Introduction
Neuronal death induced by cerebral ischemia is a
major cause of high morbidity and mortality (1). Although previous studies have yielded
encouraging results when investigating therapeutic candidates using
animal models of cerebral ischemia (2,3), no
pharmacological strategies are currently effective in the clinical
setting.
Autophagy is a pathway that is responsible for
maintaining cellular homeostasis by destroying aggregated
biomolecules (4). Upon starvation or
hypoxia, autophagosomes (APs) are formed and degraded upon fusion
with a lysosome (5). ‘Autophagic
flux’ is defined as a well-balanced cycle that consists of the
generation and degradation of APs. Notably, it has been previously
reported that autophagic flux disturbance may contribute to
ischemia-induced neuronal death (6).
This finding has prompted the hypothesis that the promotion of
autophagic flux may be a potent therapeutic strategy for cerebral
ischemia.
A previous study demonstrated that caloric
restriction (CR) alleviates neurodegeneration through the
activation of autophagy (7).
Intermittent fasting (IF), a variation of CR, is a defined regimen
that consists of intervals between meals. In the past decade, IF
has gained attention because of its beneficial role in weight loss
(8). Previous studies have indicated
that IF is neuroprotective due to its role in attenuating
neuroinflammation (9) and increasing
neurogenesis (10). However, to the
best of our knowledge, there are no reports on the possible
modulation of autophagy activity as well as apoptosis as a
therapeutic mechanism underlying IF on cerebral ischemia.
Therefore, the present study aimed to test the
hypothesis whether i) IF confers neuroprotection against cerebral
ischemia via modulation of autophagy; and ii) autophagic modulation
by IF is accompanied by an inhibition of apoptosis in a rat model
of focal cerebral ischemia.
Materials and methods
Animals
A total of 128 male Sprague-Dawley rats (weight,
220–250 g; age, 6 weeks) were purchased from a breeding colony
maintained at the Korea Research Institute of Bioscience and
Biotechnology (Daejeon, Korea) and maintained at a constant
temperature (23°C) and humidity (40–50%) with a 12-h light/dark
cycle. They were allowed free access to water and food. All
experimental procedures were carried out in accordance with the
National Institutes of Health (NIH, Bethesda, MD, USA). Rats were
randomly assigned to four groups: Control rats (n=32) were
permitted free access to food without any subsequent surgery; IF,
rats (n=32) were subjected to two weeks of IF without any surgery;
middle cerebral artery occlusion and reperfusion (MCAO/R), rats
(n=32) were permitted free access to food and subsequent MCAO/R;
and IF + MCAO/R, rats (n=32) were subjected to IF with subsequent
MCAO/R. To stimulate two weeks of IF, a diet schedule was designed
consisting of seven cycles of 24 h fasting and 24 h feeding. During
the IF period, all rats were allowed free access to water.
MCAO/R
Rats were subjected to MCAO/R as described in
previous studies (11,12). Briefly, rats were anesthetized via
intraperitoneal injection of 30 mg/kg zoletil (Virbac, Seoul,
Korea). Following a midline neck incision, the left common carotid
artery (CCA) and the external carotid artery were isolated and
ligated. A clip was placed on the internal carotid artery (ICA) to
prevent bleeding. After creating a small hole in the CCA, a 4–0
nylon monofilament with a silicon coated tip (0.35–0.37 mm) was
inserted into the hole of the left CCA. Following removal of the
clip at the ICA, the silicon-coated filament was placed into the
ICA to occlude the origin of the middle cerebral artery (MCA) until
resistance was felt. To produce reperfusion, the filament was
removed after 2 h of occlusion, and the incision was sutured.
Measurement of infarct volume
Infarct volume was assessed using the
2,3,5-triphenyltetrazolium chloride (TTC) method (13). At 24 h post-reperfusion, the brains
of rats (n=5/group) were cut into five slices (thickness, 2 mm),
and incubated in 2% TTC solution at 37°C in the dark for 30 min.
Brain slices were subsequently photographed. To calculate the
percentage of brain infarct, the following formula was used:
Infarct ratio (%) = (total contralateral hemispheric volume - total
ipsilateral hemispheric stained volume) / total contralateral
hemispheric volume × 100.
Assessment of brain edema
Brain edema was evaluated by assessing brain water
content (14). At 24 h
post-reperfusion, each ipsilateral hemisphere (n=5/group) was
weighed to obtain the wet weight. Samples were dried at 70°C for 48
h and the dried weight was subsequently measured. Brain water
content (%) was calculated using the following formula: [(wet
weight - dry weight) / wet weight] × 100.
Behavioral test
Two different behavioral tests were used in the
present study. Initially, rats (n=5/group) at post-operative day
(POD) 1 had their sensorimotor functions tested by modification of
scoring scales consisting of 18 points, as reported by Garcia et
al (15). Conversely, rats
(n=5/group) at POD 1, 3, 5, and 7 had their motor coordination
tested by rotarod test: Rats were placed on a rotarod drum and time
(sec) of latency to fall from the rod was measured thrice per day.
Rotarod speed was fixed to 15 rpm over a 5-min period. When rats
fell off the rod, trials ended. Average latency was calculated for
each testing day.
Neuronal counting
At 72 h post-reperfusion, rats (n=3/group) were
perfused with 4% paraformaldehyde (PFA). Brains were removed,
embedded in paraffin wax and serially sectioned at 5-µm thickness.
