Introduction
Osteonecrosis (ON), also known as aseptic or
avascular necrosis of the bone, is a disorder characterized by
segmental death of one or more osseous sites (1). Although non-traumatic ON is rare in
young people, it has been reported increasingly in children treated
for acute lymphoblastic leukemia (ALL), as ALL survival rates
continue to improve (1). The
symptoms of ON are highly variable, ranging from mild discomfort to
decreased mobility, severe pain and articular collapse (2). Although up to 25% of children treated
for ALL exhibit radiographic evidence of osteopenia (3) and one-third of ALL patients develop
symptomatic ON (1), the majority of
patients are asymptomatic, some showing the spontaneous regression
or even complete resolution of the disorder (1). Due to heterogenous diagnostic criteria,
the reported incidence of ON varies widely in children with cancer;
between 0.3 and 9% (2,4). The pathogenesis of ON is complex and
includes the enhanced differentiation of mesenchymal stem cells
(MSCs) into lipocytes at the expense of osteogenesis, as well as
damage to the venous system, vascular stasis and ischemia (1). Several risk factors for ON, such as
older age, white race and glucocorticoid therapy (1), have been identified in infant cancer
patients; however, their diagnostic power is limited and the
etiology of the disease remains unclear. Therefore, there is a
continuous effort to identify additional ON risk factors. In
addition to glucocorticoids (GCs), other therapeutic agents such as
methotrexate (MTX) and 6-mercaptopurine (6-MP) have been indicated
in bone morbidity during ALL treatment (5–7).
Recently, several polymorphisms have been identified as potential
risk factors for ON during ALL treatment in genes involved in
various processes, including: Bone metabolism, vitamin D receptor
(8), collagen type II, α1 (1) and acid phosphatase 1 (ACP-1)
(9); thrombosis,
5,10-methylenetetrahydrofolate reductase (MTHFR) (9) and plasminogen activator inhibitor-1
(PAI-1) (10); and ALL
pharmacogenetics, tymidylate synthetase (TYMS) (8). The present exploratory retrospective
study was conducted to assess whether common polymorphisms in the
target genes thiopurine S-methyltransferase (TPMT),
MTHFR, estrogen receptor alpha 1 (ESR1) and collagen
type I, α1 (COL1A1) are associated with ON in children
receiving ALL therapy and could be used as prognostic genetic
markers for ON. Target genes were selected to investigate all three
aspects of ON risk: Thrombotic (MTHFR, ESR1); bone
metabolism (ESR1, COL1A1 and MTHFR); and
pharmacogentic (TPMT, MTHFR). TPMT and MTHFR are key enzymes
in the metabolism of 6-MP and MTX (11), respectively, and are crucial to the
majority of ALL treatment protocols (12). They are administered in all phases of
the therapy and are the primary drugs used in the maintenance
phase, during which ON typically emerges (12). In addition, MTHFR variants
(9,13), as well as hyperestrogenemia (11), have been implicated in ON due to
thrombophilia in adults. Finally, ESR1 and COL1A1
were associated with bone mineral density (BMD) and other
osteoporotic phenotypes in candidate gene studies (14), in addition to large-scale association
(15,16) and genome-wide studies (17,18).
Additionally, MTHFR has been suggested to be associated with
osteoporotic phenotype in certain studies (17–19).
Materials and methods
Patients
Slovenian pediatric ALL patients diagnosed and
treated at the University Children's Hospital, University Medical
Centre (Ljubljana, Slovenia) between 1970 and 2004 were identified
via the national oncology patients registry. Among the 405
registered patients with childhood ALL, 62 had inadequate medical
records, 3 patients received non-Hodgkin's lymphoma
Berlin-Frankfurt-Münster (NHL-BFM) treatment, 3 underwent bone
marrow transplantation prior to maintenance therapy and 15 patients
succumbed or relapsed prior to receiving maintenance therapy. A
further 9 patients were later excluded from the study due to
unsuccessful DNA extraction. The final study group consisted of 313
patients with childhood ALL. The study was conducted in accordance
with the World Medical Association Declaration of Helsinki
regarding ethical conduct of research involving human subjects.
Ethical approval was obtained from the Medical Ethics Committee of
Slovenia.
Different therapy protocols were applied in the
study periods from 1970 to 2004. Pediatric oncology group (POG)
protocols (20) were used from 1970
to 1983, and thereafter BFM protocols [BFM-83 (21), −86 (22), −90 (23), −95 (24) and intercontinental (IC) trial-BFM
2002 (25)]. Therapy data, which
were systematically recorded by attending physicians, such as
incidence of toxic effects, were obtained for 313 childhood ALL
patients. From patients' medical records, 12 patients with
symptomatic ON were identified, corresponding to grades 3 and 4
adverse events of NCI Common Toxicity Criteria (version 2.0)
(26).
Genotyping
Genotyping was performed after the therapy data were
extracted from patients' medical files and analyzed by researchers
who were blinded to patients' medical data.
DNA was extracted from archival bone marrow smears
of patients at the diagnosis of ALL using a semi-automated ABI
PRISM™ 6100 Nucleic Acid PrepStation (Applied Biosystems; Thermo
Fisher Scientific, Inc., Foster City, CA, USA).
