Introduction
Adriamycin (ADR), an anthracycline, is an effective
chemotherapeutic agent. However, ADR is cardiotoxic and may cause
dose-dependent, progressive cardiac damage which is clinically
manifested as reduced left ventricular ejection fraction and
ultimately heart failure (1).
Moreover, chronic ADR-induced cardiomyopathy is associated with a
poor prognosis and low survival rate (1). One of the options to prevent
cardiotoxicity is a dose-limiting approach or limiting the use of
ADR, however, that may also compromise its chemotherapeutic
properties. In recent years, studies have been conducted to
elucidate the mechanisms underlying ADR-induced cardiomyopathy and
to explore interventional strategies, and several mechanisms have
been hypothesized to account for the ADR-induced cardiotoxicity,
including oxidative stress, imbalance of Ca2+ in the
cytoplasm and mitochondria, apoptosis and fibrosis (2,3).
However, a definite mechanism remains unclear.
Pyruvate, the anionic form of a simple α-keto acid,
is normally regarded as a key intermediate in the oxidative or
anaerobic metabolism of glucose. Pyruvate has been shown to
ameliorate organ damage, such as ischemic cardiomyopathy as well as
ischemic-reperfusion injury of heart (4), intestine (5), liver (6), and brain (7), although the underlying mechanisms are
not well defined. In addition, pyruvate also plays an important
role in combatting oxidative damage by scavenging hydrogen peroxide
and lipid peroxides and by increasing the antioxidant redox
potential of the endogenous glutathione system (8). Furthermore, pyruvate has also been
shown to resist apoptosis and fibrosis in various tissues (9,10).
However, due to aqueous instability, the use of pyruvate as a
therapeutic agent is limited. Ethyl pyruvate (EP), an ester form of
pyruvic acid (11), is used as a
practical pyruvate precursor for administration and has better
protective effects on some damaged organs because of its stable
chemical structure (5).
Therefore, EP may be able be to act as a metabolic
substrate and an effective reactive oxygen species (ROS) scavenger.
In the present study, it was hypothesized that EP has protective
effects against ADR-induced cardiomyopathy in an animal model.
Experiments were performed to examine the protective effects of EP
in a rat model, in which chronic myocardial injury was induced by
ADR. In this particular rat model, cardiac geometry and function,
myocardial tissue oxidative stress and fibrosis, and myocardial
cell apoptosis were assessed.
Materials and methods
Animals
All procedures involving animals were approved by
the Ethics Committee for Animal Research of Wuhan University
(Wuhan, China). All animals received humane care in compliance with
the Guide for the Care and Use of Laboratory Animals prepared by
the Institute of Laboratory Animal Resources and the National
Research Council (12). Male
Sprague-Dawley (SD) rats, aged 6–8 weeks and weighing 150–180 g,
were purchased from the Experimental Animal Center of Wuhan
University. All animals were acclimated to the laboratory for 7
days prior to the experiment and were maintained in a
light-controlled room (12-h light/dark cycle) at an ambient
temperature of 25° with free access to water and standard chow.
Sixty male SD rats were randomly divided into four
groups, namely the control group, ethyl pyruvate (EP) group,
Adriamycin (ADR) group and ADR + EP group (n=15 per group). Rats in
the ADR and ADR + EP groups were treated with ADR (Actavis Italy
SpA, Nerviano, Italy) at a dose of 2.5 mg/kg/week via
intraperitoneal injection for 6 weeks. By contrast, rats in the
control and EP groups were treated with normal saline (via
intraperitoneal injection) at the same dose as ADR for 6 weeks.
From the eighth week, rats in the EP group and the ADR + EP group
received EP (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany)
via stomach lavaging at a dose of 50 mg/kg/day for 30 days. Upon
the completion of 30 days treatment with EP, cardiac function was
accessed by echocardiography. Rats were then sacrificed by an
overdose of anesthesia (3.5 mg/100 g pentobarbital) for the
harvesting of heart tissues.
Echocardiography test
Echocardiography was performed using a
high-resolution ultrasound imaging system equipped with a 7V3 probe
with a frequency of 6.0 MHz (Acuson Sequoia; Siemens, Washington,
DC, USA). Fractional shortening (FS), left ventricular internal
dimension diastolic (LVIDD) and left ventricular internal dimension
systole (LVIDS) were recorded from the parasternal long-axis M-mode
images using averaged measurements from 3–5 consecutive cardiac
cycles. End diastolic volume (EDV) and end systolic volume (ESV)
were calculated from bi-dimensional long-axis parasternal views by
means of the single-plane area-length method. The ejection fraction
(EF) was calculated as follows: EF (%) = (LVEDV - LVESV) / LVEDV ×
100.
Histological examination and
determination of apoptosis
Myocardial tissues excised by horizontal intercept
from the middle part of the whole heart were fixed in 10% buffered
formalin for 24 h, and then embedded in paraffin and sliced into
5-µm sections. The sections were stained with picrosirius red (PSR)
to identify collagen deposition, and then were visualized using
light microscopy. Fibrillar collagen was visualized under the
microscope and the left ventricular collagen volume fraction was
measured using a quantitative digital image analysis system
(Image-Pro Plus 6.0; Media Cybernetics, Inc., Rockville, MD, USA).
The cardiomyocyte apoptosis rate was also assessed using a terminal
deoxynucleotidyl transferase-mediated dUTP nick end-labeling
(TUNEL) assay.
Briefly, sections (3-µm-thick) from formalin-fixed
and paraffin-embedded myocardial tissue were deparaffinized with
xylene and dehydrated with ethanol. Slides were rinsed twice with
PBS and treated with proteinase K (15l g/ml in 10 mM Tris/HCl; pH
7.4–8.0) for 15 min at 37°. Endogenous peroxidases were blocked
with 3% hydrogen peroxide in methanol at room temperature for 10
min. Tissue sections were analyzed with an in situ cell
death detection kit (Roche Diagnostics, Indianapolis, IN, USA), in
accordance with the manufacturer's instructions. Reactions were
visualized with fluorescence microscopy and were measured using a
quantitative Image-Pro Plus 6.0 digital image analysis system.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting
RT-qPCR and western blotting were performed as
previously described (13). Briefly,
after total RNA was extracted from ventricles using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
first strand cDNA was synthesized using a Transcriptor First Strand
cDNA Synthesis kit (Roche Diagnostics). qPCR was performed using
SYBR Green PCR Master Mix (Roche Diagnostics) to determine the
expression levels of genes of interest which were transforming
growth factor-β1 (TGFβ-1), collagen type 1 α 1 (Colla1), collagen
type 1 α 3 (Colla3), tissue inhibitor of metalloproteinase (TIMP)1,
TIMP2, matrix metalloproteinase (MMP)2 and MMP9, and the results
were normalized against GAPDH gene expression. PCR cycling
conditions were as follows: Predenaturation at 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec, 60°C for 1 min and 72°C
for 20 sec, and a final extension at 60°C for 5 min. The PCR
primers that were used are shown in Table I. PCR reactions were repeated
twice.
 | Table I.Primers for quantitative polymerase
chain reaction. |
Table I.
