with the
control (P<0.05; Fig. 2).
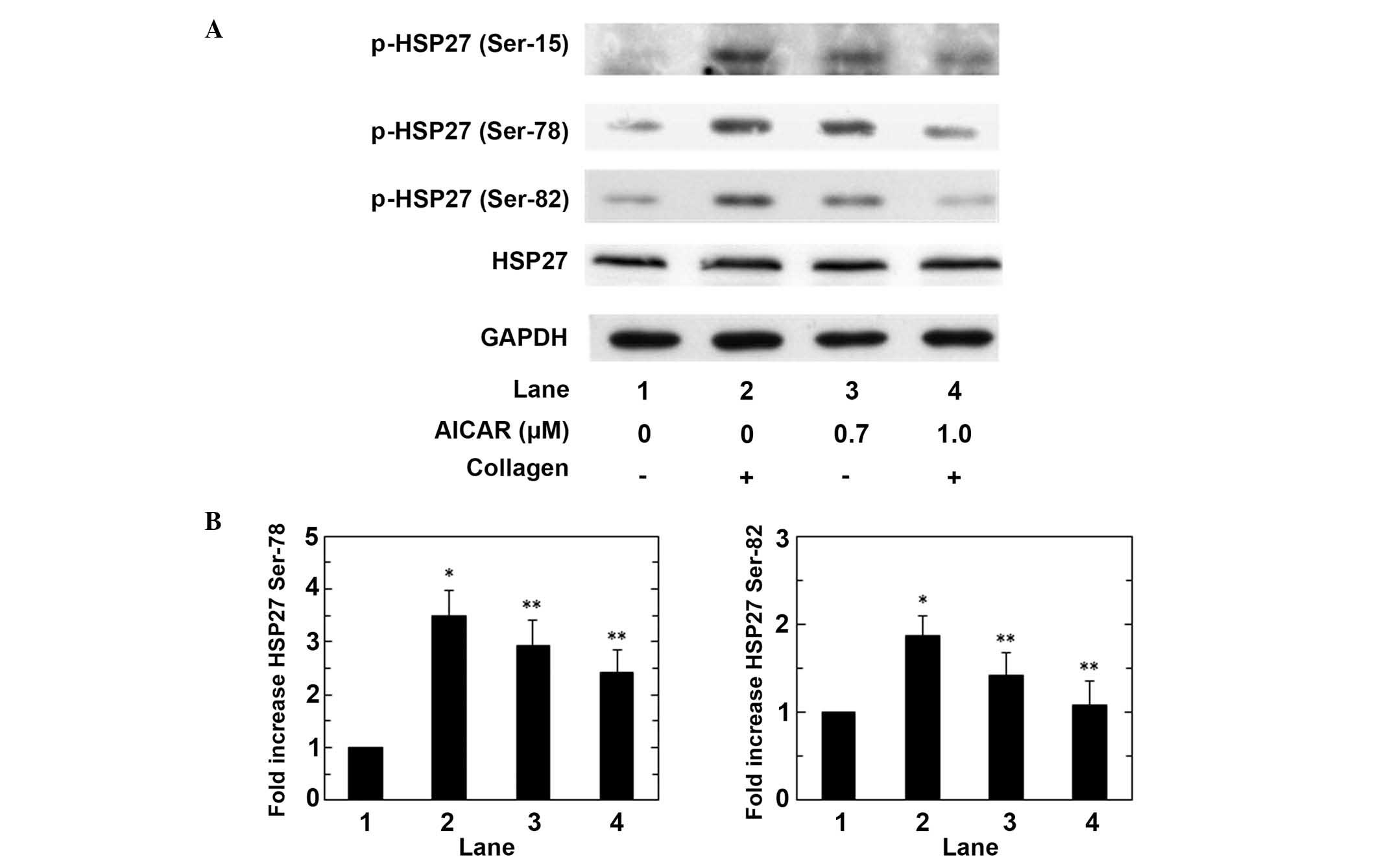
Subsequently, the effect of AICAR on the
collagen-induced phosphorylation of HSP27 was assessed in human
platelets. AICAR markedly reduced the collagen-induced
phosphorylation of HSP27 at ser-15, ser-78 and ser-82 in a
dose-dependent manner up to 1.0 µM, and significant suppression of
phosphorylation was induced by AICAR on Ser-78 and Ser-82 (both
P<0.05; Fig. 3). The maximum
suppressive effects of AICAR (1.0 µM) on the collagen-stimulated
phosphorylation of HSP27 at ser-78 and ser-82 induced a ~40% and
90% reduction in the collagen-effect, respectively. These findings
suggest that AMPK activating AICAR limits the phosphorylation of
HSP27 via the suppression of p44/p42 MAP kinase activity in
collagen stimulated human platelets.
