Introduction
Traumatic brain injury (TBI) is the leading cause of
mortality in young adults and children living in China (1). Head trauma has numerous consequences in
the brain, including axonal damage, microvascular alterations and
blood-brain barrier disruption (2).
The aforementioned outcomes are a result of both primary and
secondary mechanisms following injury (3). Primary damage is a result of mechanical
factors occurring immediately following trauma. Conversely, the
fundamental mechanisms underlying secondary damage in TBI include
inflammation, oxygen free radicals, brain edema formation and
neuronal apoptosis (4). In
particular, previous studies have demonstrated that tumor necrosis
factor-α (TNF-α), interleukin (IL)-1β and IL-6 are crucial
pro-inflammatory cytokines following trauma (5–7).
Although the potential effects of TNF-α, IL-1β and IL-6 in human
patients with TBI have yet to be elucidated, evidence from animal
models has revealed that upregulated expression levels of the
aforementioned cytokines are harmful, whereas their attenuation may
alleviate tissue damage as well as brain edema, thereby improving
the functional outcomes of patients with TBI (8).
Heat shock protein 70 (Hsp70; 72 kDa) is a highly
stress-inducible member of a chaperone family of proteins (9). Previous studies in focal and global
cerebral ischemia models have revealed that the overexpression of
Hsp70 is protective against secondary damage in TBI, such as
inflammation, oxygen free radicals and brain edema (10,11).
Conversely, downregulated expression levels of Hsp70 resulted in a
worse outcome in a rat model of stroke (12). Kim et al (13) demonstrated that Hsp70 contributed to
the modulation of immune responses by potentiating or inhibiting
them.
It is widely established that inflammation is
capable of inducing brain tissue damage following TBI. Thus, the
present study aimed to determine whether the Hsp70 activator,
17-allylamino-demethoxygeldanamycin (17-AAG), is able to upregulate
the expression levels of Hsp70 in addition to reducing the
expression levels of pro-inflammatory cytokines TNF-α, IL-1β and
IL-6, in a rat model of TBI, as observed in neuroprotective
processes.
Materials and methods
Animals and ethics statement
Male Sprague-Dawley rats (n=90; weight, 280–320 g)
obtained from the Lanzhou University Experimental Animal Center
(Lanzhou, China) were provided ad libitum access to food and
water prior to surgery under optimal conditions (12-h light/dark
cycle; 22°C). All procedures were approved by the local legislation
for ethics regarding experiments on animals (Ethics Committee of
Lanzhou University, Lanzhou, China). The rats were anaesthetized
with sodium pentobarbital (Bio-Rad Laboratories, Inc., Shanghai,
China) by intraperitoneal injection (50 mg/kg; Beijing, China).
Models of TBI
A previously described controlled cortical impact
(CCI) injury procedure was utilized (14).
Groups and drug administration
Rats were randomly assigned to the vehicle group
(n=30) or the 17-AAG group (n=30). The vehicle group received only
equal volumes of 0.9% saline solution (20 mg/ml) and the 17-AAG
group received 17-AAG dissolved in 0.9% saline that had been stored
at 4°C. Following the establishment of TBI, 17-AAG was immediately
administered by a single intraperitoneal injection in the 17-AAG
group (80 mg/kg body weight).
Evaluation of brain edema
Brain edema was evaluated by analysis of brain water
content, as previously described (15).
Recovery of motor function
Motor function was evaluated as described previously
(15). Neurologic Severity Score
(NSS) was evaluated at 1, 3, 5 and 7 days (15).
Immunofluorescence
Rats were anesthetized with sodium pentobarbital
(i.p; 50 mg/kg; Beijing, China) prior to sacrifice. Coronal
sections (10 µm) were obtained from the anterior area of the left
hemisphere. Immunofluorescence was performed as described
previously (15). Briefly, brain
tissue samples were fixed in 4% paraformaldehyde (Bio-Rad
Laboratories, Inc.) for 24 h, then transferred to a 30% sucrose
solution containing 0.1 mol/l phosphate-buffered saline (PBS; pH
7.4). Incisions were made 200 µm apart, from the anterior to the
posterior cortex (bregma −1.90 to −3.00 mm) in TBI rats then
embedded in optimal cutting temperature compound. Frozen tissue
sections (15 µm) were cut using a microtome, treated with 0.4%
Triton-100 for 10 min and blocked in normal donkey serum (both
Bio-Rad Laboratories, Inc.) for 1 h. The frozen tissue sections
were then incubated with mouse anti-neuron-specific nuclear protein
(NeuN) polyclonal antibody (diluted 1:100; cat no. sc-33684;
Bio-Rad Laboratories, Inc.) overnight at 4°C. The following day,
the tissue sections were incubated with anti-mouse immunoglobulin G
(IgG) secondary antibody (diluted 1:1,000; Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA) for 2 h at 37°C in darkness.
Images were captured using a laser scanning confocal microscope
(Olympus Corp. BX41-P, Tokyo, Japan). PBS was used in place of
primary antibodies in the negative control group.
Immunohistochemistry
Rats were anesthetized and sacrificed by
intracardiac perfusion with 0.1 mol/l PBS (pH 7.4). The brains was
rapidly isolated. Coronal sections (10 µm) were obtained from the
anterior area of the left hemisphere, washed with 0.1 M PBS, fixed
in 10% formalin (Bio-Rad Laboratories, Inc.) for 24 h at room
temperature, dehydrated in graded ethanol (70, 90, 95 and 100%),
and embedded in paraffin (Bio-Rad Laboratories, Inc.). Tissue
sections (6-mm thick) were incubated with an anti-Hsp70 antibody
(diluted 1:500; Bio-rad Laboratories, Inc.) at 4°C for 24 h. The
tissue sections were then incubated with the relevant secondary
antibodies for 90 min at room temperature. Slides were developed
with diaminobenzidine substrate (Bio-Rad Laboratories, Inc.) and
the images were captured using a microscope (BX41-P; Olympus
Corp.).