Primers for quantitative polymerase
chain reaction.
| Primer name | Forward primer | Reverse primer |
|---|
| GAPDH-rat |
ACGGGAAACCCATCACCATC |
TGGTGGTGCAGGATGCATTG |
| TGFβ1-rat |
GCGCCTGCAGAGATTCAAGTCAAC |
GTATCAGTGGGGGTCAGCAGCC |
| COL1a1-rat |
TGGCAACCTCAAGAAGTCCC |
ACAAGCGTGCTGTAGGTGAA |
| COL1a3-rat |
CAACCAGTGCAAGTGACCAA |
GCACCATTGAGACATTTTGAAG |
| MMP2-rat |
CTGATAACCTGGATGCCGTCGT |
TGCTTCCAAACTTCACGCTCTT |
| MMP9-rat |
TTATTGTGAGCATCCCTAGGG |
AGTGTCCGAGGAAGATACTTG |
| TIMP-1-rat |
ACAGCTTTCTGCAACTCGGA |
CCGGAAACCTGTGGCATTTC |
| TIMP-2-rat |
CTAATTGCAGGGAAGGCGGA |
CATAGGGCAGCGTGTGATCT |
For western blotting, cardiac tissue was lysed in
radioimmunoprecipitation assay buffer (Roche Diagnostics). Protein
extracts (30 µg per lane) were separated by 10% SDS-PAGE,
transferred to polyvinylidene difluoride (PVDF) membranes and
probed with primary antibodies overnight at 4°C. The primary
antibodies included anti-GAPDH (1:1,000; sc-365062), anti-NADPH
oxidase-4 (Nox4; 1:1,000; sc-517188), anti-Bax (1:1,000; sc-23959);
anti-Bcl-2 (1:1,000; sc-7382) and anti-caspase-3 (1:1,000;
sc-65496; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
anti-NADPH oxidase-2 (Nox2; (1:1,000; ab80508; Abcam, Cambridge,
UK). Following washing with TBST three times, the membranes were
incubated with horseradish peroxidase-conjugated goat
anti-rabbit/mouse secondary antibodies (1:100; G1201; Guge
Biotechnology Co., Ltd., Wuhan, China) for 1 h at room temperature.
Subsequently, the membranes were treated with ECL reagents (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) prior to visualization using
a FluorChem E imager (ProteinSimple, San Jose, CA, USA) according
to the manufacturer's instructions. The specific protein expression
levels were normalized to the levels of GAPDH on the same PVDF
membrane.
Evaluation of superoxide dismutase
(SOD) activity and malondialdehyde (MDA) concentration
The activity of SOD in myocardial tissue was
detected by the xanthine oxidase technique. This procedure is based
upon the inhibition of nitrite (NIT) reduction due to the
superoxide anion generated by the combination of xanthine and
xanthine oxidase. An SOD assay kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) was used to assess the
SOD activity. One unit of SOD was the amount that caused a 50%
inhibition in the rate of NIT reduction. The SOD activity was
expressed as U/mg protein in myocardial tissue homogenate.
The content of MDA in myocardial tissue was assayed
according to thiobarbituric acid (TBA) method. In this method MDA
reacts with TBA under high temperature (90–100°C) and acidic
conditions to form TBA reactive substances (TBARS). TBARS were
measured using a spectrophotometer at 532 nm. An MDA assay kit
(Nanjing Jiancheng Bioengineering Institute) was used to assess the
MDA concentration. The content of MDA was expressed in units of
nmol/mg protein in myocardial tissue homogenate.
Statistical analysis
All statistical analyses were performed using SPSS
software, version 18.0 (SPSS, Inc., Chicago, IL, USA). The
inter-group differences were analyzed by one-way analysis of
variance. The data are expressed as the mean + standard deviation.
All P-values were two-sided and P<0.05 was considered to
indicate a statistically significant difference.
Results
Mortality of rats
Out of 60 rats, 51 completed the study. The
mortality rates of the ADR group and the ADR + EP group were 33.3
and 26.7%, respectively, at the end of the interventions, while no
deaths were encountered in other groups.
Cardiac functions
Echocardiography was performed in each rat to
measure relative parameters of cardiac functions. As shown in
Fig. 1A, two-dimensional and M-mode
short-axis views of the left ventricle were acquired at the level
of the papillary muscles in rats. There was no difference in terms
of LVIDD and EVD between the four groups (data not shown). Compared
with the control group, treatment with EP did not affect FS, EF,
LVIDS and ESV (Fig. 1B-E). By
contrast, FS and EF in the ADR + EP group were significantly higher
than those in the ADR group (Fig. 1B and
C). Moreover, LVIDS and ESV in the ADR + EP group were greatly
lower than those in the ADR group (Fig.
1D and E), indicating that treatment with EP improved the
impaired cardiac functions induced by ADR in rats.