Effects of AICAR on the
collagen-induced PDGF-AB secretion or sCD40L release from human
platelets
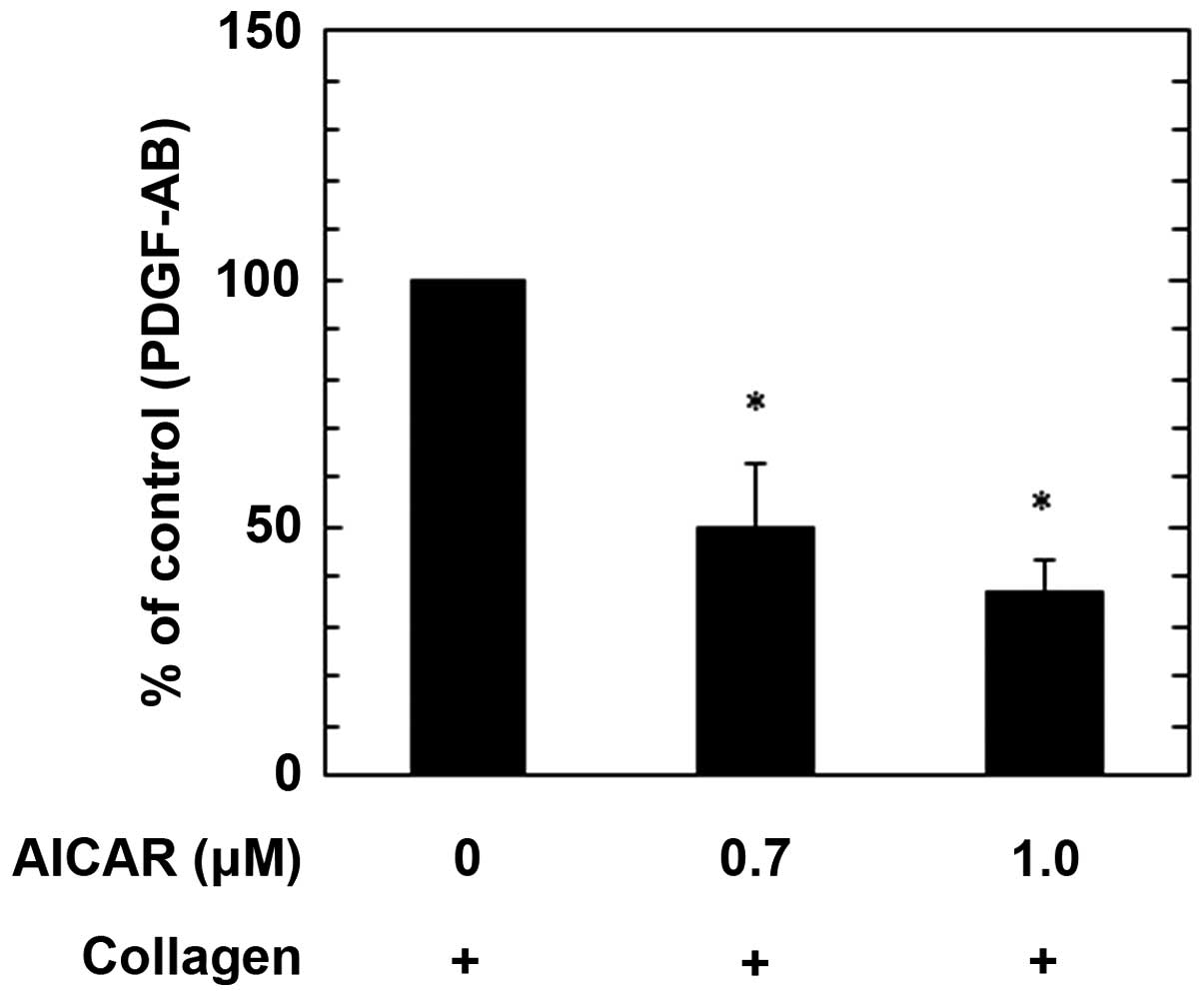
We have previously demonstrated that the
collagen-induced phosphorylation of HSP27 via p44/p42 MAP kinase in
human platelets is sufficient for PDGF-AB secretion and sCD40L
release (17). Therefore, in the
present study the effects of AICAR on the secretion of PDGF-AB and
the release of sCD40L induced by collagen were examined. AICAR
administration significantly suppressed PDGF-AB secretion in a
dose-dependent manner up to 1.0 µM, as compared with the control
(P<0.05; Fig. 4). The maximum
inhibitory effect induced by 1.0 µM AICAR on the collagen-induced
PDGF-AB secretion was ~60% a reduction in the collagen-effect.