Western blot analysis
Western blot analysis was performed as previously
described (15). Briefly, rats were
anesthetized and underwent intracardiac perfusion with 0.1 mol/l
PBS (pH 7.4). The cortex region of the brains was rapidly isolated,
homogenized (BestBio Biotechnology Co., Ltd., Shanghai, China),
total proteins were extracted and protein concentration was
determined using a Bicinchoninic Acid Protein Assay kit (Bio-Rad
Laboratories, Inc.) according to the manufacturer's protocol. The
protein samples were subjected to SDS-PAGE (Bio-Rad Laboratories,
Inc). Protein concentration was determined using a bicinchoninic
acid assay (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China). Total protein (35 µg) was separated by 20%
SDS-PAGE gel electrophoresis (80 V for 30 min at 28°C for spacer
gel, and then 120 V/1 for 1 h at 28°C for separation gel) and
transferred from the gel onto polyvinylidene fluoride membranes
(Roche Diagnostics GmbH, Mannheim, Germany). The membranes were
blocked with 5% non fat dry milk for 1 h at room temperature.
Following blocking, the membranes were incubated with the following
primary antibodies, all purchased from all Santa Cruz
Biotechnology, Inc., overnight at 4°C: Rabbit anti-Hsp70 polyclonal
antibody (diluted 1:500; cat no. sc-33575), rabbit anti-caspase-3
polyclonal antibodies diluted 1:500, rabbit anti-B-cell lymphoma 2
(Bcl-2) polyclonal antibody (diluted 1:500; cat no. sc-492), rabbit
anti-glutamate transporter-1 (GLT-1) polyclonal antibody (diluted
1:500; cat no. sc-15317) and mouse anti-β-actin monoclonal antibody
(diluted 1:500; cat no. sc-376421). Membranes were then washed
twice with tris-buffered saline with Tween-20 (TBST) for 20 min.
Subsequently, membranes were incubated with horseradish peroxidase
conjugated anti-rabbit IgG (diluted 1:5,000; cat no. sc-2027) and
anti-mouse IgG (diluted 1:5,000; cat no. sc-2025; both Santa Cruz
Biotechnology, Inc.) for 2 h at room temperature. Membranes were
washed four times with TBST for 40 min. The immunoblot on the
membrane was visualized using an enhanced chemiluminescence
detection system (ImageQuant EC; GE Healthcare Life Sciences,
Chalfont, UK) and densitometric signals were quantified using an