TPMT (*3B: 460G>A, rs1800460 and *3C:
719A>G, rs1142345), MTHFR (677C>T, rs1801133 and
1298A>C, rs1801131), ESR1 (XbaI, rs9340799) and
COL1A1 (Sp1, rs1800012) polymorphisms were determined
using TaqMan chemistry on ABI Prism® 7000 Sequence
Detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). TaqMan chemical reagents were purchased from Applied
Biosystems). As 460G>A and 719A>G polymorphisms in
TPMT are in linkage disequilibrium and inherited together in
cis, patients carrying both polymorphisms were considered to be
heterozygotes for TPMT*3A allele. All low activity TPMT
alleles were designated as TPMT*3 and wild-type as TPMT*1. We
combined MTHFR 677C>T and 1298A>C genotypes and
established that 677T rarely occurred in cis with 1298C. In the
group of 313 patients, only a single patient (0.3%) exhibited the
677TT/1298AC genotype, which is in accordance with previous results
(27). The 677CC/1298AA genotype was
designated a wild type and all other as mutated genotypes.
Statistical analysis
To assess differences in age, gender, treatment
protocol, TPMT, MTHFR, ESR1 and COL1A1 genotypes
between ON and non-ON group the Fisher exact test was used.
For multivariate analysis, we applied the
classification tree method to determine the interaction between
multiple predictive variables and to evaluate the risk of ON
associated with specific subgroups. ON was treated as the dependent
variable, while all other variables (age, gender, treatment
protocol and genotypes) were treated as independent variables. All
variables were considered when constructing the classification
tree. The classification and regression tree growing method was
used and the minimum number of cases in parent and child node was
set to 5 and 2, respectively. Due to the small number of ON cases
the results of the classification tree were confirmed using Fisher
exact test. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
using the SPSS software, version 16.0 for Windows (SPSS, Inc.,
Chicago, IL, USA).
Results
Characteristics of the patients
The study group consisted of 313 childhood ALL
patients, 12 (3.8%) of whom were found to have had symptomatic ON
(NCI grades 3 and 4). In the group of 313 childhood ALL patients,
the mean age at ALL diagnosis was 5.9±4.2 years (range, 0.2–17.0
years). There were 148 (47.3%) female and 165 (52.7%) male
patients. Furthermore, 94 (30.0%), 37 (11.8%), 55 (17.6%), 59
(18.8%), 55 (17.6%) and 13 (4.2%) of patients were treated with
POG, BFM-83, −86, −90, −95 and IC-BFM 2002 protocols, respectively
(Table I).
 | Table I.Analysis of independent risk factors
for osteonecrosis in acute lymphoblastic leukemia patients with and
without osteonecrosis. |
Table I.
Analysis of independent risk factors
for osteonecrosis in acute lymphoblastic leukemia patients with and
without osteonecrosis.
| Parameter | Osteonecrosis
(N=12) | No osteonecrosis
(N=301) | P |
|---|
| Age, years |
11.3±5.9 | 5.6±4.0 | 0.007 |
| Gender, n (%) |
|
| 0.774 |
|
Male | 7
(58.3) | 158 (52.5) |
|
|
Female | 5
(41.7) | 143 (47.5) |
|
| Treatment protocol,
n (%) |
|
| 0.018 |
|
POG | 2
(16.7) | 92
(30.6) |
|
|
BFM-83 | 0
(0.0) | 37
(12.3) |
|
|
BFM-86 | 0
(0.0) | 55
(18.3) |
|
|
BFM-90 | 5
(41.7) | 54
(17.9) |
|
|
BFM-95 | 3
(25.0) | 52
(17.3) |
|
| IC-BFM
2002 | 2
(16.7) | 11 (3.7) |
|
| TPMT
genotype, n (%) |
|
| 0.188 |
|
*1/*1 | 10
(83.3) | 282 (93.7) |
|
|
*1/*3 | 2
(16.7) | 19 (6.3) |
|
| MTHFR
genotype, n (%) |
|
| 0.620 |
| Wild
typea | 0
(0.0) | 32
(10.8) |
|
|
Mutatedb | 12 (100) | 269 (89.4) |
|
| ESR1
genotype, n (%) |
|
| 0.465 |
| GG | 4
(33.3) | 53
(19.1) |
|
| GA | 4
(33.3) | 121 (43.5) |
|
| AA | 4
(33.3) | 104 (37.4) |
|
| COL1A1 genotype, n
(%) |
|
| 0.797 |
| GG | 10
(90.9) | 191 (76.4) |
|
| GT | 1
(9.1) | 50
(20.0) |
|
| TT | 0
(0.0) | 9
(3.6) |
|
The genotyping for TPMT and MTHFR
polymorphisms was successful for all 313 patients, while
ESR1 and COL1A1 genotypes were not determined for 23
and 52 patients, respectively, due insufficient extracted DNA from
bone marrow smears. With respect to TPMT genotype, there
were no homozygous mutated (*3/*3) patients, 21 (6.7%) patients
were heterozygotes (*1/*3) and the remainder were wild-type
(*1/*1). A total of 32 (10.2%) patients were MTHFR wild
type, while 281 (89.8%) had at least one mutated allele in one of
the two MTHFR locus. Among the 261 patients that were
successfully genotyped for COL1A1, 201 (77.0%) had GG, 51
(19.5%) GT and 9 (3.4%) TT genotype. The distribution of
ESR1 genotypes in the 290 successfully genotyped patients
was as follows: GG, 57 (19.7%); AG, 125 (43.1%); and AA, 108
(37.2%). All examined polymorphisms were in Hardy-Weinberg
equilibrium.