Five sections were randomly selected according to anatomical
landmarks corresponding to ‒1.4 to ‒1.8 mm in the antero-posterior
axis of the brain atlas and stained with 0.1% cresyl-violet
solution. Mean number of morphologically intact neurons in the
1-mm2 sample of the cortex were counted and averaged by
three blinded observers using light microscopy. Only intact neurons
with clear nuclei and large cell bodies were counted.
Neuronal apoptosis assay
In order to assess neuronal apoptosis, terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
was used. Five cortical sections of each rat (n=3/group) at POD 1
were prepared, using the random selection protocols mentioned, and
stained using a commercial kit (Promega Corporation, Madison, WI,
USA) according to the manufacturer's protocol. The mean number of
TUNEL-positive cells in the cortical samples were blindly counted
and averaged.
Immunofluorescence
Randomly selected five sections from each rat
(n=3/group) were deparaffinized, hydrated in distilled water, and
exposed an antigen through antigen retrieval process using tissue
retrieval solution. Endogenous peroxidase activity was blocked with
3% H2O2 for 1 h and sections were rinsed in
phosphate-buffered saline (PBS). Subsequently, each of the sections
were incubated in a humid chamber for 24 h at 4°C with primary
rabbit polyclonal anti-light chain 3 (LC3) (PM036; MBL, Co., Ltd.,
Nagoya, Japan) and mouse monoclonal anti-NeuN (GTX30773; Gene Tex,
Inc., Irvine, CA, USA) antibodies that were diluted in blocking
solution to 1:200. Following three washes in PBS, the sections were
incubated with secondary antibodies containing polyclonal goat
anti-rabbit IgG (111–225–144) and anti-mouse IgG (115–165–003; both
Jackson Immunoresearch, West Grove, PA, USA) at a dilution of 1:200
for 4 h at room temperature, respectively. The sections were
subsequently washed with PBS and mounted with a coverslip. Images
were captured by a laser confocal microscope (LSM-700; Carl Zeiss
AG, Oberkochen, Germany).
Transmission electron microscopy
Rats (n=3/group) at 12 h post-reperfusion were
perfused with 0.1 M phosphate buffer (pH 7.4) containing 2% PFA and
2% glutaraldehyde. Brains were removed, post-fixed for 24 h,
immersed in 1% osmium tetroxide in 0.1 M cacodylate buffer and
subsequently embedded in an epoxy resin. Ultrathin sections (50 nm)
of the cortical regions were obtained, stained with 3% lead
citrate, and examined using a transmission electron microscope
(TEM; HT7700; Hitachi, Ltd., Tokyo, Japan).
Western blot analysis
At 24 h post-reperfusion, the cortical tissues of
each rat (n=5/group) in the left MCA territory were rapidly
dissected. Tissue samples were homogenized by adding lysis buffer
and a protease inhibitor cocktail prior to centrifugation at 13,800
× g for 10 min at 4°C. The total protein concentration of
the supernatant was determined using a BCA protein assay (Pierce
Biotechnology, Inc., Rockford, IL, USA). Subsequently, the protein
sample was separated by 10% SDS-PAGE and transferred onto a
polyvinylidenedifluoride membrane (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), which was blocked with 5% nonfat milk in
Tris-buffered saline with 0.1% Tween 20 (TBS-T) for 2 h at room
temperature. The membranes were incubated with the following
primary antibodies (diluted to 1:1,000) overnight at 4°C: Mouse