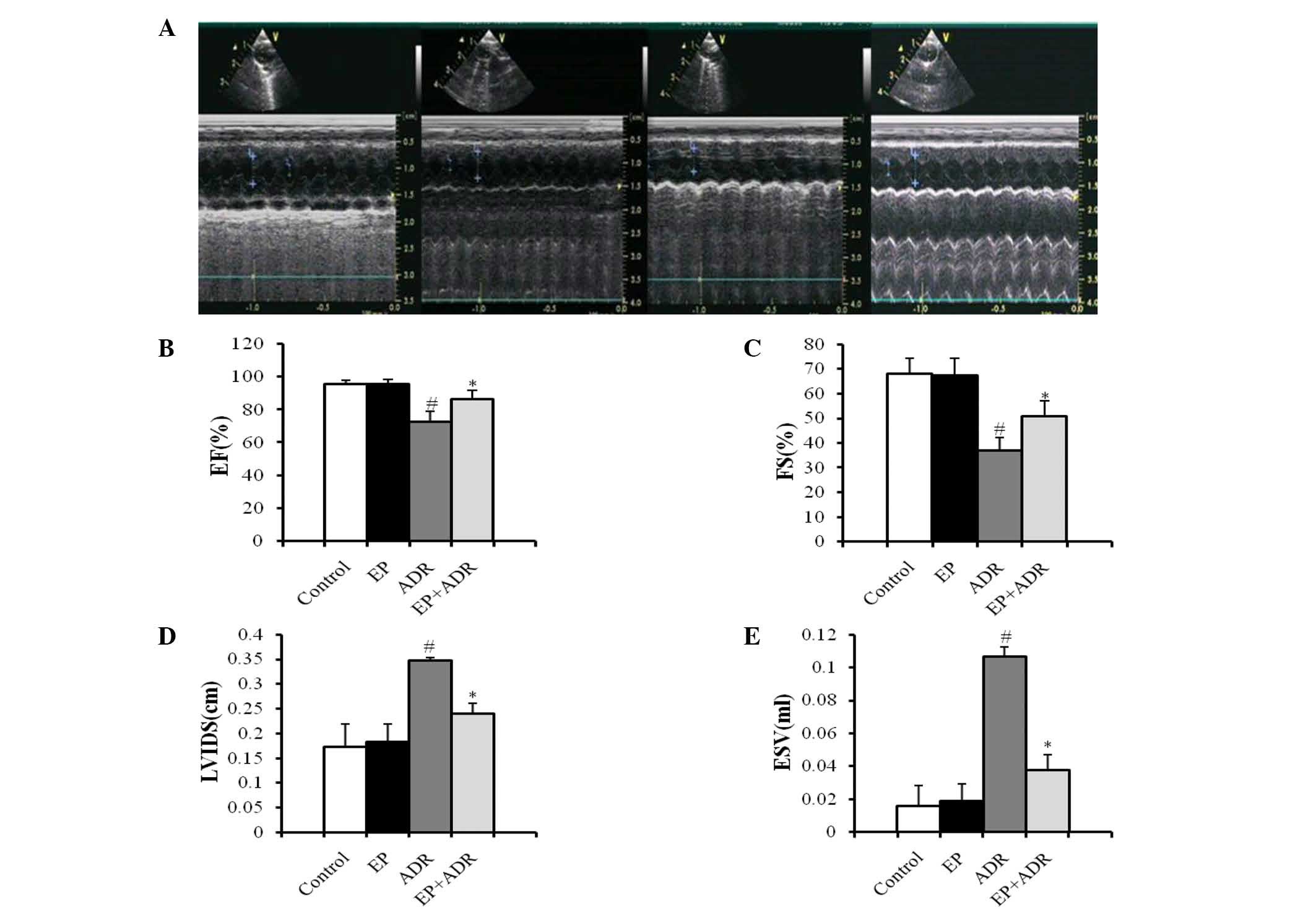 | Figure 1.Results of echocardiography indicate
that EP can improve the heart functions of rats damaged by ADR. (A)
Two-dimensional (upper panel) and M-mode (lower panel) short-axis
views of the left ventricle at the level of the papillary muscles
in four different animals from the control (first), EP (second),
ADR (third) and EP + ADR (last) groups, respectively. Results of
(B) EF, (C) FS, (D) LVIDS and (E) ESV in the four groups.
#P<0.05 vs. control and EP groups, *P<0.05 vs. ADR
group. EP, ethyl pyruvate; ADR, Adriamycin; EF, ejection fraction.
FS, fractional shortening; LVIDS, left ventricular internal
dimension systole; ESV, end systolic volume. |
Differential effects of ADR and EP on
oxidative stress-related molecules
SOD is the major defense against ROS production in
cells (14), and MDA is produced by
the actions of ROS on the lipids existing in the membranes of the
cells (15). Therefore, SOD and MDA
can be used to experimentally evaluate oxidative injury. To
understand the mechanisms underlying the protective role of EP
against ADR-induced cardiotoxicity, MDA levels and SOD activity of
myocardial tissues were measured in rats. Treatment with EP alone
did not affect the production of MDA and SOD compared with that in
the control group (Fig. 2A and B).
However, in the ADR group, the SOD activity of the myocardial
tissue was significantly lower (Fig.
2A), while the MDA level was significantly higher than those in
