Introduction
Infantile neuroaxonal dystrophy (INAD) is a rare
autosomal recessive neurodegenerative disorder, characterized by an
early symptomatic onset with rapid progression of psychomotor
regression and hypotonia, evolving into spasticity. This results in
total neurological degeneration and mortality by the age of 10.
There is no effective treatment for INAD at present and since it is
a rare disorder, involving axons in the central and peripheral
nervous system, there is little literature on the disease.
Therefore, it is important to provide accurate genetic counseling
for families with INAD.
INAD sufferers exhibit spheroid bodies in the
central nervous system and pathological swelling of axons (1). In addition, T2-weighted magnetic
resonance imaging (MRI) of patients with INAD typically reveals
cerebellar atrophy, however, MRI is not used as the primary method
of INAD diagnosis (2,3). Prior to molecular testing for
phospholipase A2 group VI (PLA2G6; 22q13.1) mutations, which have
been identified in the majority of INAD sufferers, an INAD
diagnosis could be only be confirmed through electron microscopy
identification of axon dystrophy in a tissue biopsy of conjunctiva,
skin, muscle or sural nerves (4).
Molecular diagnosis of INAD eliminates the need for invasive
biopsies, enables the detection of carriers of INAD-associated
mutations and allows for prenatal diagnosis. The present study
reports the case twins with INAD caused by PLA2G6 mutations
inherited from the mother and father, which highlights the
importance of genetic testing in the diagnosis and prevention of
INAD.
Case report
The current report presents monozygotic male twins
with INAD referred to The Children's Hospital (Zhejiang University
School of Medicine, Zhejiang, China) in December 2013 at 15 months
old with delayed development. Written informed consent for
participation in the current study was obtained from the guardians.
The parents were a non-consanguineous healthy Chinese couple, with
no significant family history and an older child (female, 8 years
old) that was healthy. The twins were born at term, following an
uneventful pregnancy, by cesarean section. The patients were
asymptomatic at birth and achieved normal developmental milestones,
including looking up, standing up and sitting independently, until
they were 6 months old. Disease onset occurred when the patients
were between 7 and 15 months old, where they could not stand
securely without support, and exhibited poor eye tracking and
listening ability. Clinical examination revealed slow reactions, a
limb muscle strength of grade IV and a muscle tension level of 1.
Electromyography identified signs of peripheral neuropathy.
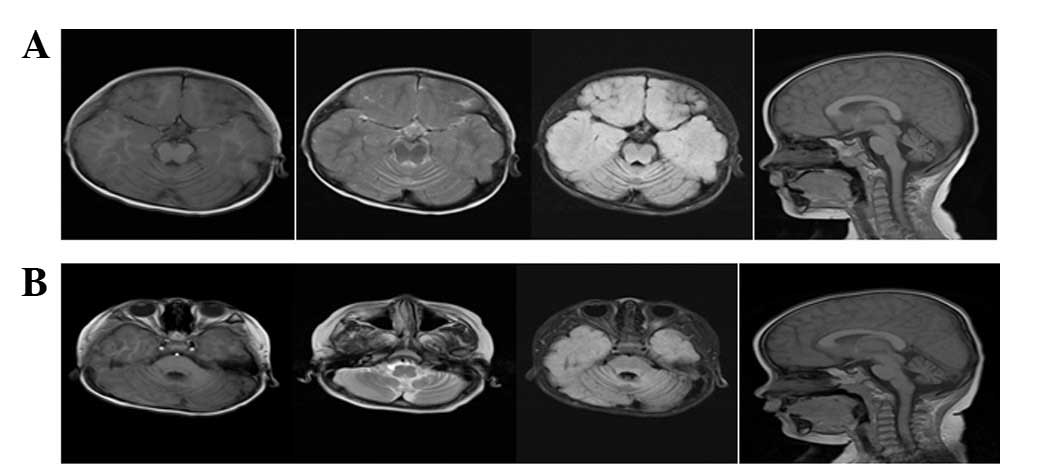
Electroencephalogram showed slow background activity. MRI imaging
of the brain revealed cerebellar atrophy (Fig. 1).
Polymerase chain reaction (PCR) amplification was
performed to screen for PLA2G6 mutation. The Whole Blood Genome DNA
Isolation kit (cat. no. 2013-1400049; Beijing ComWin Biotech Co.,
Ltd., Beijing, China) was used to obtain DNA from the patients,
according to the manufacturer's instructions. PCR was performed
using the PCR Amplification Kit (cat. no. R011) and Ex Taq DNA
polymerase (both Takara Biotechnology Co., Ltd., Dalian, China).