monoclonal anti-β-actin (sc-47778), rabbit polyclonal anti-LC3
(sc-28266), rabbit polyclonal anti-beclin1 (sc-11427), mouse
monoclonal anti-p62 (1:1,000; sc-28359), rabbit polyclonal
anti-Rab7 (sc-10767) and goat polyclonal anti-cathepsin D
(sc-6486). All primary antibodies were purchased from Santa Cruz
Biotechnology, Inc., (Dallas, TX, USA). The membranes were washed
three times for 10 min in TBS-T and further incubated for 2 h at
room temperature with horseradish peroxidase (HRP)-conjugated
rabbit anti-goat IgG (P0449), rabbit anti-mouse IgG (P0260) and
swine anti-rabbit IgG (P0217) secondary polyclonal antibodies (all
1:2,000; all Dako, Glostrup, Denmark). Following three washes in
TBS-T, the membranes were observed using the enhanced
chemiluminescent HRP substrate (WBKLS0500; EMD Millipore,
Billerica, MA, USA). Densitometric analysis was performed using
ImageJ software (Version 1.48; National Institutes of Health,
Bethesda, MA, USA).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Comparisons of the data from the different groups were
analyzed using one-way analysis of variance (PASW Statistics
version 18; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
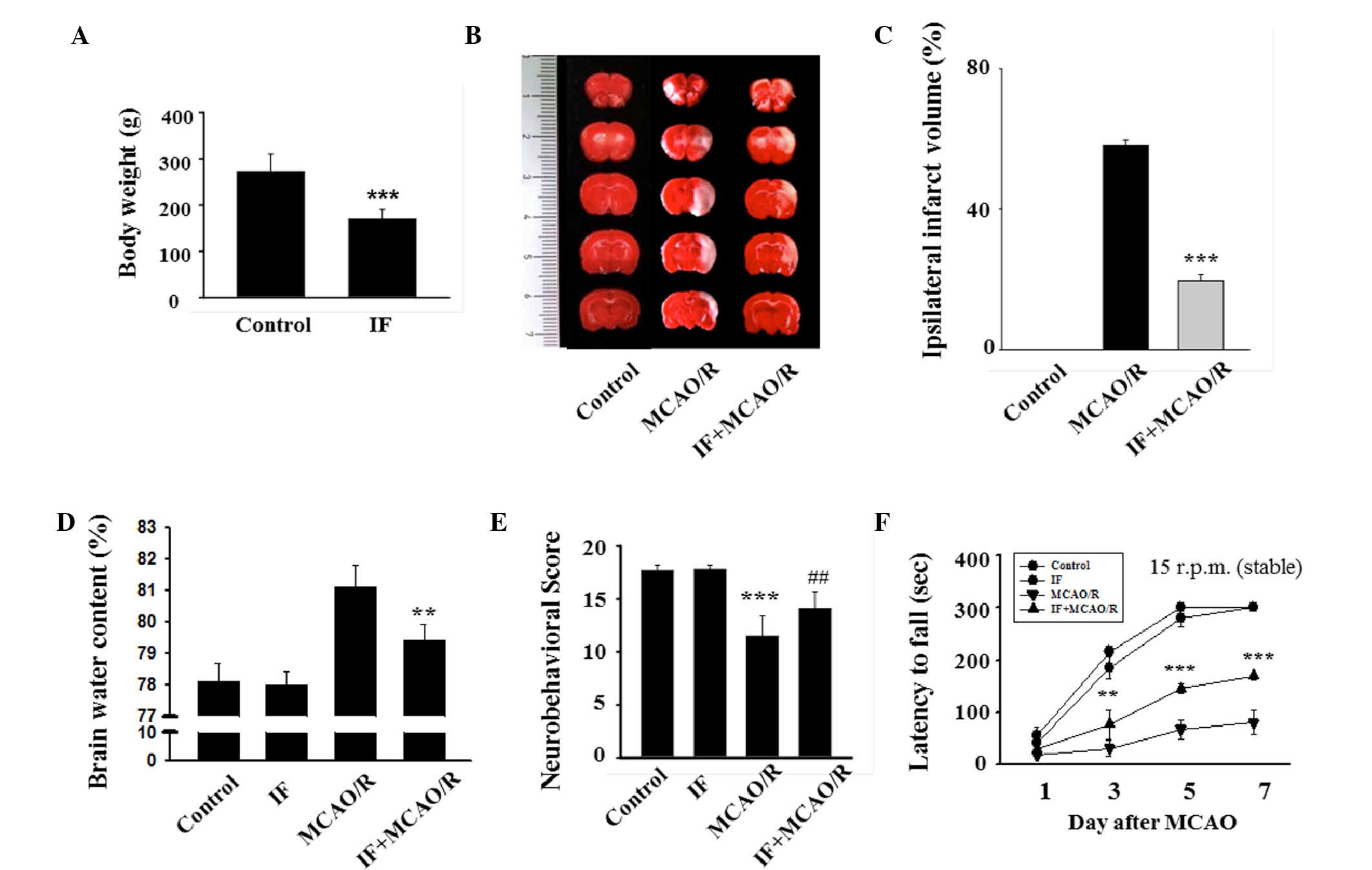
IF diminishes MCAO/R-induced infarct
volume, brain edema, and behavioral deficits
Initially, successful establishment of IF in rats
was confirmed by evaluating body weight. Similar to a previous
study (10), the body weights of IF
rats were significantly reduced compared to the control rats
(170±20.29 vs. 232±37.5 g; P<0.001; Fig. 1A). To evaluate the effects of IF on
infarct volume, brain edema and behavioral deficits after MCAO/R,
TTC staining was performed, brain water content was measured and
behavioral tests were performed, respectively. There were no
visibly infarcted areas in the control group, whereas the MCAO/R
group exhibited marked areas of infarction (Fig. 1B). The ipsilateral infarct ratio of
the IF + MCAO/R group was ~1/3 of that of the MCAO/R group
(19.59±1.40 vs. 58.23±1.71%; P<0.001; Fig. 1C). Furthermore, the degree of brain
edema in the IF + MCAO/R group was significantly decreased compared
with that of the MCAO/R group (79.4±0.5 vs. 81.1±0.7%; P<0.01;
Fig. 1D). Two different behavioral
tests were conducted to assess the role of IF in MCAO/R-associated
sensorimotor deficits and motor incoordination. The MCAO/R group
was significantly impaired in sensorimotor functions compared with
the control group (11.5±1.9 vs. 17.8±0.5; P<0.001; Fig. 1E). However, the IF + MCAO/R group
exhibited significantly improved sensorimotor function compared
with the MCAO/R group (14.1±1.6 vs. 11.5±1.9; P<0.01). In the
rotarod task performed on POD 1, 3, 5, and 7, latencies to fall
were significantly reduced in the MCAO/R group compared with the
control and IF groups at all time points except POD 1 (P<0.05
and P<0.001, respectively; Fig.
1F). As predicted, rats in the IF + MCAO/R group remained on
the rotarod longer than those in the MCAO/R group at POD 3, 5, and
7. The average latency of the IF + MCAO/R group was significantly
greater than the average latency of the MCAO/R group at POD 7
(168.8±7 vs. 79.9±23.2 sec; P<0.001). These findings
demonstrated that IF decreased the infarct volume, reduced the
degree of brain edema, and improved neurobehavioral deficits in rat
models of cerebral ischemia.