imaging program (version 1.41; National Institutes of Health,
Bethesda, MA, USA). Immunoreactive bands for all proteins were
normalized to the corresponding bands for β-actin. The western blot
results were analyzed with ImageJ Software (version 1.41; National
Institutes of Health, Bethesda, MA, USA).
Multiplex cytokine ELISA. An immunosorbent assay was
performed as described previously (16). The supernatant was then collected and
total protein was determined by the Bradford method (16). The expression levels of inflammatory
cytokines were quantified using ELISA kits specific for rats (cat
no. 5000201; and Bio-Plex Pro™ Rat Cytokine 24-plex Assay cat no.
171K1001M; Bio-Rad Laboratories, Inc.) according to the
manufacturer's protocol. The cytokine contents in the brain samples
were expressed as picograms of antigen per milligram of
protein.
Statistical analysis
All data are presented as means ± standard
deviation. SPSS software (version 16.0; SPSS, Inc., Chicago, IL,
USA) was used for the statistical analysis of all data. Statistical
analysis was performed using one-way analysis of variance and
followed by Student-Newman-Keuls post-hoc tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
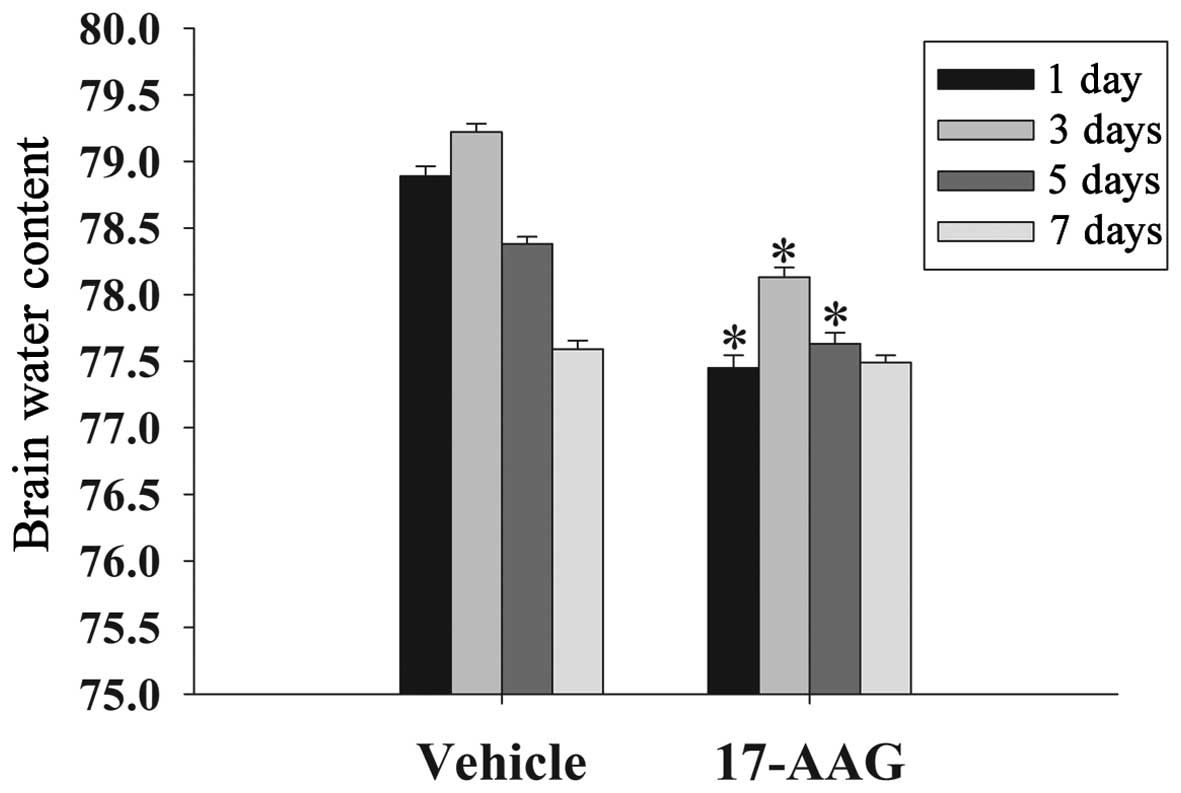
17-AAG attenuates cerebral edema
Brain edema suppresses cerebral perfusion pressure
and oxygenation in the brain, resulting in an elevation in
intracranial pressure following TBI (17). The formation of edema is an important
factor to secondary injury following TBI. In the present study, the
wet-dry weight method was used to evaluate cerebral edema at 1, 3,
5 and 7 days. As displayed in Fig.
1, the brain water content in the 17-AAG treatment group was
significantly lower on days 1, 3 and 5 compared with the vehicle
group (P<0.05).
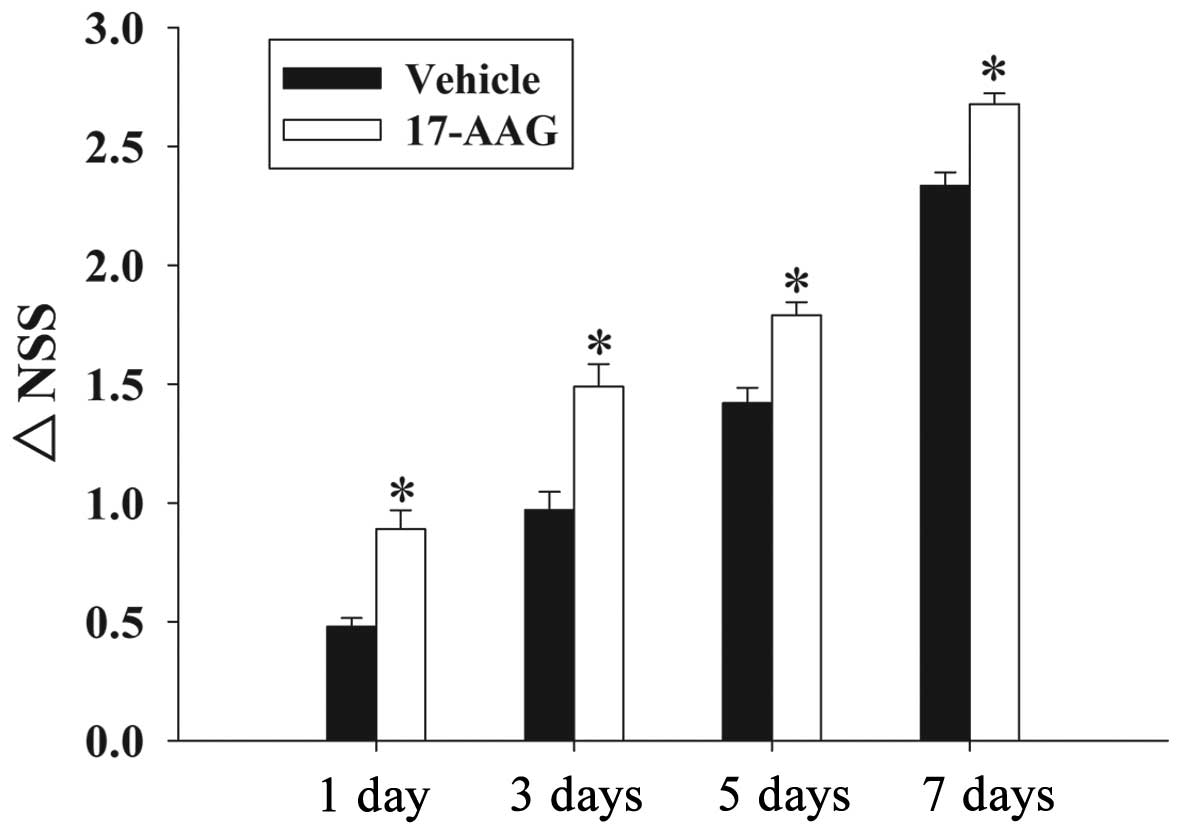
17-AAG attenuates motor deficits
Recovery of motor deficits are expressed as ΔNSS in
the present study. Fig. 2 revealed
the changes in functional recovery at 1, 3, 5 and 7 days. It is
evident that post-injury administration of 17-AAG significantly
improved (P<0.05) the motor function recovery between 1 and 7
days following trauma compared with untreated rats.