The PCR reaction included 10X Ex Taq buffer (3 µl), 2.5 mM dNTPs
(2.4 µl), Primer F (10 µM; 0.6 µl), Primer R (10 µM; 0.6 µl),
template DNA (1 µl), Ex Taq (5 U/µl; 0.15 µl), and water (22.25
µl). The thermal cycling conditions were as follows: 94°C for 3
min, 94°C for 30 sec, 55°C for 30s, 36 cycles of 55°C for 1 min,
and totally 36 cycles, and 72°C for 5 min. DNA sequencing was
performed using the ABI PRISM BigDye Terminator v3.1 Cycle
Sequencing kit (cat. no. 4337455; Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's instructions, and an Applied Biosystems 3100 Genetic
Analyzer (Thermo Fisher Scientific, Inc.) to detect the following
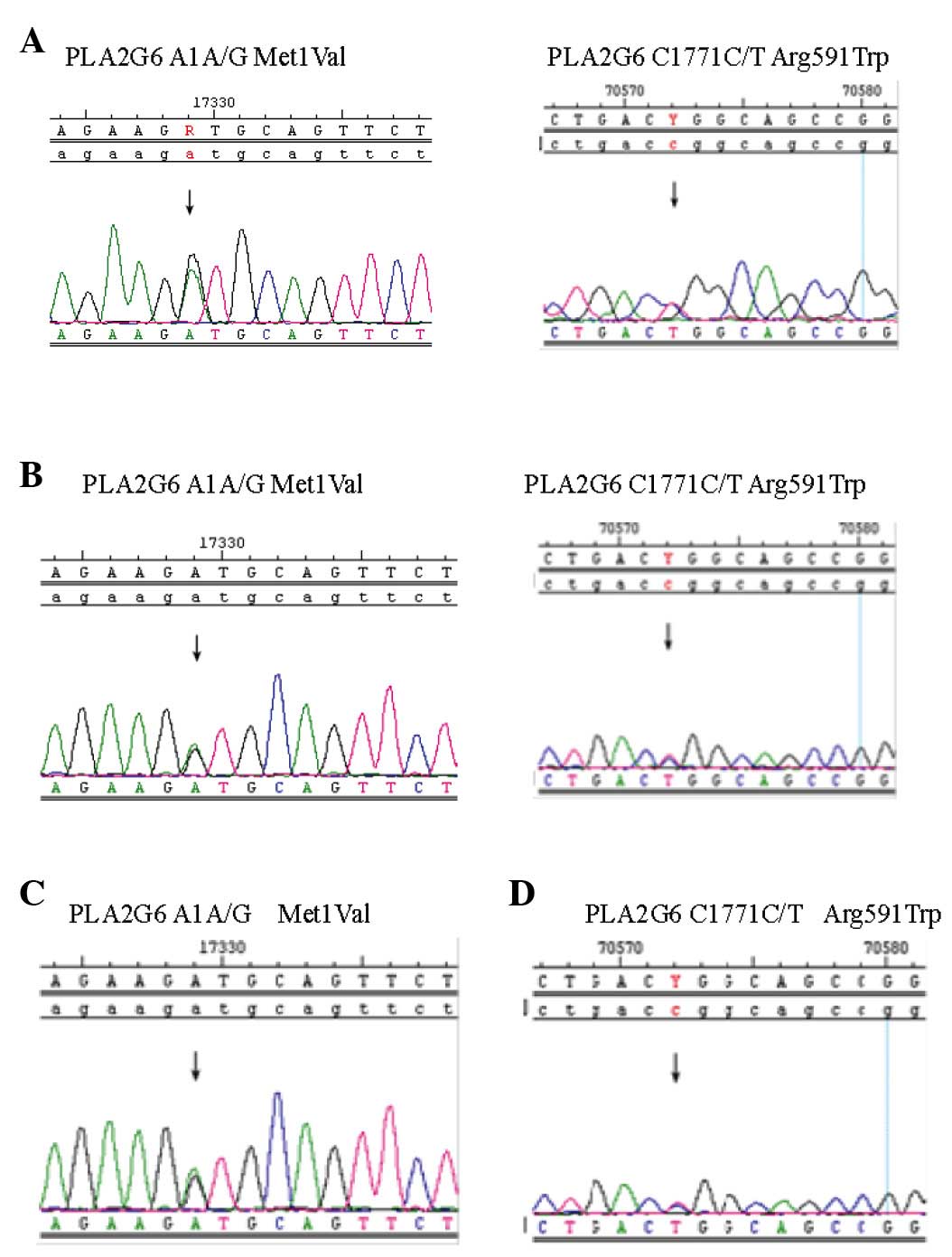
mutations: Exon 2 A1 (A/G), exon 2 G87 (G/A) and exon 13 C1771
(C/T). Interestingly, two different PLA2G6 mutations were detected
in the twins, exon 2 A1 A/G and exon 13 C1771 C/T (Fig. 2A and B). The Met1Val of A1 A/G was
from the father (Fig. 2C) and the
Arg591Trp of C1771 C/T was from mother (Fig. 2D).