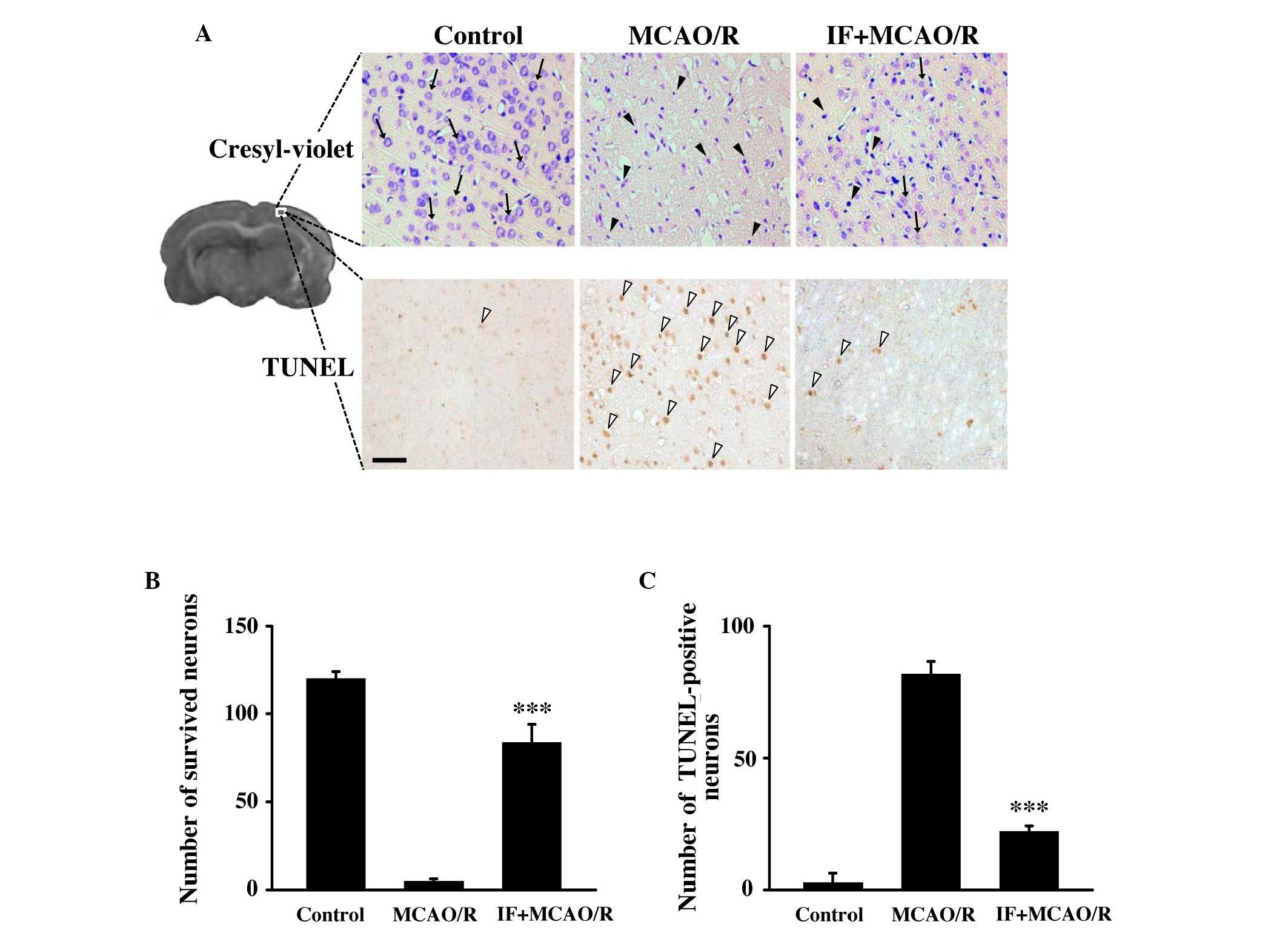
IF attenuates neuronal death and
apoptosis induced by MCAO/R
Cresyl-violet staining was used to assess the extent
of neuronal death in the cortex at 72 h post-reperfusion. Although
only minimal intact neurons were detected (black arrows; Fig. 2A), dying or dead neurons (black
arrowheads) were markedly increased in the cortices of the MCAO/R
group. However, the number of intact neurons was largely preserved
in the IF + MCAO/R group compared with the MCAO/R group. The number
of intact neurons in the cortex of the IF + MCAO/R group was
significantly increased by ~20 fold (83.5±10.4 vs. 4.5±1.5;
P<0.001; Fig. 2B) compared with
the MCAO/R group. To assess neuronal apoptosis, the TUNEL assay was
used. Neurons undergoing apoptosis (white arrowheads; Fig. 2A) in the IF + MCAO/R group were
markedly decreased in the cortex compared with the MCAO/R group.
Apoptotic cortical neurons in the IF + MCAO/R group were
significantly decreased by ~25% compared with those in the MCAO/R
group (22±2.2 vs. 81.7±4.8; P<0.001; Fig. 2C). This data suggests that the
neuroprotection induced by IF involves, at least in part, an
attenuation of neuronal apoptosis.