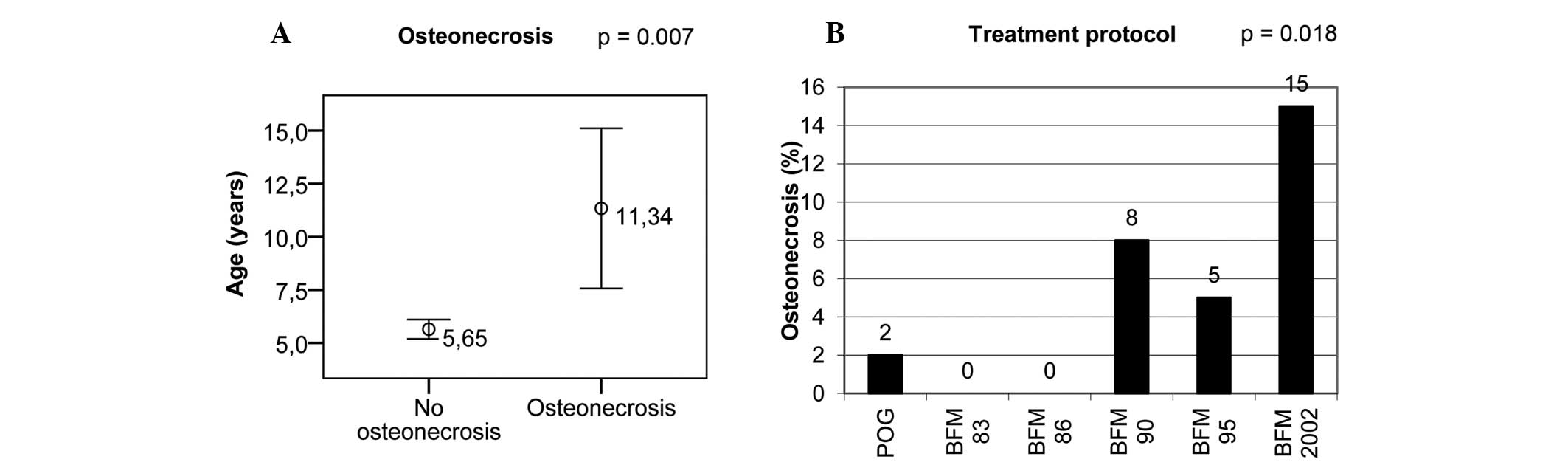
Independent risk factors for ON
To identify independent risk factors for ON, we
compared all variables (age, gender, treatment protocol and studied
genotypes) between ON patients and non-ON patients. Higher age at
diagnosis and more recent treatment protocols were identified as
independent risk factors for ON (Table
I and Fig. 1).
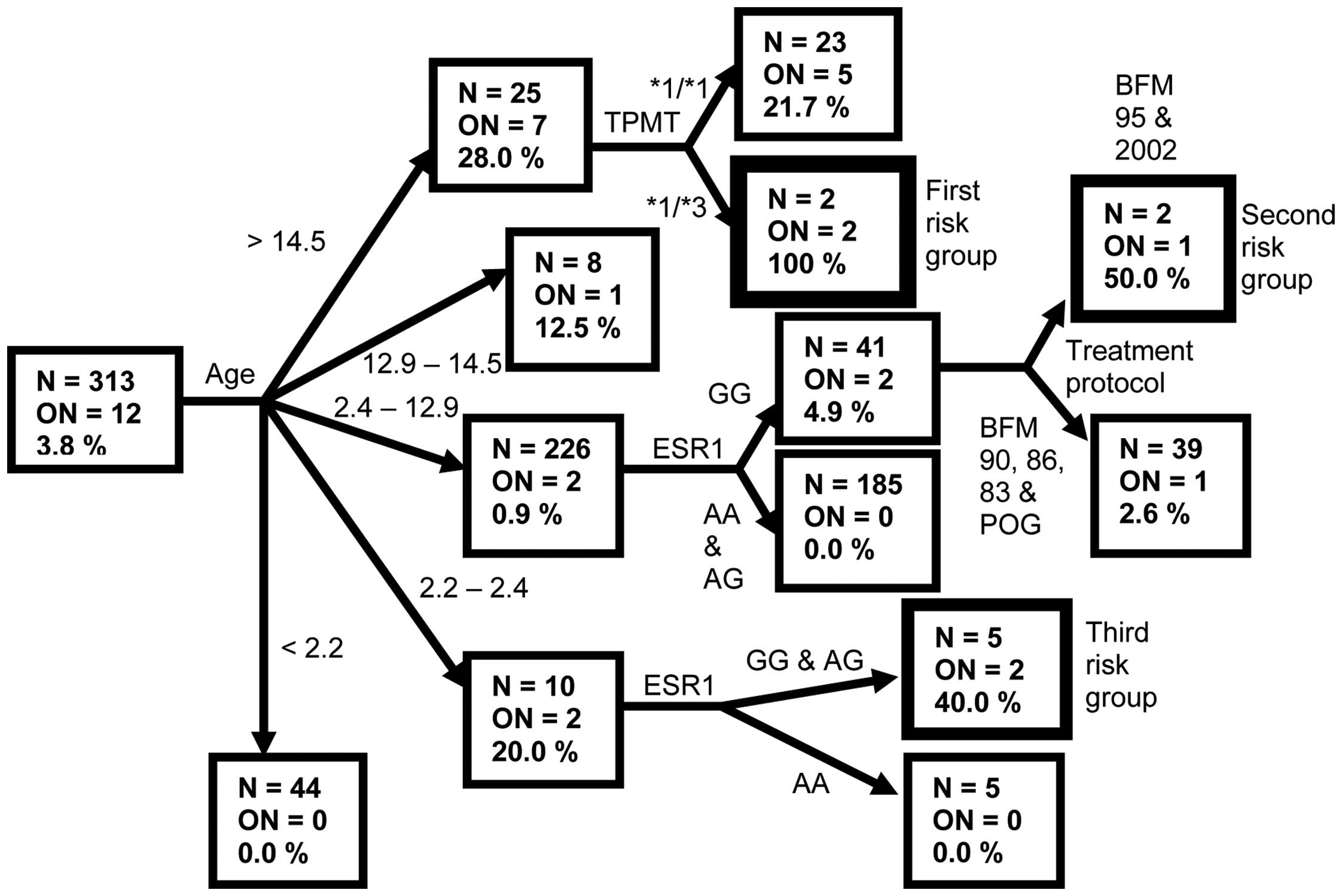
Combined risk factors for ON
To investigate the interaction between multiple risk
factors, we employed classification tree analysis (Fig. 2). The terminal nodes of the
classification tree with the highest incidence of ON represented
the patient subgroups with the highest risk for ON.