control group, EP group and ADR + EP group (Fig. 2B).
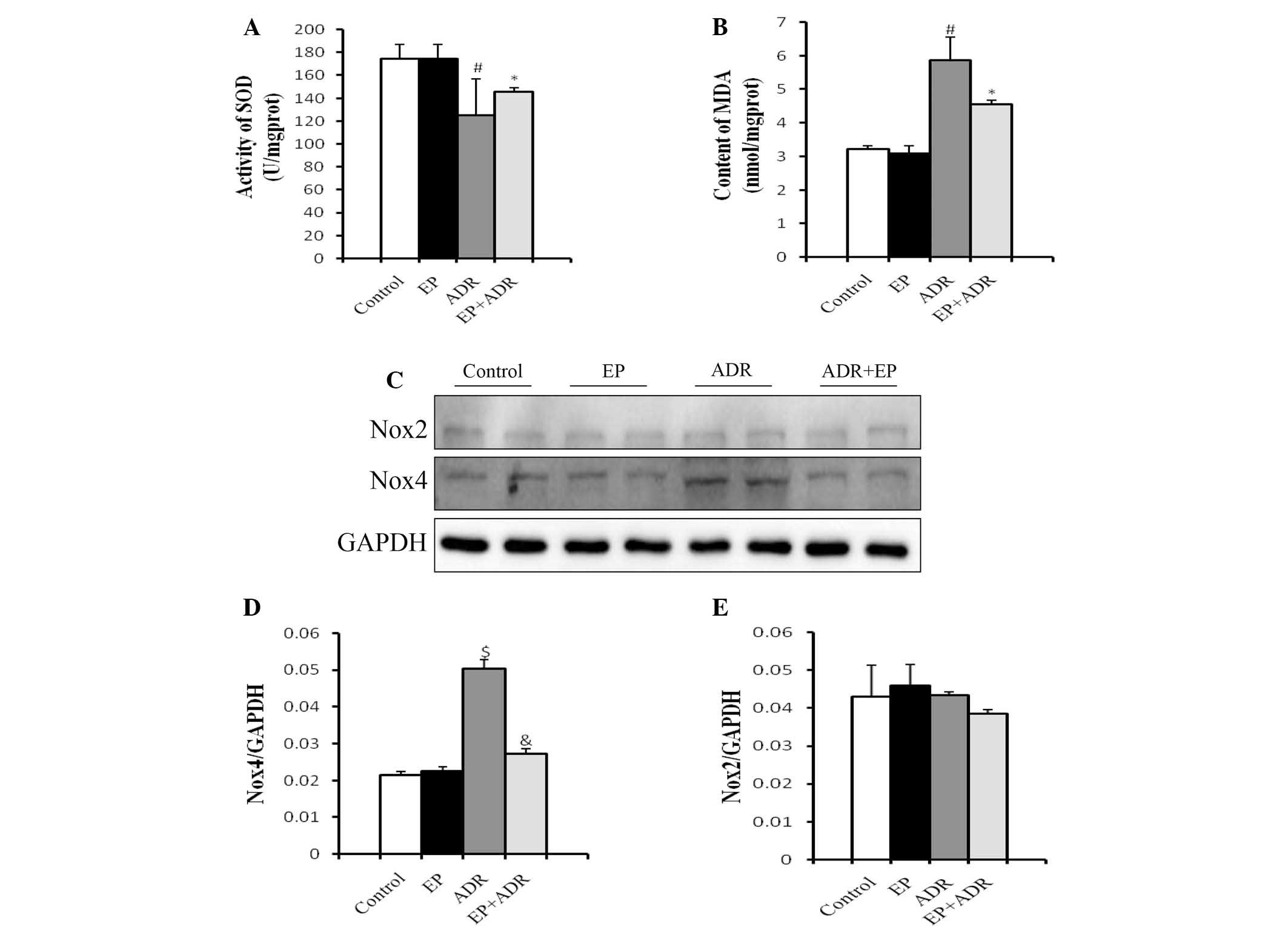 | Figure 2.Differential effects of ADR and EP on
oxidative stress-related molecules. (A) SOD activity and (B) MDA
levels in myocardial tissue. (C) Western blot results for Nox4 and
Nox2 in each group, and quantified results for (D) Nox4 and (E)
Nox2. #P<0.05 vs. control and EP groups, *P<0.05
vs. ADR group, $P<0.01 vs. control and EP groups,
&P<0.01 vs. ADR group. EP, ethyl pyruvate; ADR,
Adriamycin; SOD, superoxide dismutase; MDA, malondialdehyde; Nox2,
NADPH oxidase-2; Nox4, NADPH oxidase-4. |
Another two molecules associated with oxidative
stress, namely Nox2 and Nox4, were also investigated in rats using
western blot analysis. The protein level of Nox4 in the
cardiomyocytes in the ADR group was significantly higher than those
in the control group, EP group and ADR + EP group, and was reduced
by EP (Fig. 2C and D). By contrast,
there was no significant difference in the protein levels of Nox2
among the four groups (Fig. 2C and
E).
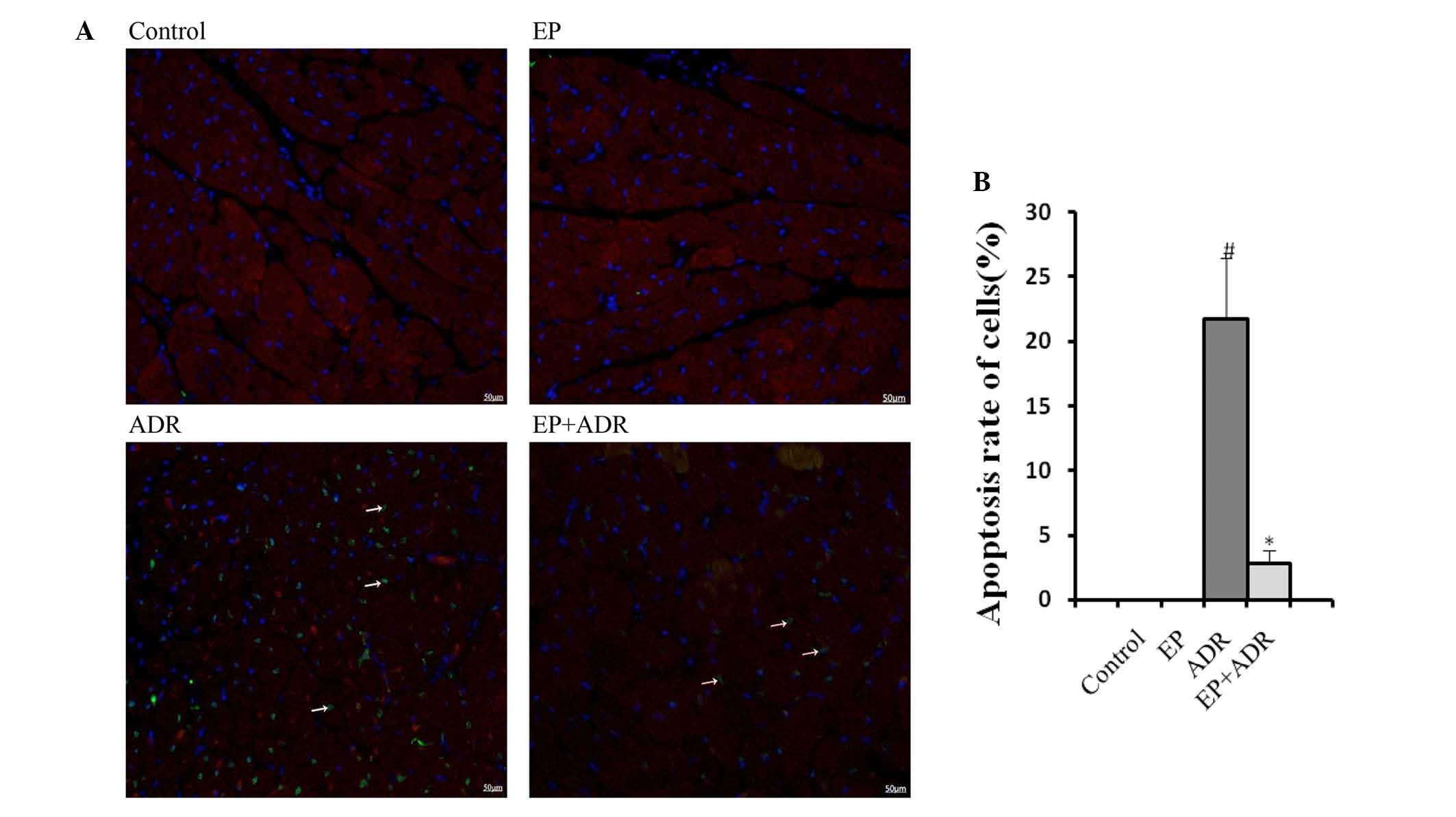
EP reduces ADR-induced myocardial cell
apoptosis
To assess the effects of ADR and EP on myocardial
cell apoptosis, markers of apoptosis were assessed in this study.
TUNEL staining showed an increase in cardiomyocyte apoptosis rate
in the ADR group, whereas the increased apoptosis rate was reduced
by EP treatment in the ADR + EP group (Fig. 3). No apoptosis was observed in the
control and EP only groups (Fig. 3).