Discussion
INAD is a severe early onset neurodegenerative
disease. Clinical and neurophysiological findings alone may be
insufficient for early diagnosis of INAD. MRI examination
(T2-weighted) of patients with INAD typically reveals marked
cerebellar atrophy and a signal hyperintensity of the cerebellar
cortex. Nardocci et al (1)
proposed clinical diagnostic criteria for INAD. However,
developments in molecular diagnosis have showed that the majority
of INAD cases are associated with mutations in the PLA2G6 gene
(4). PLA2G6, which encodes the
Ca2+-independent phospholipase A2β, iPLA2β, has been
identified as a causative gene of INAD (5). The enzyme iPLA2β serves a key role in
membrane composition, via hydrolysis of peroxidized fatty acid, and
in signal transduction. The determination that PLA2G6 mutations are
the cause of INAD has revolutionized the ability to diagnosis this
disorder (6). Khateeb et al
(7) demonstrated the role of
phospholipase mutation in neurodegenerative disorders. Malik et
al (8) indicated that loss of
iPLA2β function triggers age-dependent impairment of protein
degradation signaling pathways and homeostasis of the axonal
membranes, resulting in age-related neurological impairment.
Polster et al (9) suggested
that PLA2G6 serves a role in neuronal proliferation in the
developing brain, neurons, cortical plate and hindbrain. PLA2G6
expression is widespread in neuronal tissues, however, the
expression pattern changes dynamically over time, which suggests
that INAD pathogenesis begins prenatally. Mutations in the gene
encoding iPLA2β are found in ~85% of patients with INAD and Strokin
et al (10) suggests that
altered Ca2+ signaling is the key mechanism in the
development of INAD. In addition, Biancheri et al (11) reported that a 2-year-old boy
presenting with hypotonia and psychomotor regression, whose MRI
showed cerebellar atrophy with normal cerebellar cortex signal
intensity, had a homozygous 5′ splice site PLA2G6 mutation
(12). This indicates that the
absence of cerebellar cortex signal hyperintensity does not rule
out an INAD diagnosis (13).
Previous studies containing the phrase ‘INAD and
PLA2G6’ were identified in PubMed (http://www.ncbi.nlm.nih.gov/pubmed), resulting in 61
papers from 2006 to 2015. Since 2006, just 11 genetic studies of
patients with INAD have been published (2,6,12–17). In
China, only 2 studies reported PLA2G6-associated neurodegeneration.
The first study discussed 10 patients with INAD, who had been
diagnosed through neuropathology, that were analyzed for PLA2G6
mutations (17). The second was a
follow-up study of 25 Chinese children with PLA2G6-associated
neurodegeneration that found 27 different mutations, of which 13
were novel (18).
In conclusion, the results of present study
highlight that in children who present with early, rapid cognitive
and motor regression, and axial hypotonia, INAD is an important
differential diagnosis. In this regard, neurogenetic disease should
be considered in children with developmental stagnation of an
unknown cause, particularly when similar cases have appeared within
their family. Furthermore, INAD should be considered when children
have delayed central and peripheral nerve development. In future,
genetic testing will be the primary method used for INAD diagnosis.
In addition, this testing could be used for carrier detection and
prenatal diagnosis in affected families, in order to facilitate a
more precise assessment of the recurrence risks, to guide prenatal
care and to prevent genetic diseases.
Acknowledgements
The present study was supported by Zhejiang Province
Science and Technology Fund (grant no. 2015C33178).