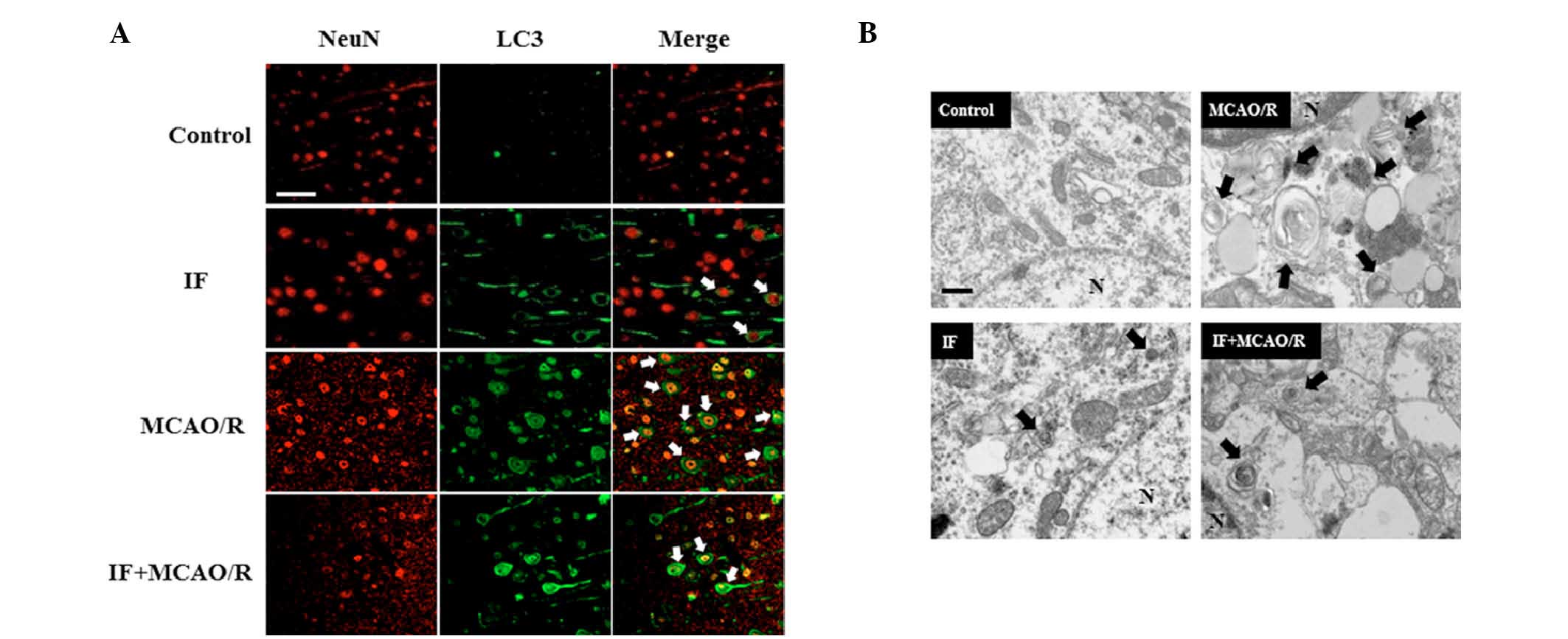
IF activates neuronal autophagy and
attenuates the MCAO/R-induced accumulation of APs
It was hypothesized that IF may be neuroprotective
in focal cerebral ischemia by modulating autophagy activity. While
neuronal APs (white arrows; Fig.
3A), as indicated by LC3 puncta surrounding the
NeuN-immunopositive neuronal nuclei, were rarely detected in the
cortices of the control group, these were markedly increased by IF.
Furthermore, as expected, APs were abundantly present in cortical
neurons in the MCAO/R group compared with the control group.
However, APs in the IF + MCAO/R group were decreased compared with
the MCAO/R group. Subsequently, in order to visualize APs directly,
the cortical neuronal ultrastructure was observed with the aid of
TEM (Fig. 3B). APs (black arrows)
were identified by the presence of digested or partially digested
organelles confined within a double membrane. APs in cortical
neurons were increased in the IF group compared with the control
group. The results of immunofluorescence staining and TEM also
indicated that MCAO/R induced AP accumulation, whereas IF
attenuated this. This data revealed that IF increased the basal
activity of autophagy, and upon MCAO/R, IF attenuated the
pathological accumulation of APs in cortical neurons.
IF attenuates MCAO/R-induced
autophagic flux disturbance
The pathological accumulation of APs by MCAO/R may
be triggered by two mechanisms: i) An increase in autophagy
induction and ii) the disturbance of autophagic flux (16). To elucidate the mechanism, initially
the amount of the conversion of LC3-I to its lipidated form was
quantified, as LC3-II is a reliable marker for autophagy induction
(17). In the present study, the
ratio of LC3-II/LC3-I was increased in the IF, MCAO/R and IF +
MCAO/R groups compared with the control group (Fig. 4A and B). As no difference was
detected in LC3-II levels between the MCAO/R and IF + MCAO/R
groups, this suggests that neuronal autophagy was not significantly
altered during autophagy induction in the IF + MCAO/R group,
compared with the MCAO/R group. The effect of IF on autophagic flux
in rat cortical neurons, with or without MCAO/R, was subsequently
tested by measuring the levels of p62, cathepsin D, and Rab7
(18). Whereas p62, a cargo protein
that undergoes autophagic degradation, is a negative-correlative
marker for autophagic flux (19),
cathepsin D and Rab7, an intralysosomal enzyme involved in the
degradation of cellular waste and an essential protein for
autophagosome-lysosome fusion, respectively, are both
positive-correlative markers for autophagic flux (20,21).