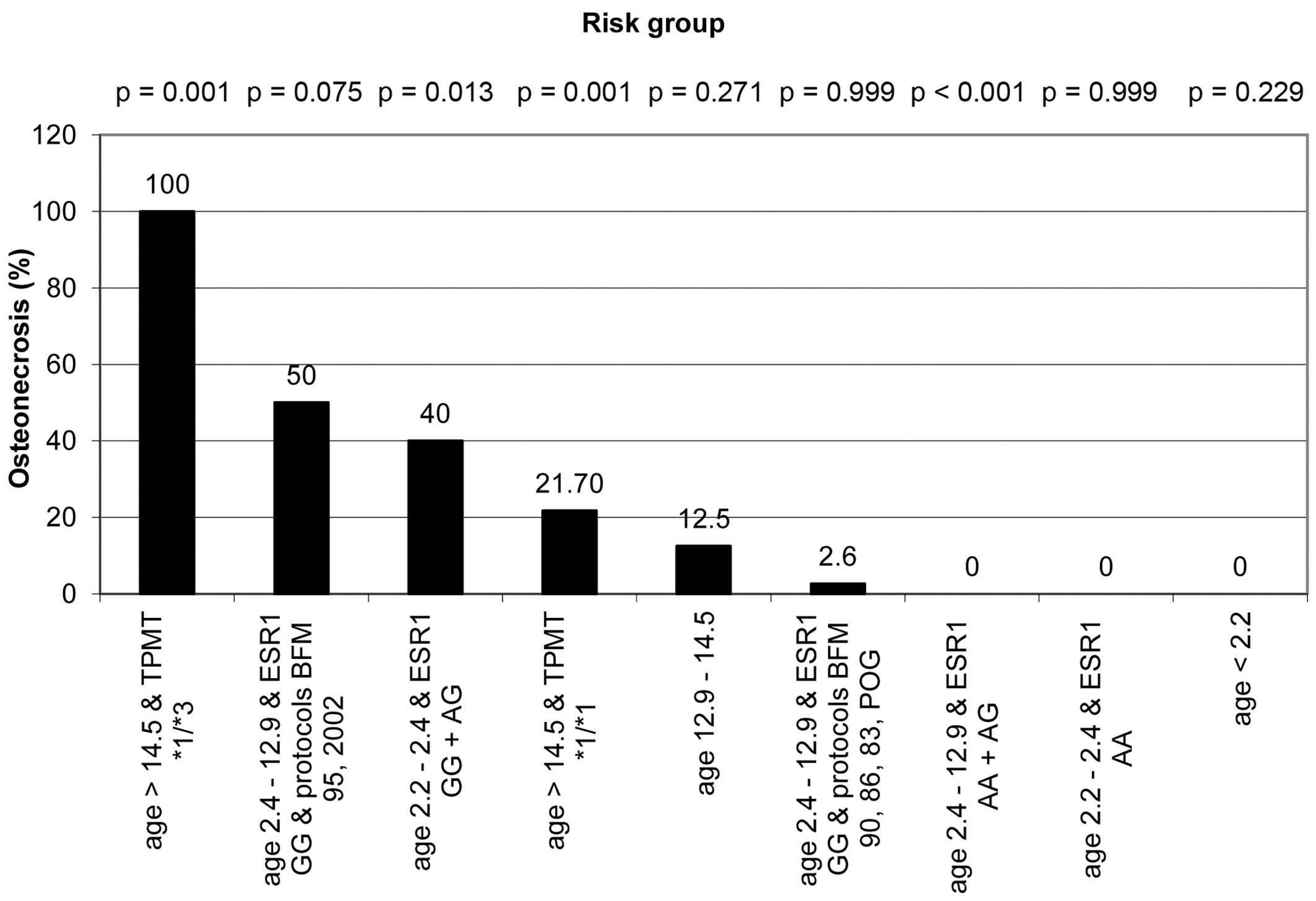
The subgroup with highest (100%) risk for ON
consisted of ALL patients >14.5 years old with the mutated
TPMT genotype. The ON incidence in this subgroup was
compared with its incidence in the remainder of the observed
population by means of Fisher exact test, which showed a
statistically significant difference (P=0.001) (Fig. 3). The group with the second highest
risk (50%) for ON consisted of patients 2.4–12.9 years old, who had
GG ESR1 genotype and were treated with protocols BFM-95 and
IC-BFM 2002 (P=0.075). Patients aged 2.2–2.4 years with ESR1
GG and AG genotypes represented the third risk group with ON
incidence of 40% (P=0.013). The incidence of ON in all terminal
nodes and its comparison to the remainder of the observed
population is shown in Fig. 3.
Notably, a combination of age 2.4–12.9 years and ESR1 AA and
AG genotypes had a protective effect, as none of the 185 patients
in this subgroup developed symptomatic ON (P<0.001).
Discussion
In the studied population of childhood ALL patients,
the incidence of ON between 1970 and 2004 was 3.8%. Greater age and
later treatment protocols were independent risk factors for ON.
Furthermore, in patients >14.5 years, a mutated TPMT
genotype was associated with increased ON risk, while for children
younger than 12.9 years ESR1 GG and AG genotypes were the
main risk factor for ON.