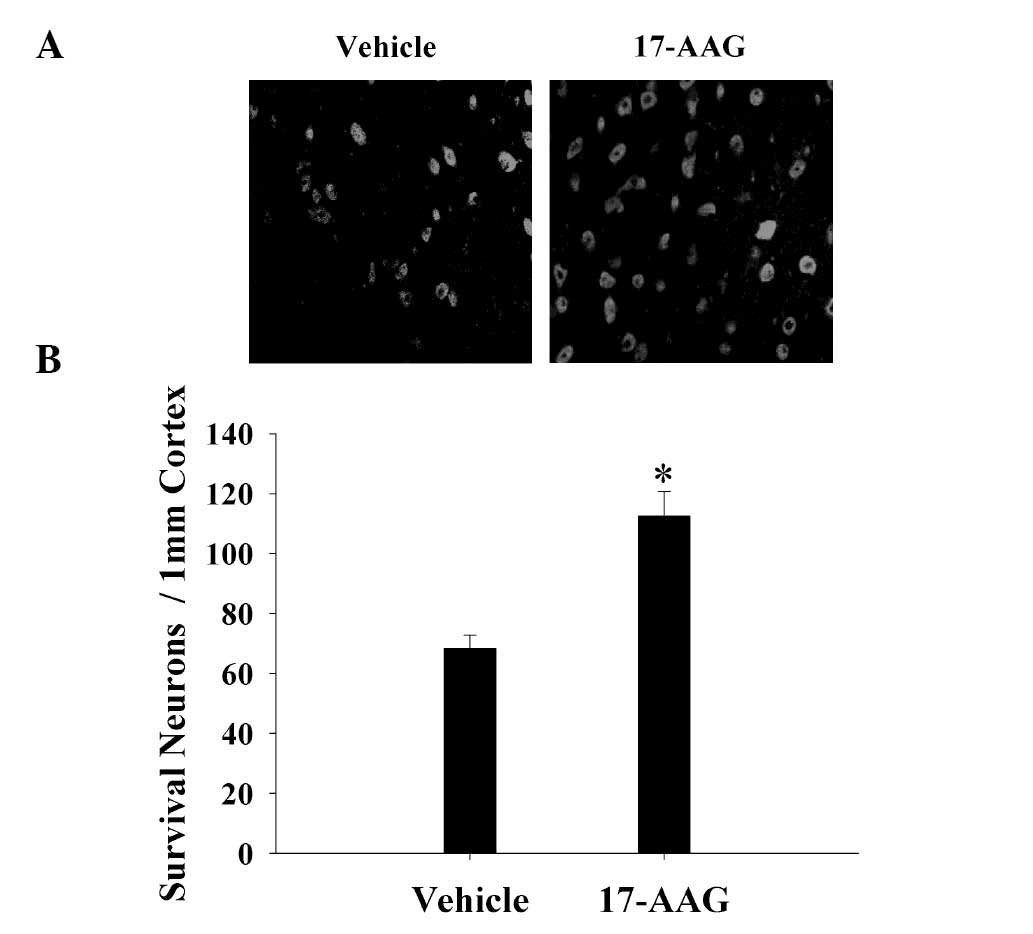
17-AAG increases neuronal survival in
the cortex
The cortex region of the brains was collected and
immunostaining with the NeuN neuronal marker 24 h after TBI.
Neuronal survival was quantified by counting the number of
NeuN-positive cells in 1 mm of the rat cortex region. As presented
in Fig. 3, 24 h after TBI treatment
with 17-AAG significantly increased neuronal survival in the cortex
region of the brain following trauma (P<0.05).
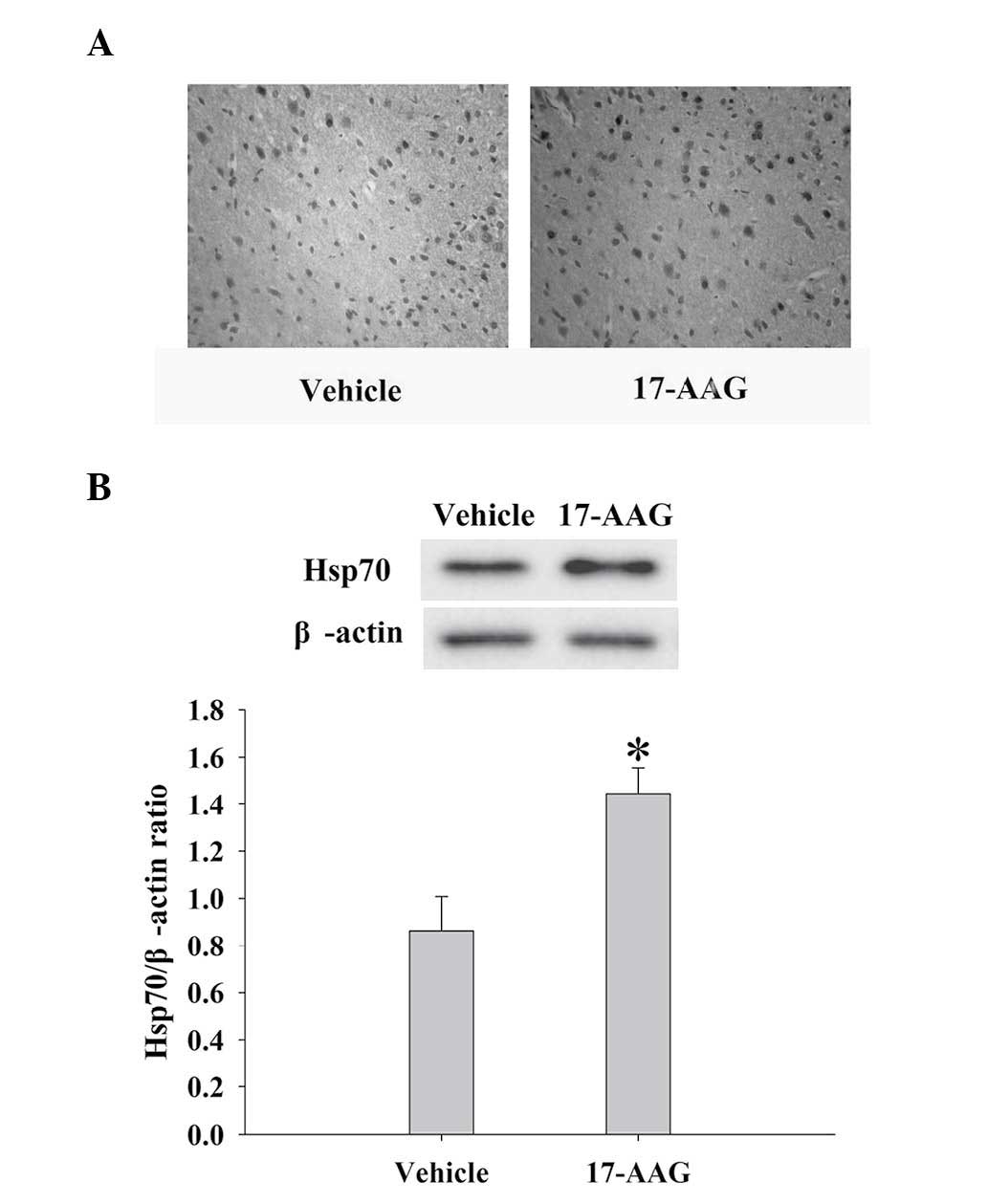
17-AAG induces Hsp70 expression in the
cortex
The expression levels of Hsp70 in the cortex at the
24 h time-point were measured by immunohistochemistry and western