Notably, the level of p62 was not altered in the IF group, but was
significantly increased in the MCAO/R group compared with the
control group (P<0.01; Fig. 4C).
This suggests that autophagic flux was maintained in the IF group,
and disturbed in the MCAO/R group. Additionally, the IF + MCAO/R
group exhibited a significant decrease in neuronal p62 accumulation
compared with the MCAO/R group (P<0.01; Fig. 4A and C). Furthermore, cathepsin D and
Rab7 levels were increased in the IF + MCAO/R group compared with
the MCAO/R group (Fig. 4A, D and E).
These results suggest that IF attenuates MCAO/R-induced autophagic
flux disturbance by promoting autophagic clearance, rather than by
activating autophagy induction.
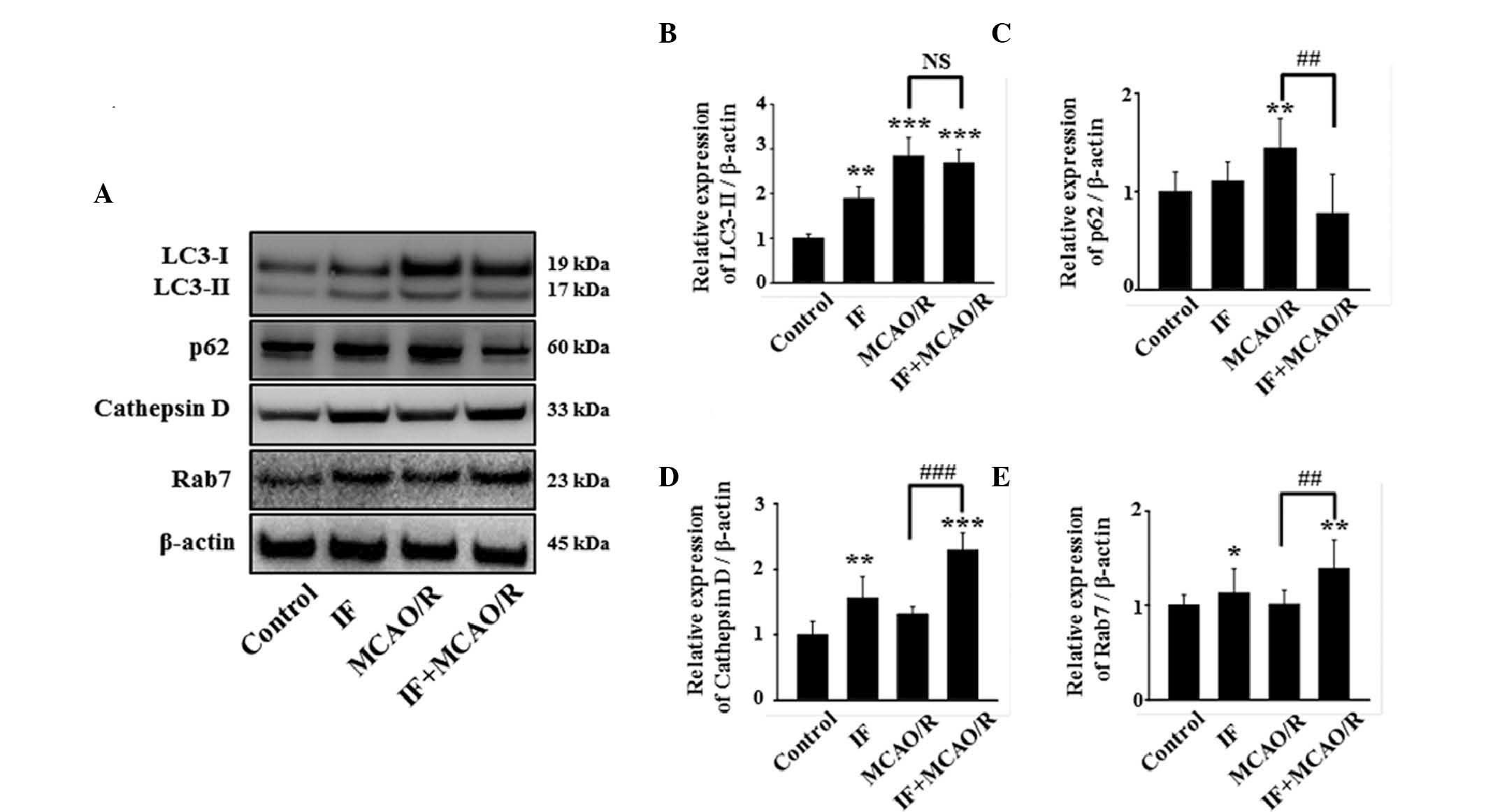 | Figure 4.IF enhances the autophagic flux in
cortical neurons after MCAO/R. (A) Representative image of
immunoblots for LC3, p62, cathepsin D, and Rab7 in cortical
homogenates in different groups at 12 h post-MCAO/R. (B)
Quantification of LC3-II protein level. These data were quantified
as a proportion of LC3-I intensity (n=5/group; **P<0.01 and
***P<0.001 vs. control). (C-E) Quantification of p62, cathepsin
D, and Rab7 protein levels, respectively. β-actin used as a loading
control. (n=5/group; *P<0.05, **P<0.01 and ***P<0.001 vs.
control; ##P<0.01 and ###P<0.001 vs.