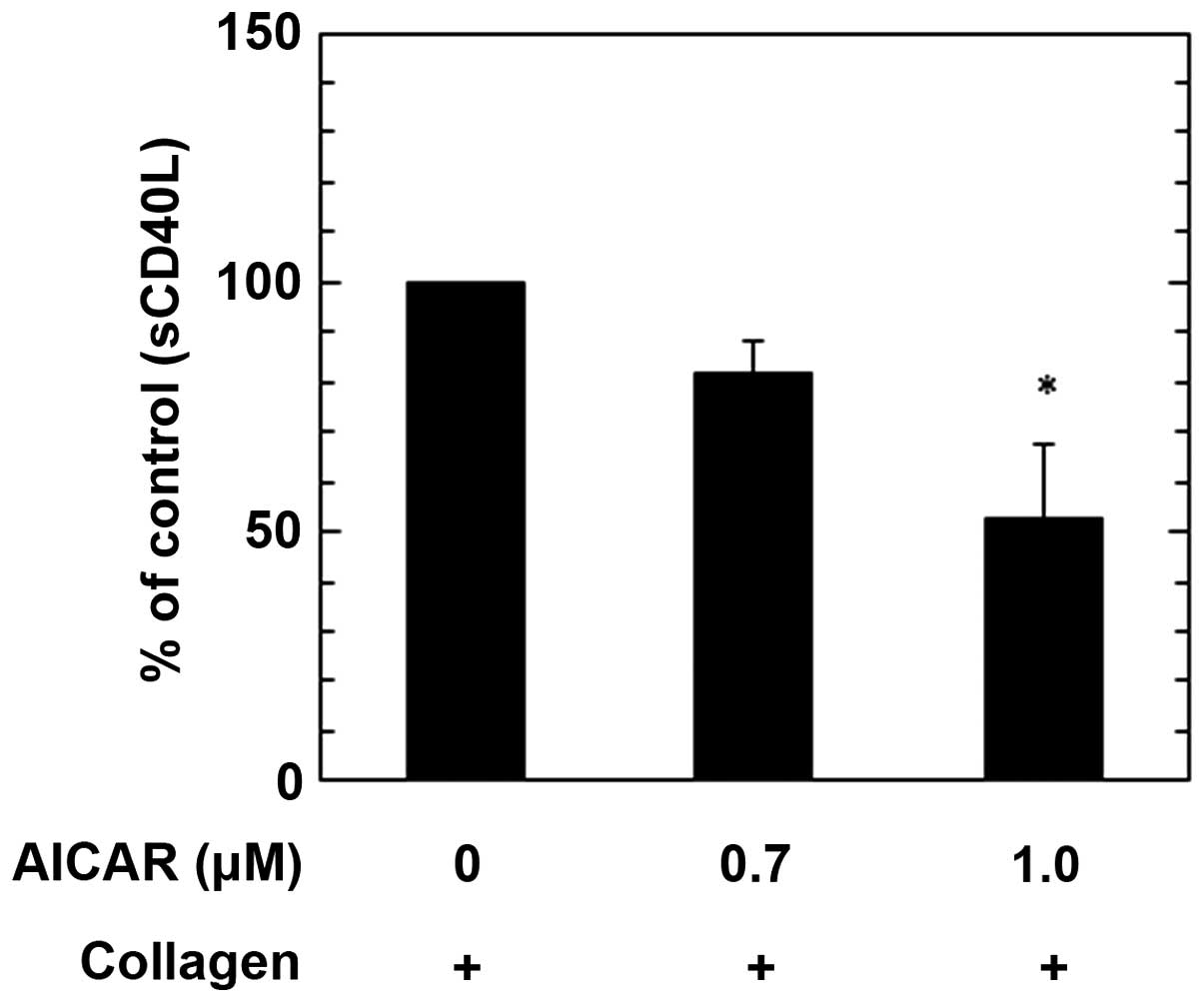
Furthermore, treatment with 1.0 µM AICAR significantly reduced the
collagen-stimulated release of sCD40L from human platelets, as
compared with the control (P<0.05); however treatment with 1.0
µM AICAR did not induce a significant reduction. The maximum
suppressive effect induced by 1.0 µM AICAR was a ~40% reduction in
the collagen-effect (Fig. 5). These
findings indicate that AMPK activates AICAR, simultaneously
inhibiting PDGF-AB secretion and sCD40L release from human
platelets, stimulated by collagen.