blotting. As depicted in Fig. 4,
administration of 17-AAG produced significant elevations in Hsp70
protein expression levels 24 h following TBI, as compared with the
untreated group (P<0.05).
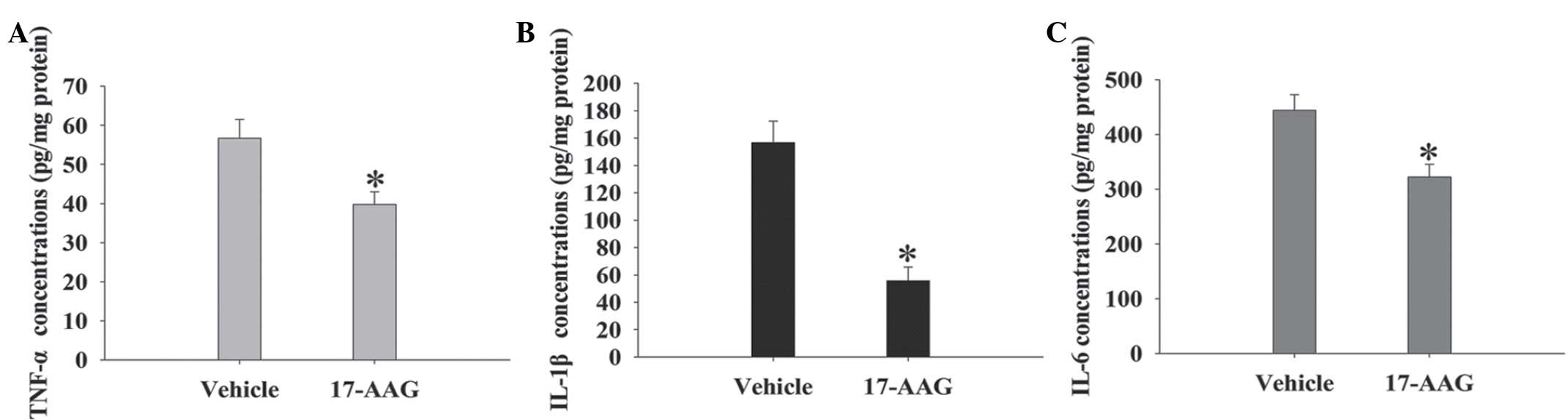
17-AAG induces significant reductions
in multiplex cytokine levels in the cortex
The expression levels of pro-inflammatory cytokines
(IL-1β, IL-6, and TNF-α) in the cortex region of the rats were
measured using commercially available ELISA kits at 24 h (Fig. 5). All three cytokine expression
levels exhibited significant decreases following 17-AAG treatment
compared with the vehicle group (P<0.05).