In the present cohort age was a strong independent
risk factor for ON, which is in accordance with numerous studies
(2,4,8,28,29). It
has been established that adolescents, particularly those >15
years, are at highest risk for ALL therapy-induced ON. This may
reflect the greater vulnerability of rapidly growing bone in
adolescent period (1).
Another independent risk factor in the present study
was treatment protocol. More recent treatment protocols (BFM-90,
−95 and IC-BFM 2002) had higher incidence of ON compared with older
protocols. This could be explained by the differences in drug
administration schemes in different protocols. All chemotherapeutic
agents that are used in the treatment of ALL were found to reduce
the number of osteoblast-like cells in vitro (5), and combinations of different agents had
more toxic effects compared with individual agents (30). GCs were found to exhibit the
strongest risk of ON in the clinical setting during ALL therapy
(2), although MTX and alkylating
agents were also indicated to be associated with ON risk in certain
studies (2,3,6).
Therefore, we reviewed all used protocols to detect differences in
administration schedule and doses of GC and MTX. The main
difference between recent (BFM-90, −95 and IC-BFM 2002) and older
(POG, BFM-83 and −86) protocols were higher cumulative doses of GC
(particularly dexamethasone) and MTX in recent protocols. This is
in accordance with the results of two large childhood cancer
survivor studies, in which GC therapy was strong risk factor for ON
(2) and patients receiving
dexamethasone were more likely to develop ON compared with patients
who received prednisone alone (2,28).
Another important difference in the IC-BFM 2002 was the
introduction of intermittent maintenance therapy (IMT) consisting
of weekly MTX (20 mg/m2) and daily 6-MP (50
mg/m2). IMT was administered in all patient risk groups
between the blocks of reinduction therapy (25). Introduction of 2 IMT blocks for
standard risk and 3 blocks for intermediate risk group into the
reinduction therapy resulted in higher cumulative doses of
dexamethasone compared to standard reinduction therapy. In
accordance with this, the incidence of ON was highest in the IC-BFM
2002 group (15%), markedly exceeding ON incidence in BFM-90 (8%)
and BFM-95 (5%) protocols. Notably, all ON patients treated with
the IC-BFM 2002 protocol were assigned to the intermediate risk
group.
Deactivating polymorphisms in the TPMT gene
were a risk factor for ON in patients >14.5 years. TPMT is a
S-Adenosyl methionine (SAM)-dependent methyltransferse that
catalyses the deactivation of thiopurine drugs, such as 6-MP and
6-thioguanine (31). If the TPMT
activity is low (such as in the presence of polymorphisms in the
TPMT gene), this will result in high concentrations of
cytotoxic thioguanine nucleotides (TGN) due to decreased conversion
of thiopurine drugs to inactive methylated metabolites (31). The toxic effects of 6-MP on bone and
bone cells have been demonstrated in vitro (5) and in vivo (7). 6-MP reduced cell numbers in
osteoblast-like cell line MG63 at therapeutically relevant
concentrations (5), and in a
retrospective study of childhood ALL survivors number of weeks of
6-MP exposure were strongly correlated with hip BMD reduction
(7). High concentrations of TGN
exert toxic effects on majority of cells and this effect is most
pronounced in bone marrow cells (32). Since newly formed osteoblasts derive
from MSCs residing in bone marrow, we hypothesize that higher
levels of TGNs in individuals deficient in TPMT may lead to
decreased numbers of MSCs and consequently osteoblasts, leading to
the increased ON risk. In this manner, 6-MP could aggravate the
effects of GC on MSCs, which include the reduction of MSC number in
bone marrow (33) and increased
differentiation of MSCs into adipocytes at the expense of
osteogenesis (34). Recently it has
also been implicated that MSCs may have an important role in the
repair process of osteonecrotic lesion (35). MSCs, cultured in the low-oxygen
conditions, resembling those in the osteonecrotic lesion, were
found to produce high levels of vasculogenic cytokines (35). Since vascular damage is speculated to
be one of the primary ON causes, this induction of the
vascularization by MSCs may lead to ON improvement (35). Thus, any additional factors
decreasing MSC number or differentiation into osteoblasts may
worsen the osteonecrotic process, induced by GC. This could explain
why only a small fraction of patients receiving GC therapy develop
ON and why a relatively small percentage of patients with positive
radiograph and MRI osteonecrotic results are symptomatic (35). In the case of 6-MP therapy of the
present study, decreased TPMT activity leads to greater bone marrow
toxicity and decreased MSC number, which may impair bone formation
and repair the forming osteonecrotic lesion. In addition to
increased 6-MP toxicity, the impaired TPMT activity may influence
bone toxicity through changes in methylation potential. It has been
demonstrated in vitro that decreased activity of
SAM-dependent methyltransferases lead to impaired osteoblast
differentiation via DNA-methylation independent mechanism (36). Reduced global methylation decreases
the activity of transcription factor Runx2, a key regulator of
osteoblast differentiation (36).