Discussion
The present study investigated the effect of AICAR,
which is an AMPK activator (23), on
the collagen-induced activation of human platelets. Using an
aggregometer with a laser light scattering system, which can
simultaneously detect both the light-transmittance and the size of
platelet aggregates, the effect of AICAR on the aggregation of
human platelets was assessed. The results demonstrated that AICAR
administration inhibited collagen-induced platelet aggregation and
decreased the collagen-induced formation of large aggregates (50–70
µm) and increased the formation of small aggregates (9–25 µm).
Alterations in aggregate size are recognized to be more sensitive
than the change of transmittance in platelet reactivity (26). Therefore, these findings suggested
that AMPK-activating AICAR may inhibit collagen-stimulated human
platelet aggregation.
We have previously reported that the
collagen-induced phosphorylation of HSP27 via p44/p42 MAP kinase is
directly proportional to the secretion of PDGF-AB and the release
of sCD40L from human platelets (17). On the basis of these findings, the
present study examined the effects of AICAR on the phosphorylation
of HSP27 and p44/p42 MAP kinase induced by collagen in human
platelets. The results of the present study demonstrated that AICAR
markedly suppressed the expression levels of phosphorylated p44/p42
MAP kinase, suggesting that AMPK-activating AICAR negatively
regulates the collagen-stimulated activation of p44/p42 MAP kinase
in human platelets. Furthermore, the present study demonstrated
that AICAR administration attenuated the phosphorylation of HSP27.
These findings suggested that AMPK-activating AICAR may limit the
phosphorylation of HSP27 via the suppression of p44/p42 MAP kinase
activity in collagen-stimulated human platelets. The results of the
present study also showed that the collagen-induced granule
secretion of PDGF-AB was significantly reduced by pretreatment with
AICAR. In addition, the collagen-stimulated sCD40L release was also
suppressed by AICAR. Therefore, these findings indicated that
AMPK-activating AICAR may simultaneously inhibit PDGF-AB secretion
and sCD40L release from human platelets stimulated by collagen. It
is most likely that AICAR negatively regulates the
collagen-stimulated secretion of PDGF-AB and the release of sCD40L
from human platelets through the inhibition of HSP27
phosphorylation via p44/p42 MAP kinase and platelet
aggregation.
It is well recognized that PDGF-AB, which is
released from α-granules in activated-platelets, is a potent growth
factor that promotes atherosclerosis (27). Furthermore, it has been demonstrated
that sCD40L, which is released from activated platelets (28,29),
induces inflammatory responses in vascular endothelial cells and
neutrophils via CD40 (30),
resulting in the progression of atherosclerosis (27). It has been reported that the
elevation of plasma sCD40L is a risk factor for cardiovascular
events in patients with unstable coronary artery disease (31). Therefore, a simultaneous reduction in
PDGF-AB secretion, sCD40L release and platelet aggregation may be
beneficial to the prohibition of atherosclerosis-related diseases,
including acute coronary syndrome and stroke. The results of the
present study may provide a novel therapeutic strategy for the
treatment of these diseases. Notably, it has previously been
demonstrated metformin, which is an AMPK-activating substance that
is widely used to treat DM, is capable of inducing a reduction in
cardiovascular events and ischemic stroke characterized by
atherosclerosis (32). It is well
known that DM is a risk factor of cardiovascular and
cerebrovascular ischemic diseases (33). Regarding the function of platelets in
DM, we have previously reported that the phosphorylation levels of
collagen-induced p38 MAP kinase and p44/p42 MAP kinase are
correlated with the hyperaggregability of platelets derived from
patients (34). Therefore, the
ameliorating effect of an AMPK-activating agent on vascular events
in DM may be due to the suppression of p44/p42 MAP kinase in human
platelets; this suppression would result in the reduction of HSP27
phosphorylation associated with aggregation and the release of
pro-inflammatory substances from platelets. Further investigation
is required in order to clarify the molecular mechanisms underlying
the effects of AMPK on human platelets.