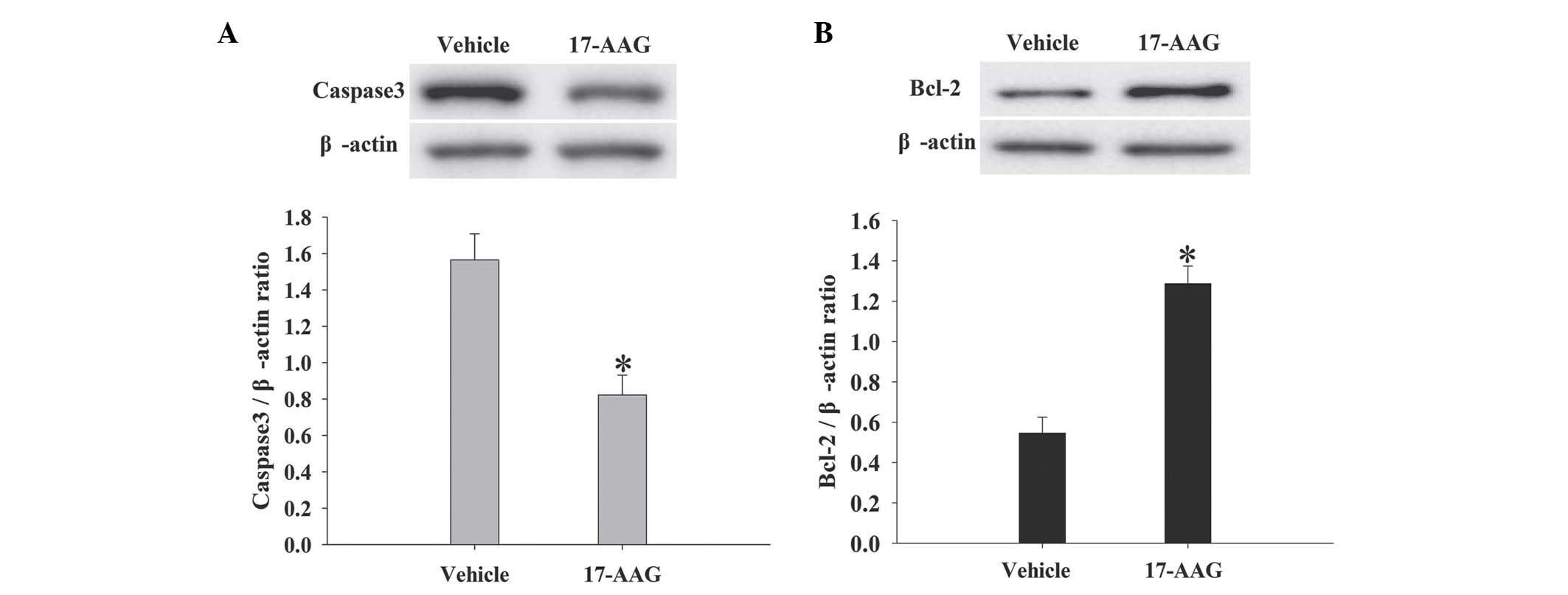
17-AAG attenuates neuronal apoptosis
death in the cortex
The expression levels of caspase-3 and Bcl-2 in the
cortex at 24 h were assessed by western blotting (Fig. 6). As demonstrated in Fig. 6, caspase-3 expression levels in the
17-AAG treatment group exhibited significant decreases compared
with the vehicle group (P<0.05). Conversely Bcl-2 expression
levels were significantly elevated (P<0.05). The present results
indicate that 17-AAG treatment leads to the reduction of neuronal
apoptosis in the cortex following TBI.
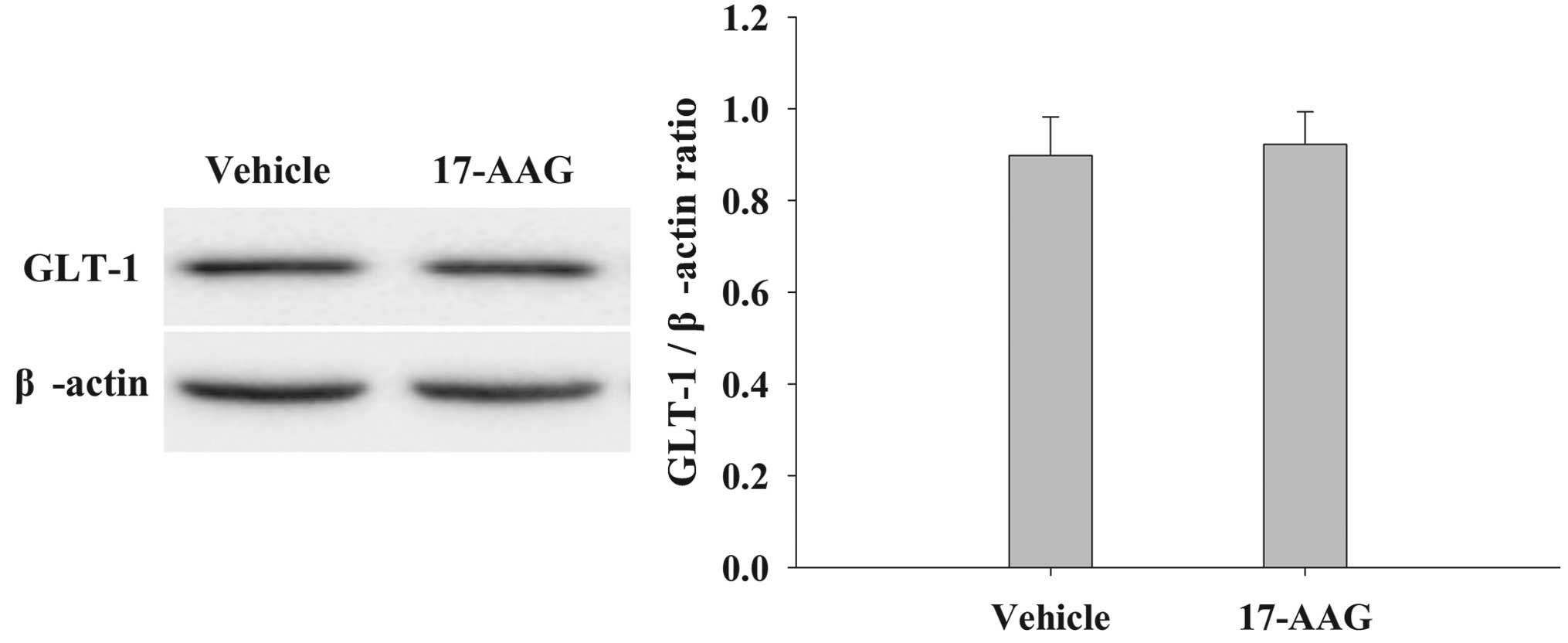
17-AAG induces no significant changes
in GLT-1 expression in the cortex of the brain
The protein expression levels of GLT-1 in the cortex
were analyzed by western blotting 24 h after TBI (Fig. 7). Treatment with 17-AAG induced no
significant changes in GLT-1 expression compared with the vehicle
group (P>0.05).