In the present study, the XbaI polymorphism
in the ESR1 gene represents an ON risk factor only in
pre-pubertal children. In children <12.9 years the GG genotype
increased ON risk, while AA genotype was protective of ON. ESR1
plays an important role in the regulation of skeletal growth and
maintenance of bone mass (37). We
investigated a common polymorphism in the ESR1 gene
(XbaI), which has already been extensively studied (15,38–40,42). A
meta-analysis of association studies involving 5,834 subjects
showed a correlation of G allele with higher BMD and decreased risk
of fracture (38), while in another
large-scale genome-wide study a correlation with fracture that was
independent of BMD was observed (15). However, all of the aforementioned
studies were performed on adults and not in the context of cancer
therapy. In a study by Boot et al, the XbaI AA
genotype was associated with lower lumbar spine BMD in healthy
children (39). Furthermore, the
effect of ESR1 was more pronounced in pre-pubertal children, which
is in accordance with the present results. This may be due to the
fact that defects in estrogen receptor have more pronounced
clinical consequences in the setting of low estrogen
concentrations, such as in pre-pubertal children or post-menopausal
women (39). However, the present
results indicating XbaI GG genotype as risk factor for ON
are not in accordance with the results of a previous study on
healthy children where the same genotype was associated with higher
lumbar spine BMD. Notably, in the aforementioned study (33), XbaI was associated with lumbar
spine BMD, but not with total body BMD. This may be because the
spine is rich in trabecular bone, which has a high bone turnover
rate, while the body primarily consists of cortical bone with low
turnover (39). Notably, patients in
the present study had ON of the hip, where the percent of
trabecular bone is lower compared with the spine. There is evidence
that estrogens have a more marked effect on cortical than on
trabecular bone, possibly due to increased expression of ERa in
cortical bone (39). The molecular
mechanism by which ESR1 XbaI affects osteoporosis and BMD is
unclear; however, there is evidence that it may affect gene
transcription. Results of Maruyama et al suggest that the
presence of XbaI G allele can decrease ESR1 activity
(40). This is in accordance with
the present results that indicate that GG genotype is a risk factor
for ON, as decreased levels of estrogen are known to be associated
with low BMD and osteoporosis (41).
In the context of childhood ALL therapy, polymorphisms in
ESR1 were not correlated with BMD, although they were
associated with the recovery of lean body mass after the treatment
(42). Finally, all of the discussed
previous studies examining the effects of ESR1 on BMD were
performed on osteoporotic populations. Although osteoporosis and ON
are characterized by low BMD, the pathological mechanism of each
diseases is different (1). In
osteoporosis, bone resorption and formation are accelerated, while
the resorption/formation ratio is increased (43). In ALL therapy-related ON the bone
formation is inhibited due to the decreased number of precursor
MSCs and their increased differentiation to adipocytes, while at
the same time all bone cells are dying at an increased rate due to
the decreased level of oxygen and nutrients caused by thrombotic
complications (1). This may underlie
the discrepancy between the present results regarding osteonecrotic
patients and the result of osteoporosis studies in connection to
ESR1.
The present exploratory study contained a number of
limitations, the first point that needs to be addressed is the
number of cases. The study included all pediatric ALL patients from
Slovenia from a period of over 30 years. As severe ON is a very
rare condition, the number of cases is low despite the lengthy
study period. To confirm the present findings, a replication study
on another ALL population is required. Secondly, we did not measure
BMD in ALL patients as this was not routinely performed during
diagnosis and treatment of ALL patients. BMD measurements may offer
an additional insight into ON; however, due to the retrospective
nature of the study, this was not possible. Thirdly, we employed a
hypothesis-based approach and thus did not fully investigate all
the possible genetic markers. However, studied genes were selected
to cover all three aspects that contribute to the ON development:
Bone metabolism (COL1A1, ESR1, MTHFR), thrombosis (MTHFR,
ESR1) and ALL pharmacogenetics (TPMT, MTHFR).
Besides these selected genes, there are numerous other genes that
may warrant addition study, such as ACP-1, PAI-1 and
TYMS; however, due to the limited quantity of the samples
these were impossible to perform. From the same reason, we did not
fully cover all genetic variations in the studied genes, but
included only those that were clinically and epidemiologically most
relevant.
In conclusion, the present retrospective exploratory
study of 313 childhood ALL patients suggests that greater age and
more recent treatment protocols were independent risk factors for
ON. Furthermore, different genetic risk factors were identified in
specific age groups. While in patients >14.5 years TPMT
genotype modulated the risk of ON, in children <12.9 years
ESR1 genotypes were implicated in the pathogenesis of ON. As
these genetic markers have potential prognostic value for the
prediction of ON in pediatric ALL therapy, these findings require
confirmation in a replication study with a larger number of ON
patients.
Acknowledgements
The authors thank Miss Lil Klaas (Erasmus exchange
student) for technical assistance and Professor Roger Pain for
assistance with English language. The study was funded by the
Slovenian Research Agency (grant nos. J3-3615 and L3-2330).
Glossary
Abbreviations
Abbreviations:
|
ACP-1
|
acid phosphatase 1
|
|
ALL
|
acute lymphoblastic leukemia
|
|
BFM
|
Berlin-Frankfurt-Münster
|
|
BMD
|
bone mineral density
|
|
COL1A1
|
collagen, type I, α1
|
|
ESR1
|
estrogen receptor alpha 1
|
|
GC
|
glucocorticoide
|
|
6-MP
|
6-mercaptopurine
|
|
MSC
|
mesenchimal stem cell
|
|
MTHFR
|
5,10-methylenetetrahydrofolate
reductase
|
|
MTX
|
methotrexate
|
|
ON
|
osteonecrosis
|
|
PAI-1
|
plasminogen activator inhibitor-1
|
|
POG
|
pediatric oncology group
|
|
TPMT
|
thiopurine S-methyltransferase
|
|
TYMS
|
tymidylate synthetase
|
References
|
1
|
Sala A, Mattano LA Jr and Barr RD:
Osteonecrosis in children and adolescents with cancer-an adverse
effect of systemic therapy. Eur J Cancer. 43:683–689. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kadan-Lottick NS, Dinu I,
Wasilewski-Masker K, Kaste S, Meacham LR, Mahajan A, Stovall M,
Yasui Y, Robison LL and Sklar CA: Osteonecrosis in adult survivors
of childhood cancer: A report from the childhood cancer survivor
study. J Clin Oncol. 26:3038–3045. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davies JH, Evans BA, Jenney ME and Gregory
JW: Skeletal morbidity in childhood acute lymphoblastic leukaemia.