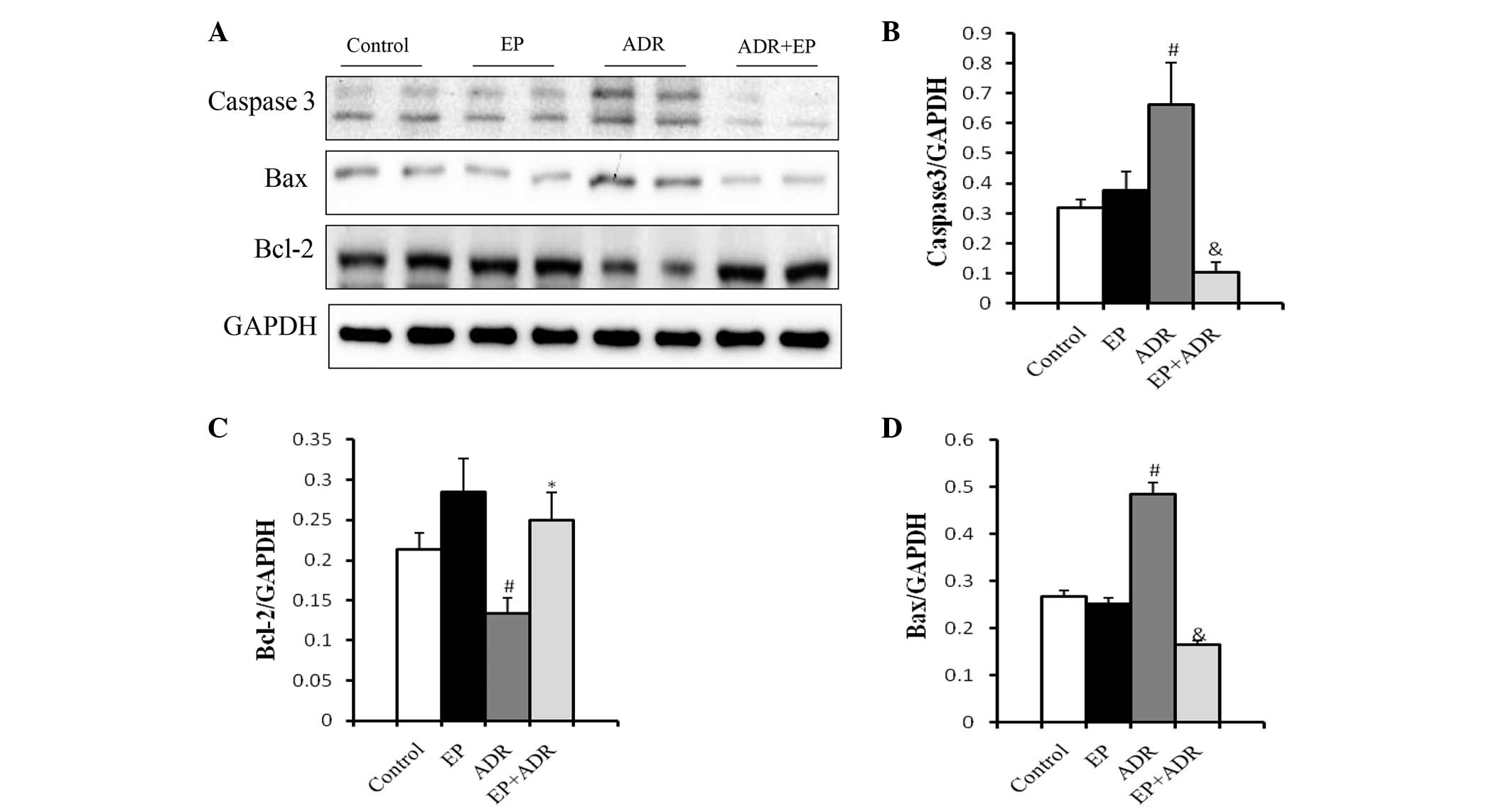
To confirm these findings, western blot analysis was applied to
assess the expression levels of the apoptosis-related proteins
caspase-3, Bax and Bcl-2. As shown in Fig. 4, the expression levels of casepase-3
and Bax in myocardial tissue were significantly higher in the ADR
group than in the control group, EP group and ADR + EP group. By
contrast, the expression level of Bcl-2 was significantly lower in
the ADR group than in the other three groups (Fig. 4C).
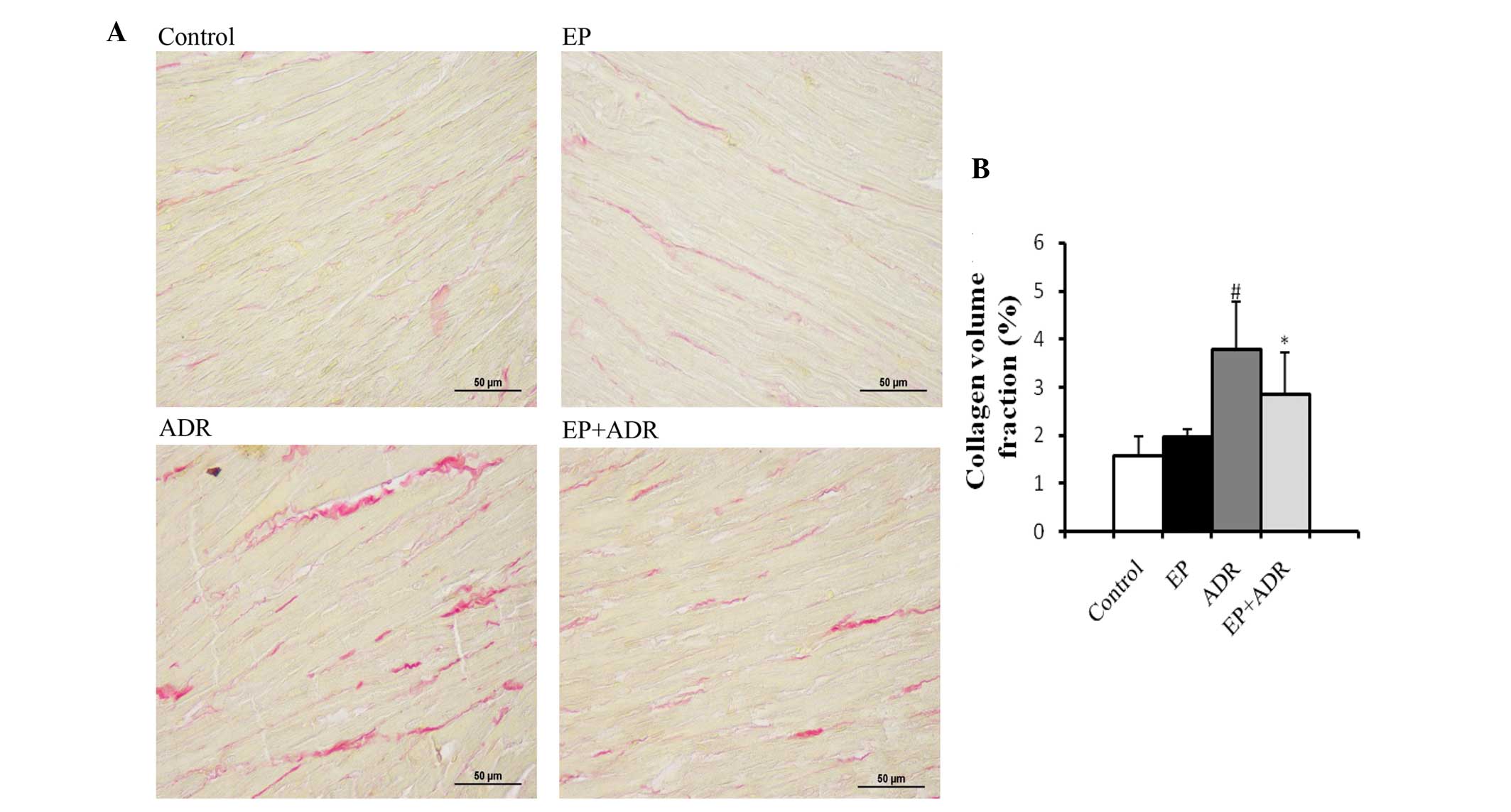
EP attenuates ADR-induced myocardial
tissue fibrosis
The consequence for ADR-induced chronic damage is
myocardial tissue fibrosis. Thus, to explore whether treatment with
EP can attenuate ADR-induced myocardial tissue fibrosis, PSR
staining and RT-qPCR assays were applied to evaluate the fibrosis
in myocardial tissues. PSR staining in left ventricular tissue
sections showed that the collagen volume fraction (%) in the ADR
group was significantly higher than those in the control group, EP
group and ADR + EP group (Fig. 5).