Discussion
TBI is the primary cause of mortality and disability
in young adults living in China (18). In the present study, a CCI model of
brain trauma was established. The advantages of a CCI model include
the ability to control deformation parameters, such as depth and
velocity of the impact (19).
Additionally, this model is able to imitate the spectrum of diffuse
axonal injury and focal-type damage observed in TBI (19). In the present study, any rats that
did not display moderate to severe neurological deficits following
the surgery were excluded from further investigations.
Hsps are a family of chaperones that control the
synthesis, folding and degradation of proteins within cells
(20). The Hsp family constitutes a
major control system for protein quality. Previous studies have
demonstrated that overexpression of chaperones may attenuate the
formation of protein aggregates in transgenic mouse models, thus
reducing oxidative stress levels in addition to enhancing the
antioxidant activity of enzymes (20–23).
Furthermore, studies have revealed that the overexpression of Hsp70
may exert neuroprotective effects in animal models of neurological
diseases (22,23). As 17-AAG is an Hsp70 activator in
vivo and displays little hepatotoxicity (24), the present study hypothesized that
17-AAG may be a promising approach for the treatment of TBI. The
results of the present study demonstrated that administration of
17-AAG immediately following TBI significantly reduced brain edema
and motor neurological deficits, in addition to increasing neuronal
survival. Furthermore, the aforementioned findings were associated
with the downregulation of pro-inflammatory cytokines. Previous
studies have demonstrated that 17-AAG provides neuroprotective
effects in various animal models of neurological diseases,
including neurodegenerative disorders, epilepsy, ischemia and acute
brain injury (13,25). The results of the present study were
concordant with those of the previous studies and consolidated upon
previous evidence to demonstrate that post-injury treatment of
17-AAG provides neuroprotective properties through the attenuation
of proinflammatory cytokines in a rat CCI model.
The inflammatory response induced following TBI is
an important therapeutic target for reducing tissue damage
following trauma (26).
Additionally, it is a major contributing factor to secondary injury
in TBI (26). A previous
investigation revealed that when Hsp70 is overexpressed, the
expression levels of the inducible isoform of nitric oxide synthase
are reduced in astrocytes, whereas NF-κB is activated (27). Furthermore, in an in vitro
study, Hsp70 prevented the lipopolysaccharide-induced upregulation
of proinflammatory cytokines (28).
Additionally, in a rat model of experimental stroke, overexpression
of Hsp70 inhibited the production of IL-1β and TNF-α (29). The present study did not establish
the existence of a direct association between TBI-induced
functional deficits and pro-inflammatory factors, however, in
vivo evidence that 17-AAG can provide a neuroprotective effect
partly by inhibiting proinflammatory cytokine activation (8) has been demonstrated.
Excitotoxicity is another crucial process in nervous
cell death following TBI (30).
Astrocytes may protect against glutamate excitotoxicity via
glutamate transporters, and GLT-1 is the most important glutamate
transporter in the brains of rats (31). However, in the present study, no
significant change in GLT-1 expression levels following post-TBI
17-AAG treatment were observed. It was there hypothesized that the
neuroprotective effect of 17-AAG is not associated with the
regulation of GLT-1 protein expression.
In summary, the present study demonstrated that the
administration of Hsp70 activator 17-AAG reduced brain edema and
motor neurological deficits, in addition to increasing neuronal
survival, in a rat model of TBI. Furthermore, 17-AAG reduced TNF-α,
IL-1β and IL-6 protein expression levels. Conversely, no
significant changes in GLT-1 expression levels were observed. The
findings of the present study emphasize that 17-AAG provides
neuroprotection via anti-inflammatory effects in a rat model of
TBI.
Glossary
Abbreviations
Abbreviations:
|
17-AAG
|
17-allylamino-demethoxygeldanamycin
|
|
TBI
|
traumatic brain injury
|
|
CCI
|
controlled cortical impact
|
|
NSS
|
Neurologic Severity Score
|
|
NeuN
|
neuron-specific nuclear protein
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL-6
|
interleukin-6
|
|
IL-1β
|
interleukin-1β
|
|
GLT-1
|
glutamate transporter-1
|
References
|
1
|
Hyder AA, Wunderlich CA, Puvanachandra P,
Gururaj G and Kobusingye OC: The impact of traumatic brain
injuries: A global perspective. NeuroRehabilitation. 22:341–353.