MCAO/R). Data are presented as the mean ± standard error of the
mean. IF, intermittent fasting; MCAO/R, middle cerebral artery
occlusion and reperfusion; LC3, protein light chain 3; NS, no
statistical significance. |
Discussion
In the present study, the involvement of autophagy
in a rat model of MCAO/R after IF was investigated. The findings
demonstrated that IF decreased MCAO/R-induced damages, including an
increased infarct volume, brain edema, neurobehavioral deficits,
and apoptotic neuronal death. In addition, IF was demonstrated to
elevate basal autophagic activity and attenuate MCAO/R-induced
autophagic flux disturbance using various methods. These results
provide a potential direct mode of action of the neuroprotective
role of IF, rather than indirect effects (hormonal or emotional
influences). Therefore, preventing the deterioration of autophagic
clearance, thereby minimizing autophagic flux disturbance, may be
one of the direct effects of IF-induced neuroprotection.
Programmed cell death (PCD), which includes
apoptosis (PCD type I) and autophagy (PCD type II), has an
important role in cerebral ischemia (22). Whereas PCD type I always contributes
to cell death, autophagy can lead to either cell survival or cell
death, depending on the duration and severity of the process
(23). The beneficial roles of
autophagy in cellular survival are due to the elimination of
damaged organelles and protein aggregates, as well as the
facilitation of bioenergetic homeostasis (24). Previous studies have demonstrated
that autophagy triggered by nutrient and growth factor deprivation
has a pro-survival function at the cellular and organismal levels
during instances of endoplasmic reticulum stress, development,
microbial infection, and diseases characterized by the accumulation
of protein aggregates (25,26). In contrast, it has been demonstrated
that autophagy can also promote cell death (27). When cells receive continuous or
extremely stressful stimuli, PCD type II may occur (28). PCD type II has been implicated in
various neurodegenerative diseases, including Alzheimer's disease
and Parkinson's disease, as well as in acute injuries, such as
stroke and trauma (29).
Although the key determinant of the paradoxical
roles of autophagy in nervous system diseases remains to be
addressed, it is generally accepted that ‘autophagic stress’, which
is the sustained impairment of the balance between AP formation and
degradation, contributes to PCD type II (30). If APs are efficiently degraded in
autolysosomes, neurons can maintain homeostasis, leading to
survival. However, when the late stages of the autophagic process
(maturation or degradation) are defective, autophagic degradation
is impaired, which can cause the pathologic accumulation of APs and
subsequent neurodegeneration. In the present study, it was
demonstrated that IF and MCAO/R increased APs in neurons, both
morphologically and biochemically; whereas IF increased the degree
of autophagic degradation, and MCAO/R led to a decrease. Notably,
IF prior to MCAO/R significantly increased the extent of autophagic
degradation when compared with MCAO/R alone. These observations
support the hypothesis that IF preconditioning may confer an
attenuation of MCAO/R-induced autophagic stress, and thus induce
neuroprotection.
If it is true that IF attenuates autophagic stress
and thereby confers neuroprotection, the underlying mechanism of
this phenomenon requires investigation. Furthermore, the possible
relationship between autophagic flux restoration and apoptosis
inhibition in neurons, both of which were observed as effects of IF
on ischemic neurons, are yet to be elucidated. Recent studies have
focused on the neuroprotective effects of ischemic preconditioning
(IPC) to investigate these mechanisms. It has been reported that
the autophagy inhibitor 3-methyladenine significantly suppressed
IPC-induced neuroprotection and molecular chaperone upregulation in
a rat model of cerebral ischemia (31). This data suggests that autophagy
exerts IPC-induced neuroprotection via an upregulation of molecular
chaperones. Furthermore, Carloni et al (32) revealed that the upregulation of
autophagy was neuroprotective in a cerebral hypoxia-induced
ischemia model in neonatal rats. According to their reports, an
upstream autophagic activator, Beclin1, was markedly enhanced after
hypoxia-ischemia and co-localized with TUNEL-positive cells,
suggesting that the upregulation of autophagy and the promotion of
apoptosis occurred simultaneously during ischemic injury. Moreover,
inhibiting autophagy caused rapid cell death by apoptosis.
Considering these previous reports, the findings of the present
study suggest that IF increases basal activity of autophagy,
thereby strengthening neuronal defense mechanisms, including
molecular chaperone upregulation. Once toxic conditions are imposed
on neurons, such as via MCAO/R, IF-induced autophagy activation
confers neuroprotection through an inverse relationship with
apoptosis.
In conclusion, the present study demonstrated that
IF prevents neuronal damage in focal cerebral ischemia and that
this protection is, at least partly, mediated by minimizing
autophagic flux disturbance and the inhibition of apoptosis.
Nevertheless, the possibility that IF induces neuroprotective
effects via additional mechanisms such as systemic effects,
(emotional and hormonal influences) cannot be excluded. Therefore,
further studies with a more isolated approach may be necessary to
examine the preventive role of IF in cerebral ischemia.
Acknowledgements
This study was equally supported by the Development
of Forest Science and Technology (grant no. S111414L030100) and the
Korea Research Foundation (grant no. NRF-2016R1C1B2012351).