References
|
1
|
Nardocci N, Zorzi G, Farina L, Binelli S,
Scaioli W, Ciano C, Verga L, Angelini L, Savoiardo M and Bugiani O:
Infantile neuroaxonal dystrophy: Clinical spectrum and diagnostic
criteria. Neurology. 52:1472–1478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurian MA, Morgan NV, MacPherson L, Foster
K, Peake D, Gupta R, Philip SG, Hendriksz C, Morton JE, Kingston
HM, et al: Phenotypic spectrum of neurodegeneration associated with
mutations in the PLA2G6 gene (PLAN). Neurology. 70:1623–1629. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Farina L, Nardocci N, Bruzzone MG,
D'Incerti L, Zorzi G, Verga L, Morbin M and Savoiardo M: Infantile
neuroaxonal dystrophy: Neuroradiological studies in 11 patients.
Neuroradiology. 41:376–380. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gregory A, Westaway SK, Holm IE, Kotzbauer
PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G,
et al: Neurodegeneration associated with genetic defects in
phospholipase A(2). Neurology. 71:1402–1409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wada H, Kojo S and Seino K: Mouse models
of human INAD by Pla2g6 deficiency. Histol Histopathol. 28:965–969.
2013.PubMed/NCBI
|
|
6
|
Morgan NV, Westaway SK, Morton JE, Gregory
A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, et
al: PLA2G6, encoding a phospholipase A2, is mutated in
neurodegenerative disorders with high brain iron. Nat Genet.
38:752–754. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khateeb S, Flusser H, Ofir R, Shelef I,
Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K and Birk
OS: PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J
Hum Genet. 79:942–948. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malik I, Turk J, Mancuso DJ, Montier L,
Wohltmann M, Wozniak DF, Schmidt RE, Gross RW and Kotzbauer PT:
Disrupted membrane homeostasis and accumulation of ubiquitinated
proteins in a mouse model of infantile neuroaxonal dystrophy caused
by PLA2G6 mutations. Am J Pathol. 172:406–416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Polster B, Crosier M, Lindsay S and
Hayflick S: Expression of PLA2G6 in human fetal development:
Implications for infantile neuroaxonal dystrophy. Brain Res Bull.
83:374–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Strokin M, Seburn KL, Cox GA, Martens KA
and Reiser G: Severe disturbance in the Ca2+ signaling in
astrocytes from mouse models of human infantile neuroaxonal
dystrophy with mutated Pla2g6. Hum Mol Genet. 21:2807–2814. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Biancheri R, Rossi A, Alpigiani G,
Filocamo M, Gandolfo C, Lorini R and Minetti C: Cerebellar atrophy
without cerebellar cortex hyperintensity in infantile neuroaxonal
dystrophy (INAD) due to PLA2G6 mutation. Eur J Paediatr Neurol.
11:175–177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frattini D, Nardocci N, Pascarella R,
Panteghini C, Garavaglia B and Fusco C: Downbeat nystagmus as the
presenting symptom of infantile neuroaxonal dystrophy: A case
report. Brain Dev. 37:270–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Solomons J, Ridgway O, Hardy C, Kurian MA,
Jayawant S, Hughes S, Pretorius P and Németh AH: Infantile
neuroaxonal dystrophy caused by uniparental disomy. Dev Med Child
Neurol. 56:386–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Riku Y, Ikeuchi T, Yoshino H, Mimuro M,
Mano K, Goto Y, Hattori N, Sobue G and Yoshida M: Extensive
aggregation of α-synuclein and tau in juvenile-onset neuroaxonal
dystrophy: An autopsied individual with a novel mutation in the
PLA2G6 gene-splicing site. Acta Neuropathol Commun. 1:122013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tonelli A, Romaniello R, Grasso R,
Cavallini A, Righini A, Bresolin N, Borgatti R and Bassi MT: Novel
splice-site mutations and a large intragenic deletion in PLA2G6
associated with a severe and rapidly progressive form of infantile
neuroaxonal dystrophy. Clin Genet. 78:432–440. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carrilho I, Santos M, Guimarães A,
Teixeira J, Chorão R, Martins M, Dias C, Gregory A, Westaway S,
Nguyen T, et al: Infantile neuroaxonal dystrophy: What's most
important for the diagnosis? Eur J Paediatr Neurol. 12:491–500.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y,
Xiong H, Chang X, Bao X, Zhang Y, Xiao J and Wu X: Clinical study
and PLA2G6 mutation screening analysis in Chinese patients with
infantile neuroaxonal dystrophy. Eur J Neurol. 16:240–245. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang P, Gao Z, Jiang Y, Wang J, Zhang F,
Wang S, Yang Y, Xiong H, Zhang Y, Bao X, et al: Follow-up study of
25 Chinese children with PLA2G6-associated neurodegeneration. Eur J
Neurol. 20:322–330. 2013. View Article : Google Scholar : PubMed/NCBI
|