Furthermore, the results of RT-qPCR indicated that the mRNA levels
of TGFβ-1, Colla1 and Colla3, TIMP1, TIMP2, MMP2 and MMP9 in
myocardial tissue in the ADR group were significantly higher than
those in the other three groups (Fig.
6A-E). The ratios MMP9/TIMP1 and MMP2/TIMP2 in the ADR group
were notably lower than those in the ADR + EP group (Fig. 6F and G), however, they were not
significantly different from those in the control group and the EP
group (Fig. 6F and G).
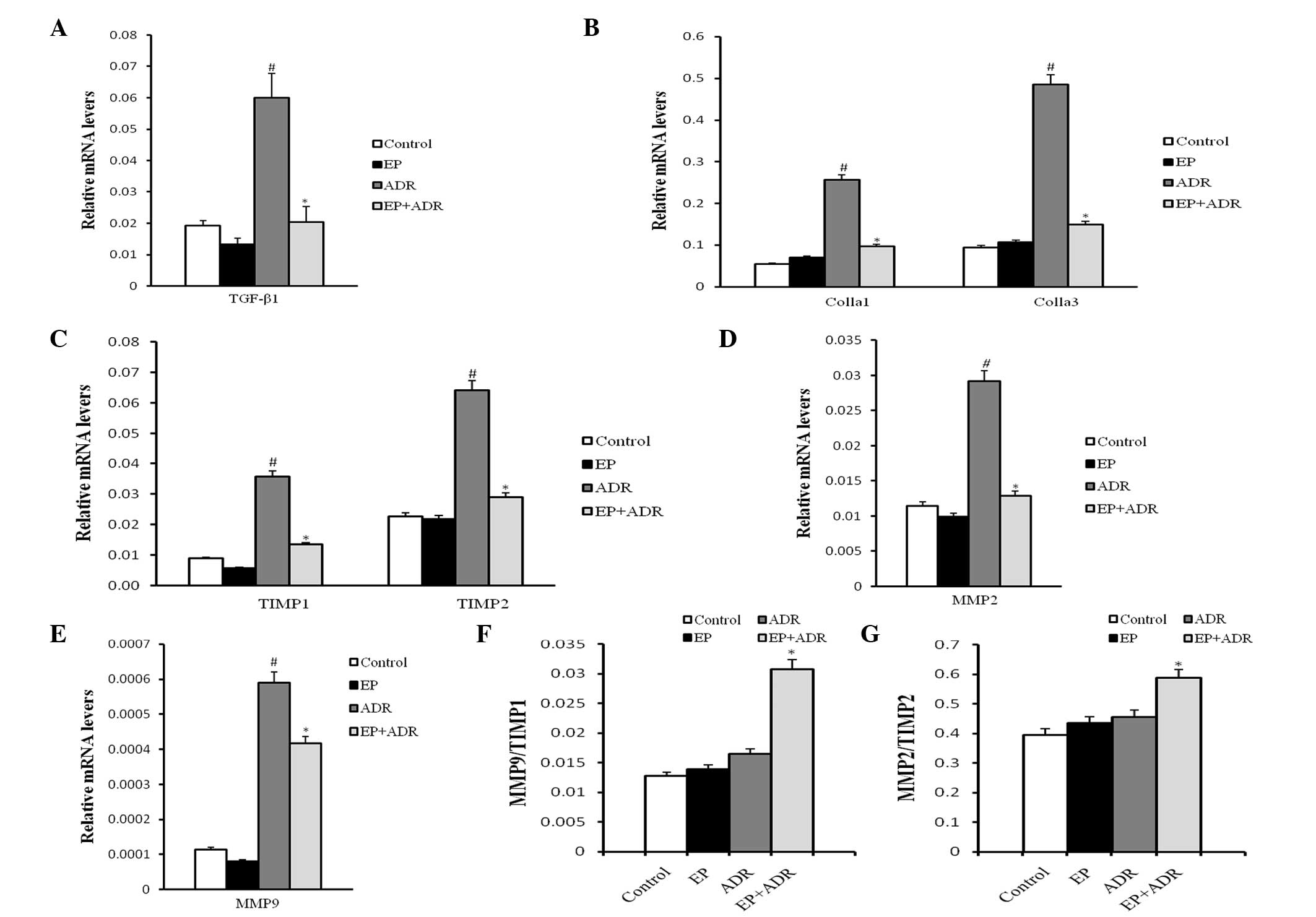 | Figure 6.Results of RT-PCR analysis. The mRNA
level of (A) TGF-β1, (B) Colla1 and Colla3, (C) TIMP1 and TIMP2,
(D) MMP2 and (E) MMP9, and the ratios of (F) MMP9/TIMP1 and (G)
MMP2/TIMP2, respectively. #P<0.05 vs. control and EP
groups, *P<0.05 vs. ADR group. EP, ethyl pyruvate; ADR,
Adriamycin; TGFβ-1, transforming growth factor-β1; Colla1, collagen
type 1 α 1; Colla3, collagen type 1 α 3; MMP2, matrix
metalloproteinase-2; MMP9, matrix metalloproteinase-9; TIMP1,
tissue inhibitor of metalloproteinase-1; TIMP2, inhibitor of
metalloproteinase-2. |
Discussion
ADR is known to be cardiotoxic because it induces
degenerative myocardial lesions and cardiac dysfunction. Therefore,
ADR has been used to establish models of dilated cardiomyopathy
(16) and acute (17) or chronic (18) heart failure in many studies. In the
present study, lower FS% and decreased EF% were observed in the ADR
group, while the LVIDS and ESV were distinctly larger than those in
the control group. By contrast, treatment with EP reversed the
abnormalities in the indices FS%, EF%, LVIDS and ESV. However,
there were no significant differences of LVIDD and EDV among the
four groups.