In conclusion, the results of the present study
suggested that AICAR-activated AMPK may reduce the collagen-induced
secretion of PDGF-AB and release of sCD40L by inhibiting HSP27
phosphorylation via p44/p42 MAP kinase in human platelets.
Acknowledgements
The authors of the present study would like to thank
Mrs. Yumiko Kurokawa for her skillful technical assistance. The
present study was supported by grants from the Ministry of
Education, Science, Sports and Culture of Japan (grant no.
23592249), and the Research Fund for Longevity Sciences (25–4) from
National Center for Geriatrics and Gerontology in Japan (grant no.
26462335).
References
|
1
|
Davì G and Patrono C: Platelet activation
and atherothrombosis. N Eng J Med. 357:2482–2494. 2007. View Article : Google Scholar
|
|
2
|
Nieswandt B and Watson SP:
Platelet-collagen interaction: Is GPVI the central receptor? Blood.
102:449–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moroi M and Jung SM: Platelet glycoprotein
VI: Its structure and function. Thromb Res. 114:221–233. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng YM, Liu C, Chen H, Locke D, Ryan JC
and Kahn ML: Expression of the platelet receptor GPVI confers
signaling via the Fc receptor gamma-chain in response to the snake
venom convulxin but not to collagen. J Biol Chem. 276:12999–13006.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berlanga O, Tulasne D, Bori T, Snell DC,
Miura Y, Jung S, Moroi M, Frampton J and Watson SP: The Fc receptor
gamma-chain is necessary and sufficient to initiate signalling
through glycoprotein VI in transfected cells by the snake C-type
lectin, convulxin. Eur J Biochem. 269:2951–2960. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blake RA, Schieven GL and Watson SP:
Collagen stimulates tyrosine phosphorylation of phospholipase
C-gamma 2 but not phospholipase C-gamma 1 in human platelets. FEBS
Lett. 353:212–216. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yanaga F, Poole A, Asselin J, Blake R,
Schieven GL, Clark EA, Law CL and Watson SP: Syk interacts with
tyrosine-phosphorylated proteins in human platelets activated by
collagen and cross-linking of the Fc gamma-IIA receptor. Biochem J.
311:471–478. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shattil SJ and Newman PJ: Integrins:
Dynamic scaffolds for adhesion and signaling in platelets. Blood.
104:1606–1615. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hendrick JP and Hartl FU: Molecular
chaperone functions of heat-shock proteins. Annu Rev Biochem.
62:349–384. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kampinga HH, Hageman J, Vos MJ, Kubota H,
Tanguay RM, Bruford EA, Cheetham ME, Chen B and Hightower LE:
Guidelines for the nomenclature of the human heat shock proteins.
Cell Stress Chaperones. 14:105–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kato K, Ito H and Inaguma Y: Expression
and phosphorylation of mammalian small heat shock proteins. Prog
Mol Subcell Biol. 28:129–150. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koteiche HA and McHaourab HS: Mechanism of
chaperone function in small heat-shock proteins.
Phosphorylation-induced activation of two-mode binding in alpha
B-crystallin. J Biol Chem. 278:10361–10367. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Acunzo J, Katsogiannou M and Rocchi P:
Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and
HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol.
44:1622–1631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Merendino AM, Paul C, Vignola AM, Costa
MA, Melis M, Chiappara G, Izzo V, Bousquet J and Arrigo AP: Heat
shock protein-27 protects human bronchial epithelial cells against
oxidative stress-mediated apoptosis: Possible implication in
asthma. Cell Stress Chaperones. 7:269–280. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rogalla T, Ehrnsperger M, Preville X,
Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieshe M, Arrigo AP,
Buchner J and Gaestel M: Regulation of Hsp27 oligomerization,
chaperone function, and protective activity against oxidative
stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem.