Clin Endocrinol (Oxf). 63:1–9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Faraci M, Calevo MG, Lanino E, Caruso S,
Messina C, Favr C, Iori A, Santaron S, Bonanomi S, Rondelli R, et
al: Osteonecrosis after allogeneic stem cell transplantation in
childhood. A case-control study in Italy. Haematologica.
91:1096–1099. 2006.PubMed/NCBI
|
|
5
|
Davies JH, Evans BA, Jenney ME and Gregory
JW: In vitro effects of chemotherapeutic agents on human
osteoblast-like cells. Calcif Tissue Int. 70:408–415. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tillmann V, Darlington AS, Eiser C, Bishop
NJ and Davies HA: Male sex and low physical activity are associated
with reduced spine bone mineral density in survivors of childhood
acute lymphoblastic leukemia. J Bone Miner Res. 17:1073–1080. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Warner JT, Evans WD, Webb DK, Bell W and
Gregory JW: Relative osteopenia after treatment for acute
lymphoblastic leukemia. Pediatr Res. 45:544–551. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Relling MV, Yang W, Das S, Cook EH, Rosner
GL, Neel M, Howard S, Ribeiro R, Sandlund JT, Pui CH and Kaste SC:
Pharmacogenetic risk factors for osteonecrosis of the hip among
children with leukemia. J Clin Oncol. 22:3930–3936. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zalavras CG, Malizos KN, Dokou E and
Vartholomatos G: The 677C->T mutation of the
methylene-tetrahydrofolate reductase gene in the pathogenesis of
osteonecrosis of the femoral head. Haematologica. 87:111–112.
2002.PubMed/NCBI
|
|
10
|
French D, Hamilton LH, Mattano LA Jr,
Sather HN, Devidas M, Nachman JB and Relling MV: Children's
Oncology Group: A PAI-1 (SERPINE1) polymorphism predicts
osteonecrosis in children with acute lymphoblastic leukemia: A
report from the Children's Oncology Group. Blood. 111:4496–4499.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mei L, Ontiveros EP, Griffith EA, Thompson
JE, Wang ES and Wetzler M: Pharmacogenetics predictive of response
and toxicity in acute lymphoblastic leukemia therapy. Blood Rev.
29:243–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hunger SP and Mullighan CG: Acute
lymphoblastic leukemia in children. N Engl J Med. 373:1541–1552.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Glueck CJ, Freiberg RA, Fontaine RN, Tracy
T and Wang P: Hypofibrinolysis, thrombophilia, osteonecrosis. Clin
Orthop Relat Res. 19–33. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ralston SH and de Crombrugghe B: Genetic
regulation of bone mass and susceptibility to osteoporosis. Genes
Dev. 20:2492–2506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ioannidis JP, Ralston SH, Bennett ST,
Brandi ML, Grinberg D, Karassa FB, Langdahl B, van Meurs JB,
Mosekilde L, Scollen S, et al: Differential genetic effects of ESR1
gene polymorphisms on osteoporosis outcomes. JAMA. 292:2105–2114.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ralston SH, Uitterlinden AG, Brandi ML,
Balcells S, Langdahl BL, Lips P, Lorenc R, Obermayer-Pietsch B,
Scollen S, Bustamante M, et al: Large-scale evidence for the effect
of the COLIA1 Sp1 polymorphism on osteoporosis outcomes: The
GENOMOS study. PLoS Med. 3:e902006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Steer CD, Emmett PM, Lewis SJ, Smith GD
and Tobias JH: Methylenetetrahydrofolate reductase (MTHFR) C677T
polymorphism is associated with spinal BMD in 9-year-old children.
J Bone Miner Res. 24:117–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Styrkarsdottir U, Halldorsson BV,
Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T,
Jonsdottir T, Saemundsdottir J, Center JR, Nguyen TV, et al:
Multiple genetic loci for bone mineral density and fractures. N
Engl J Med. 358:2355–2365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abrahamsen B, Jørgensen HL, Nielsen TL,
Andersen M, Haug E, Schwarz P, Hagen C and Brixen K: MTHFR
c.677C>T polymorphism as an independent predictor of peak bone
mass in Danish men-results from the odense androgen study. Bone.
38:215–219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maloney KW, Shuster JJ, Murphy S, Pullen J
and Camitta BA: Long-term results of treatment studies for
childhood acute lymphoblastic leukemia: Pediatric oncology group
studies from 1986–1994. Leukemia. 14:2276–2285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riehm H, Reiter A, Schrappe M, Berthold F,
Dopfer R, Gerein V, Ludwig R, Ritter J, Stollmann B and Henze G:
The in vivo response on corticosteroid therapy as an additional
prognostic factor in childhood acute lymphoblastic leukemia
(therapy study ALL-BFM 83). Klin Padiatr. 199:151–160. 1987.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reiter A, Schrappe M, Ludwig WD, Hiddemann
W, Sauter S, Henze G, Zimmermann M, Lampert F, Havers W and
Niethammer D: Chemotherapy in 998 unselected childhood acute
lymphoblastic leukemia patients. Results and conclusions of the
multicenter trial ALL-BFM 86. Blood. 84:3122–3133. 1994.PubMed/NCBI
|
|
23
|
Schrappe M, Reiter A, Ludwig WD, Harbott
J, Zimmermann M, Hiddemann W, Niemeyer C, Henze G, Feldges A, Zintl
F, et al: Improved outcome in childhood acute lymphoblastic
leukemia despite reduced use of anthracyclines and cranial
radiotherapy: Results of trial ALL-BFM 90. German-Austrian-Swiss
ALL-BFM study group. Blood. 95:3310–3322. 2000.PubMed/NCBI
|
|
24
|
Möricke A, Reiter A, Zimmermann M, Gadner
H, Stanulla M, Dördelmann M, Löning L, Beier R, Ludwig WD, Ratei R,
et al: Risk-adjusted therapy of acute lymphoblastic leukemia can
decrease treatment burden and improve survival: Treatment results
of 2169 unselected pediatric and adolescent patients enrolled in
the trial ALL-BFM 95. Blood. 111:4477–4489. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fronkova E, Mejstrikova E, Avigad S, Chik
KW, Castillo L, Manor S, Reznickova L, Valova T, Zdrahalova K,
Hrusak O, et al: Minimal residual disease (MRD) analysis in the
non-MRD-based ALL IC-BFM 2002 protocol for childhood ALL: Is it
possible to avoid MRD testing? Leukemia. 22:989–997. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
National Cancer Institute Common toxicity
criteria manual. version 2.0. Available on-line at. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcmanual_v4_10-4-99.pdfAccessed.