It has previously been suggested that the severity
and timing of degenerative myocardial lesions and cardiac
dysfunction affect the mortality of rats (19). In the present study, the highest
mortality rate was observed in the ADR group, followed by the ADR +
EP group, indicating that EP reduced the ADR-induced mortality.
However, the reports of ADR-induced mortality by different groups
are heterogeneous. A mortality rate of 14.3% was reported in one
study, in which the same dose and frequency for ADR was used
(16), while another study reported
no mortality with the same total dose (20). These different results for mortality
could be explained by the differences in dosage and protocols of
ADR administration, experimental methods and animal conditions.
Despite inconsistent reports, the data from the present study
suggest that treatment with EP is able to alleviate the
cardiotoxicity of ADR and thus improve the cardiac function and
survival rate of rats.
Although ADR is known to be cardiotoxic, the
mechanisms are not well established. Several theories are
postulated and one of them is associated with the formation of ROS
and myocardial oxidative injury. The NADPH oxidases are important
sources of cellular ROS (21). In
the Nox family, there are seven different homologs of Nox enzymes
in mammalian genomes (22), named as
Nox1 to Nox5 and Duox1 and Duox2. Moreover, Nox2 and Nox4 are
abundantly expressed in cardiomyocytes (23). Zhao et al (24) found that Nox2-deficient mice
exhibited less oxidative injury of myocardial tissue in response to
ADR. Moreover, Ortiz et al (25) reported that ADR induces ROS
production by upregulating the expression of Nox4. Consistent with
this, it was observed in the present study that ADR upregulates the
protein expression of Nox4 in myocardial tissue, which was
suppressed by EP. However, no effects of ADR or EP on the protein
expression of Nox2 were identified. Furthermore, it was observed
that ADR reduced the activity of SOD and increased the levels of
MDA in cardiomyocytes, while EP reversed these changes. Thus, these
findings indicate that EP is able to inhibit ADR-induced oxidative
injury, which might occur through downregulation of the expression
of Nox4 and improvement of anti-oxidant activity.
Some studies have reported that ADR induces
apoptosis in cardiomyocytes by upregulating the expression of Bax
(a pro-apoptotic molecule) and caspase-3 and downregulating the
expression of Bcl-2 (an anti-apoptotic molecule) (26,27),
which was further supported by the current study. In addition, the
cardiomyocyte apoptosis rate in the EP + ADR group was reduced,
which was accompanied by downregulated protein expression of Bax
and caspase-3 and upregulated protein expression of Bcl-2. Thus, EP
can protect against ADR-induced cardiotoxicity via changes in the
expression of apoptosis-related proteins and reducing myocardial
cell apoptosis.
Fibrosis is characterized as the excessive
deposition of extracellular matrix components, which mainly consist
of collagen (28), and can be
regulated by many factors, including TGFβ-1, MMPs and TIMPs. TGFβ-1
can directly induce the expression of collagen proteins (29) and promote extracellular matrix
deposition. MMPs are able to degrade extracellular matrix
components, while TIMPs inhibit the degradation of extracellular
matrix components (30). Therefore,
upregulating the mRNA level of TIMPs or downregulating the mRNA
levels of TGFβ-1 and MMPs may inhibit fibrosis. Fibrosis is
reportedly induced by ADR in myocardial tissue (31), which is supported by the present
study findings. The present results indicate that EP can alleviate
ADR-induced fibrosis by upregulating the mRNA levels of MMPs
relative to those of TIMPs as well as downregulating the mRNA
levels of TGFβ-1 and TIMPs, since it was observed that rats in the
ADR + EP group had a lower collagen volume fraction in myocardial
tissue, lower mRNA levels of TGFβ-1, TIMP1, TIMP2, Colla1 and
Colla3, and higher ratios of MMP9/TIMP1 and MMP2/TIMP2, compared
with those in the ADR group. However, the mRNA levels of MMP2 and
MMP9 in the ADR group were higher than those in the other groups,
which may due to the compensatory response of the myocardium to ADR
treatment.
In conclusion, this study showed that EP could
alleviate ADR-induced myocardial damage by preserving the diastolic
relaxation and systolic contractile force of the heart. In
addition, it blocked the source of ROS and thus resisted oxidative
injury in myocardial tissue. Furthermore, EP prevented the
apoptosis and fibrosis of myocardial tissues in ADR-treated rats.
These findings indicate that EP is a potential novel therapeutic
agent for ADR-induced cardiomyopathy.
Acknowledgements
This study was supported by grants to Dr J Wan from
the National Natural Science Foundation of China (grant nos.
81170208 and 30871050), and the Natural Science Foundation of Hubei
province, China (grant no. 302-131725).