274:18947–18956. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saklatvala J, Rawlinson L, Waller RJ,
Sarsfield S, Lee JC, Morton LF, Barnes MJ and Farndale RW: Role for
p38 mitogen-activated protein kinase in platelet aggregation caused
by collagen or a thromboxane analogue. J Biol Chem. 271:6586–6589.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kato H, Adachi S, Doi T,
Matsushima-Nishiwaki R, Minamitani C, Akamatsu S, Enomoto Y, Tokuda
H, Otsuka T, Iwama T, et al: Mechanism of collagen-induced release
of 5-HT, PDGF-AB and sCD40L from human platelets: Role of HSP27
phosphorylation via p44/p42 MAPK. Thromb Res. 126:39–43. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hardie DG: AMP-activated protein kinase as
a drug target. Annu Rev Pharmacol Toxicol. 47:185–210. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steinberg GR and Kemp BE: AMPK in Health
and Disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wong AK, Howie J, Petrie JR and Lang CC:
AMP-activated protein kinase pathway: A potential therapeutic
target in cardiometabolic disease. Clin Sci (Lond). 116:607–620.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang BB, Zhou G and Li C: AMPK: An
emerging drug target for diabetes and the metabolic syndrome. Cell
Metab. 9:407–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Oh SJ, Chang KH, Kim YG and Lee MY:
Antiplatelet effect of AMP-activated protein kinase activator and
its potentiation by the phosphodiesterase inhibitor dipyridamole.
Biochem Pharmacol. 86:914–925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cool B, Zinker B, Chiou W, Kifle L, Cao N,
Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al:
Identification and characterization of a small molecule AMPK
activator that treats key components of type 2 diabetes and the
metabolic syndrome. Cell Metab. 3:403–416. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kato K, Ito H, Hasegawa K, Inaguma Y,
Kozawa O and Asano T: Modulation of the stress-induced synthesis of
hsp27 and alpha B-crystallin by cyclic AMP in C6 rat glioma cells.
J Neurochem. 66:946–950. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fusegawa Y and Handa S: Platelet
aggregation induced by ADP or epinephrine is enhanced in habitual
smokers. Thromb Res. 97:287–295. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heldin CH and Westermark B: Mechanism of
action and in vivo role of platelet-derived growth factor. Physiol
Rev. 79:1283–1316. 1999.PubMed/NCBI
|
|
28
|
Hermann A, Rauch BH, Braun M, Schrör K and
Weber AA: Platelet CD40 ligand (CD40L)-subcellular localization,
regulation of expression, and inhibition by clopidogrel. Platelets.
12:74–82. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
André P, Nannizi-Alaimo L, Prasad SK and
Phillips DR: Platelet-derived CD40L: The switch-hitting player of
cardiovascular disease. Circulation. 106:896–899. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Henn V, Slupsky JR, Gräfe M,
Anagnostopoulos I, Förster R, Müller-Berghaus G and Kroczek RA:
CD40 ligand on activated platelets triggers an inflammatory
reaction of endothelial cells. Nature. 391:591–594. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Heeschen C, Dimmeler S, Hamm CW, van den
Vrand MJ, Boersma E, Zeiher AM and Simoons ML: CAPTURE study
Investigators: Soluble CD40 ligand in acute coronary syndrome. N
Eng J Med. 348:1104–1111. 2003. View Article : Google Scholar
|
|
32
|
Chang CH, Chang YC, Lin JW, Chen ST,
Chuang LM and Lai MS: Cardiovascular risk associated with acarbose
versus metformin as the first-line treatment in patients with type
2 diabetes: A Nationwide Cohort Study. J Clin Endocrinol Metab.
100:1121–1129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Emerging Risk Factors Collaboration.
Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio
E, Ingelsson E, Lawlor DA, Selvin E, et al: Diabetes mellitus,
fasting blood glucose concentration, and risk of vascular disease:
A collaborative meta-analysis of 102 prospective studies. Lancet.
375:2215–2222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hanai Y, Adachi S, Yasuda I, Takai S,
Matsushima-Nishiwaki R, Kato H, Enomoto Y, Akamatsu S, Sakakibara
S, Ogura S, et al: Collagen-induced p38 MAP kinase activation is a
biomarker of platelet hyper-aggregation in patients with diabetes
mellitus. Life Sci. 85:386–394. 2009. View Article : Google Scholar : PubMed/NCBI
|