August 03–2014
|
|
27
|
Ogino S and Wilson RB: Genotype and
haplotype distributions of MTHFR677C>T and 1298A>C single
nucleotide polymorphisms: A meta-analysis. J Hum Genet. 48:1–7.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aricò M, Boccalatte MF, Silvestri D,
Barisone E, Messina C, Chiesa R, Santoro N, Tamaro P, Lippi A,
Gallisai D, et al: Osteonecrosis: An emerging complication of
intensive chemotherapy for childhood acute lymphoblastic leukemia.
Haematologica. 88:747–753. 2003.PubMed/NCBI
|
|
29
|
Burger B, Beier R, Zimmermann M, Beck JD,
Reiter A and Schrappe M: Osteonecrosis: A treatment related
toxicity in childhood acute lymphoblastic leukemia
(ALL)-experiences from trial ALL-BFM 95. Pediatr Blood Cancer.
44:220–225. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davies JH, Evans BA, Jenney ME and Gregory
JW: In vitro effects of combination chemotherapy on osteoblasts:
Implications for osteopenia in childhood malignancy. Bone.
31:319–326. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karas-Kuzelicki N and Mlinaric-Rascan I:
Individualization of thiopurine therapy: Thiopurine
S-methyltransferase and beyond. Pharmacogenomics. 10:1309–1322.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maltzman JS and Koretzky GA: Azathioprine:
Old drug, new actions. J Clin Invest. 111:1122–1124. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hernigou P, Beaujean F and Lambotte JC:
Decrease in the mesenchymal stem-cell pool in the proximal femur in
corticosteroid-induced osteonecrosis. J Bone Joint Surg Br.
81:349–355. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yin L, Li YB and Wang YS:
Dexamethasone-induced adipogenesis in primary marrow stromal cell
cultures: Mechanism of steroid-induced osteonecrosis. Chin Med J
(Engl). 119:581–588. 2006.PubMed/NCBI
|
|
35
|
Muller I, Vaegler M, Holzwarth C,
Tzaribatchev N, Pfister SM, Schütt B, Reize P, Greil J,
Handgretinger R and Rudert M: Secretion of angiogenic proteins by
human multipotent mesenchymal stromal cells and their clinical
potential in the treatment of avascular osteonecrosis. Leukemia.
22:2054–2061. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vaes BL, Lute C, van der Woning SP, Piek
E, Vermeer J, Blom HJ, Mathers JC, Müller M, de Groot LC and
Steegenga WT: Inhibition of methylation decreases osteoblast
differentiation via a non-DNA-dependent methylation mechanism.
Bone. 46:514–523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakamura T, Imai Y, Matsumoto T, Sato S,
Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, et
al: Estrogen prevents bone loss via estrogen receptor alpha and
induction of Fas ligand in osteoclasts. Cell. 130:811–823. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ioannidis JP, Stavrou I, Trikalinos TA,
Zois C, Brandi ML, Gennari L, Albagha O, Ralston SH and Tsatsoulis
A: ER-alpha Genetics Meta-Analysis: Association of polymorphisms of
the estrogen receptor alpha gene with bone mineral density and
fracture risk in women: A meta-analysis. J Bone Miner Res.
17:2048–2060. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boot AM, van der Sluis IM, de Muinck
Keizer-Schrama SM, van Meurs JB, Krenning EP, Pols HA and
Uitterlinden AG: Estrogen receptor alpha gene polymorphisms and
bone mineral density in healthy children and young adults. Calcif
Tissue Int. 74:495–500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maruyama H, Toji H, Harrington CR, Sasaki
K, Izumi Y, Ohnuma T, Arai H, Yasuda M, Tanaka C, Emson PC, et al:
Lack of an association of estrogen receptor alpha gene
polymorphisms and transcriptional activity with Alzheimer disease.
Arch Neurol. 57:236–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Riggs BL: The mechanisms of estrogen
regulation of bone resorption. J Clin Invest. 106:1203–1204. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
te Winkel ML, van Beek RD, de Muinck
Keizer-Schrama SM, Uitterlinden AG, Hop WC, Pieters R and van den
Heuvel-Eibrink MM: Pharmacogenetic risk factors for altered bone
mineral density and body composition in pediatric acute
lymphoblastic leukemia. Haematologica. 95:752–759. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Raisz LG: Pathogenesis of osteoporosis:
Concepts, conflicts, and prospects. J Clin Invest. 115:3318–3325.
2005. View Article : Google Scholar : PubMed/NCBI
|