References
|
1
|
Felker GM, Thompson RE, Hare JM, Hruban
RH, Clemetson DE, Howard DL, Baughman KL and Kasper EK: Underlying
causes and long-term survival in patients with initially
unexplained cardiomyopathy. N Engl J Med. 342:1077–1084. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu
YL, Liu LF and Yeh ET: Identification of the molecular basis of
doxorubicin-induced cardiotoxicity. Nat Med. 18:1639–1642. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jones LW, Haykowsky MJ, Swartz JJ, Douglas
PS and Mackey JR: Early breast cancer therapy and cardiovascular
injury. J Am Coll Cardiol. 50:1435–1441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arya DS, Bansal P, Ojha SK, Nandave M,
Mohanty I and Gupta SK: Pyruvate provides cardioprotection in the
experimental model of myocardial ischemic reperfusion injury. Life
Sci. 79:38–44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sims CA, Wattanasirichaigoon S, Menconi
MJ, Ajami AM and Fink MP: Ringer's ethyl pyruvate solution
ameliorates ischemia/reperfusion-induced intestinal mucosal injury
in rats. Crit Care Med. 29:1513–1518. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sileri P, Schena S, Morini S, Rastellini
C, Pham S, Benedetti E and Cicalese L: Pyruvate inhibits hepatic
ischemia-reperfusion injury in rats. Transplantation. 72:27–30.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yi JS, Kim TY, Kyu Kim D and Koh JY:
Systemic pyruvate administration markedly reduces infarcts and
motor deficits in rat models of transient and permanent focal
cerebral ischemia. Neurobiol Dis. 26:94–104. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marcengill MB, Puri S, Puri SK, Mohanram
A, Leonova E, Raymond RM and Watts JA: Antioxidant effects of
pyruvate in isolated rat hearts. J Mol Cell Cardiol. 27:2059–2067.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kao KK and Fink MP: The biochemical basis
for the anti-inflammatory and cytoprotective actions of ethyl
pyruvate and related compounds. Biochem Pharmacol. 80:151–159.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo JL, Zhang J, Luo XY, Luo WM, Lin CY,
Zhang KL and Ji YM: Effects of ethyl pyruvate on cardiac function
recovery and apoptosis reduction after global cold ischemia and
reperfusion. Exp Ther Med. 7:1197–1202. 2014.PubMed/NCBI
|
|
11
|
Kao KK and Fink MP: The biochemical basis
for the anti-inflammatory and cytoprotective actions of ethyl
pyruvate and related compounds. Biochem Pharmacol. 80:151–159.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: The 1996 Guide for the Care and Use of Laboratory
Animals. ILAR J. 38:41–48. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang DS, Li L, Huang L, Gong J, Xia H,
Liu X, Wan N, Wei X, Zhu X, Chen Y, et al: Interferon regulatory
factor 1 is required for cardiac remodeling in response to pressure
overload. Hypertension. 64:77–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Desagher S, Glowinski J and Premont J:
Astrocytes protect neurons from hydrogen peroxide toxicity. J
Neurosci. 16:2553–2562. 1996.PubMed/NCBI
|
|
15
|
Shadab M, Agrawal DK, Aslam M, Islam N and
Ahmad Z: Occupational health hazards among sewage workers:
Oxidative stress and deranged lung functions. J Clin Diagn Res.
8:BC11–BC12. 2014.PubMed/NCBI
|
|
16
|
Leontyev S, Schlegel F, Spath C, Schmiedel
R, Nichtitz M, Boldt A, Rübsamen R, Salameh A, Kostelka M, Mohr FW
and Dhein S: Transplantation of engineered heart tissue as a
biological cardiac assist device for treatment of dilated
cardiomyopathy. Eur J Heart Fail. 15:23–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li K, Sung RY, Huang WZ, Yang M, Pong NH,
Lee SM, Chan WY, Zhao H, To MY, Fok TF, et al: Thrombopoietin
protects against in vitro and in vivo cardiotoxicity induced by
doxorubicin. Circulation. 113:2211–2220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lau DH, Psaltis PJ, Carbone A, Kelly DJ,
Mackenzie L, Worthington M, Metcalf RG, Kuklik P, Nelson AJ, Zhang
Y, et al: Atrial protective effects of n-3 polyunsaturated fatty
acids: A long-term study in ovine chronic heart failure. Heart
Rhythm. 8:575–582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Andreadou I, Mikros E, Ioannidis K, Sigala
F, Naka K, Kostidis S, Farmakis D, Tenta R, Kavantzas N, Bibli SI,
et al: Oleuropein prevents doxorubicin-induced cardiomyopathy
interfering with signaling molecules and cardiomyocyte metabolism.
J Mol Cell Cardiol. 69:4–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ohkura K, Lee JD, Shimizu H, Nakano A,
Uzui H, Horikoshi M, Fujibayashi Y, Yonekura Y and Ueda T:
Mitochondrials complex I activity is reduced in latent
adriamycin-induced cardiomyopathy of rat. Mol Cell Biochem.
248:203–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brandes RP, Weissmann N and Schröder K:
Nox family NADPH oxidases: Molecular mechanisms of activation. Free
Radic Biol Med. 76:208–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
West AP, Brodsky IE, Rahner C, Woo DK,
Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS and
Ghosh S: TLR signalling augments macrophage bactericidal activity
through mitochondrial ROS. Nature. 472:476–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maejima Y, Kuroda J, Matsushima S, Ago T
and Sadoshima J: Regulation of myocardial growth and death by NADPH
oxidase. J Mol Cell Cardiol. 50:408–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao Y, McLaughlin D, Robinson E, Harvey
AP, Hookham MB, Shah AM, McDermott BJ and Grieve DJ: Nox2 NADPH
oxidase promotes pathologic cardiac remodeling associated with
doxorubicin chemotherapy. Cancer Res. 70:9287–9297. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ortiz C, Caja L, Sancho P, Bertran E and
Fabregat I: Inhibition of the EGF receptor blocks autocrine growth
and increases the cytotoxic effects of doxorubicin in rat hepatoma
cells: Role of reactive oxygen species production and glutathione
depletion. Biochem Pharmacol. 75:1935–1945. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao S, Li H, Feng XJ, Li M, Liu ZP, Cai Y,
Lu J, Huang XY, Wang JJ, Li Q, et al: α-Enolase plays a
catalytically independent role in doxorubicin-induced cardiomyocyte
apoptosis and mitochondrial dysfunction. J Mol Cell Cardiol.
79:92–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Janeesh PA and Abraham A: Robinin
modulates doxorubicin-induced cardiac apoptosis by TGF-β1 signaling
pathway in Sprague Dawley rats. Biomed Pharmacother. 68:989–998.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wight TN and Potter-Perigo S: The
extracellular matrix: An active or passive player in fibrosis? Am J
Physiol Gastrointest Liver Physiol. 301:G950–G955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sethi A, Jain A, Zode GS, Wordinger RJ and
Clark AF: Role of TGFbeta/Smad signaling in gremlin induction of
human trabecular meshwork extracellular matrix proteins. Invest
Ophthalmol Vis Sci. 52:5251–5259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu J, Wang P, Wang T, Wang M, Chen S, Yu P
and Yu D: Effects of reduced β2-glycoprotein I on the expression of
aortic matrix metalloproteinases and tissue inhibitor matrix
metalloproteinases in diabetic mice. BMC Cardiovasc Disord.
14:1142014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arafa MH, Mohammad NS, Atteia HH and
Abd-Elaziz HR: Protective effect of resveratrol against
doxorubicin-induced cardiac toxicity and fibrosis in male
experimental rats. J Physiol Biochem. 70:701–711. 2014. View Article : Google Scholar : PubMed/NCBI
|