References
|
1
|
Lipton P: Ischemic cell death in brain
neurons. Physiol Rev. 79:1431–568. 1999.PubMed/NCBI
|
|
2
|
Kim JH, Yu KS, Jeong JH, Lee NS, Lee JH,
Jeong YG, Yoo YC and Han SY: All-trans-retinoic acid rescues
neurons after global ischemia by attenuating neuroinflammatory
reactions. Neurochem Res. 38:2604–2615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu L, Zhang W, Wang L, Li Y, Tan B, Lu X,
Deng Y, Zhang Y, Guo X, Mu J and Yu G: Curcumin prevents cerebral
ischemia reperfusion injury via increase of mitochondrial
biogenesis. Neurochem Res. 39:1322–1331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Son JH, Shim JH, Kim KH, Ha JY and Han JY:
Neuronal autophagy and neurodegenerative diseases. Exp Mol Med.
44:89–98. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang F, Chu X, Yin M, Liu X, Yuan H, Niu Y
and Fu L: mTOR and autophagy in normal brain aging and caloric
restriction ameliorating age-related cognition deficits. Behav
Brain Res. 264:82–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barnosky AR, Hoddy KK, Unterman TG and
Varady KA: Intermittent fasting vs daily calorie restriction for
type 2 diabetes prevention: A review of human findings. Transl Res.
164:302–311. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fann DY, Santro T, Manzanero S,
Widiapradja A, Cheng YL, Lee SY, Chunduri P, Jo DG, Stranahan AM,
Mattson MP and Arumugam TV: Intermittent fasting attenuates
inflammasome activity in ischemic stroke. Exp Neurol. 257:114–119.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Manzanero S, Erion JR, Santro T, Steyn FJ,
Chen C, Arumugam TV and Stranahan AM: Intermittent fasting
attenuates increases in neurogenesis after ischemia and reperfusion
and improves recovery. J Cereb Blood Flow Metab. 34:897–905. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Calloni RL, Winkler BC, Ricci G, Poletto
MG, Homero WM, Serafini EP and Corleta OC: Transient middle
cerebral artery occlusion in rats as an experimental model of brain
ischemia. Acta Cir Bras. 25:428–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Güzel A, Rölz R, Nikkhah G, Kahlert UD and
Maciaczyk J: A microsurgical procedure for middle cerebral artery
occlusion by intraluminal monofilament insertion technique in the
rat: A special emphasis on the methodology. Exp Transl Stroke Med.
6:6. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li F, Irie K, Anwer MS and Fisher M:
Delayed Triphenyltetrazolium chloride staining remains useful for
evaluating cerebral infarct volume in a rat stroke model. J Cereb
Blood Flow Metab. 17:1132–1135. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ito U, Ohno K, Nakamura R, Suganuma F and
Inaba Y: Brain edema during ischemia and after restoration of blood
flow. Measurement of water, sodium, potassium content and plasma
protein permeability. Stroke. 10:542–547. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garcia JH, Wagner S, Liu KF and Hu XJ:
Neurological Deficit and Extent of neuronal necrosis attributable
to middle cerebral artery occlusion in rats: Statistical
validation. Stroke. 26:627–635. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gu Z, Sun Y, Liu K, Wang F, Zhang T, Li Q,
Shen L, Zhou L, Dong L, Shi N, et al: The role of autophagic and
lysosomal pathways in ischemic brain injury. Neural Regen Res.
8:2117–2125. 2013.PubMed/NCBI
|
|
17
|
Tanida I, Ueno T and Kominami E: LC3 and
Autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Su H, Li F, Ranek MJ, Wei N and Wang X:
COP9 Signalosome regulates autophagosome maturation. Circulation.
124:2117–2128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sarkar C, Zhao Z, Aungst S, Sabirzhanov B,
Faden AI and Lipinski MM: Impaired autophagy flux is associated
with neuronal cell death after traumatic brain injury. Autophagy.
10:2208–2222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hyttinen JM, Niittykoski M, Salminen A and
Kaarniranta K: Maturation of autophagosomes and endosomes: A key
role for Rab7. Biochim Biophys Acta. 1833:503–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hossmann KA: Viability thresholds and the
penumbra of focal ischemia. Ann Neurol. 36:557–565. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das G, Shravage BV and Baehrecke EH:
Regulation and function of autophagy during cell survival and cell
death. Cold Spring Harb Perspect Biol. 4(pii):
a0088132012.PubMed/NCBI
|
|
25
|
Dalby KN, Tekedereli I, Lopez-Berestein G
and Ozpolat B: Targeting the prodeath and prosurvival functions of
autophagy as novel therapeutic strategies in cancer. Autophagy.
6:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bursch W, Ellinger A, Gerner CH, Fröhwein
U and Schulte-Hermann R: Programmed cell death (PCD). Apoptosis,
autophagic PCD, or Others? Ann N Y Acad Sci. 926:1–12. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yue Z, Friedman L, Komatsu M and Tanaka K:
The cellular pathways of neuronal autophagy and their implication
in neurodegenerative diseases. Biochim Biophys Acta.
1793:1496–1507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chu CT: Autophagic stress in neuronal
injury and disease. J Neuropathol Exp Neurol. 65:423–432. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meriin AB and Sherman MY: Role of
molecular chaperones in neurodegenerative disorders. Int J
Hyperthermia. 21:403–419. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carloni S, Girelli S, Scopa C, Buonocore
G, Longini M and Balduini W: Activation of autophagy and Akt/CREB
signaling play an equivalent role in the neuroprotective effect of
rapamycin in neonatal hypoxia-ischemia. Autophagy. 6:366–377. 2010.
View Article : Google Scholar : PubMed/NCBI
|