2007.PubMed/NCBI
|
|
2
|
Johnson VE, Stewart W and Smith DH:
Widespread τ and amyloid-β pathology many years after a single
traumatic brain injury in humans. Brain Pathol. 22:142–149. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Werner C and Engelhard K: Pathophysiology
of traumatic brain injury. Br J Anaesth. 99:4–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loane DJ and Faden AI: Neuroprotection for
traumatic brain injury: Translational challenges and emerging
therapeutic strategies. Trends Pharmacol Sci. 31:596–604. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kamm K, Vanderkolk W, Lawrence C, Jonker M
and Davis AT: The effect of traumatic brain injury upon the
concentration and expression of interleukin-1β and interleukin-10
in the rat. J Trauma. 60:152–157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aibiki M, Maekawa S, Ogura S, Kinoshita Y,
Kawai N and Yokono S: Effect of moderate hypothermia on systemic
and internal jugular plasma IL-6 levels after traumatic brain
injury in humans. J Neurotrauma. 16:225–232. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hang C-H, Shi J-X, Li J-S, Li W-Q and Wu
W: Expressions of intestinal NF-kappaB, TNF-α, and IL-6 following
traumatic brain injury in rats. J Surg Res. 123:188–193. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen G, Shi JX, Hang CH, Xie W, Liu J and
Liu X: Inhibitory effect on cerebral inflammatory agents that
accompany traumatic brain injury in a rat model: A potential
neuroprotective mechanism of recombinant human erythropoietin
(rhEPO). Neurosci let. 425:177–182. 2007. View Article : Google Scholar
|
|
9
|
Mayer MP and Bukau B: Hsp70 chaperones:
Cellular functions and molecular mechanism. Cell Mol Life Sci.
62:670–684. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nowak TS Jr and Jacewicz M: The heat
shock/stress response in focal cerebral ischemia. Brain Pathol.
4:67–76. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kelly S and Yenari MA: Neuroprotection:
Heat shock proteins. Curr Med Res Opin. 18:(Suppl 2). s55–s60.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee S-H, Kim M, Yoon B-W, Kim YJ, Ma SJ,
Roh JK, Lee JS and Seo JS: Targeted hsp70. 1 disruption increases
infarction volume after focal cerebral ischemia in mice. Stroke.
32:2905–2912. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim N, Kim JY and Yenari MA:
Anti-inflammatory properties and pharmacological induction of Hsp70
after brain injury. Inflammopharmacology. 20:177–185. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen SF, Hsu CW, Huang WH and Wang JY:
Post-injury baicalein improves histological and functional outcomes
and reduces inflammatory cytokines after experimental traumatic
brain injury. Br J Pharmacol. 155:1279–1296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui CM, Gao JL, Cui Y, Sun LQ, Wang YC,
Wang KJ, Li R, Tian YX and Cui JZ: Chloroquine exerts
neuroprotection following traumatic brain injury via suppression of
inflammation and neuronal autophagic death. Mol Med Rep.
12:2323–2328. 2015.PubMed/NCBI
|
|
16
|
Wei J, Pan X, Pei Z, Wang W, Qiu W, Shi Z
and Xiao G: The beta-lactam antibiotic, ceftriaxone, provides
neuroprotective potential via anti-excitotoxicity and
anti-inflammation response in a rat model of traumatic brain
injury. J Trauma Acute Care Surg. 73:654–660. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ikeda Y and Long DM: The molecular basis
of brain injury and brain edema: The role of oxygen free radicals.
Neurosurgery. 27:1–11. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McKinlay A, Grace RC, Horwood LJ,
Fergusson DM, Ridder EM and MacFarlane MR: Prevalence of traumatic
brain injury among children, adolescents and young adults:
Prospective evidence from a birth cohort. Brain Inj. 22:175–181.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
O'Connor WT, Smyth A and Gilchrist MD:
Animal models of traumatic brain injury: A critical evaluation.
Pharmacol Ther. 130:106–113. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Outeiro TF, Klucken J, Strathearn KE, Liu
F, Nguyen P, Rochet JC, Hyman BT and McLean PJ: Small heat shock
proteins protect against α-synuclein-induced toxicity and
aggregation. Biochem Biophys Res Commun. 351:631–638. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Adachi H, Katsuno M, Minamiyama M, Sang C,
Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu
M and Sobue G: Heat shock protein 70 chaperone overexpression
ameliorates phenotypes of the spinal and bulbar muscular atrophy
transgenic mouse model by reducing nuclear-localized mutant
androgen receptor protein. J Neurosci. 23:2203–2211.
2003.PubMed/NCBI
|
|
22
|
Hollander JM, Martin JL, Belke DD, Scott
BT, Swanson E, Krishnamoorthy V and Dillmann WH: Overexpression of
wild-type heat shock protein 27 and a nonphosphorylatable heat
shock protein 27 mutant protects against ischemia/reperfusion
injury in a transgenic mouse model. Circulation. 110:3544–3552.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hutter JJ, Mestril R, Tam EK, Sievers RE,
Dillmann WH and Wolfe CL: Overexpression of heat shock protein 72
in transgenic mice decreases infarct size in vivo. Circulation.
94:1408–1411. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fumo G, Akin C, Metcalfe DD and Neckers L:
17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in
down-regulating mutated, constitutively activated KIT protein in
human mast cells. Blood. 103:1078–1084. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harrison EM, Sharpe E, Bellamy CO, McNally
SJ, Devey L, Garden OJ, Ross JA and Wigmore SJ: Heat shock protein
90-binding agents protect renal cells from oxidative stress and
reduce kidney ischemia-reperfusion injury. Am J Physiol Renal
Physiol. 295:F397–F405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morganti-Kossmann MC, Rancan M, Stahel PF
and Kossmann T: Inflammatory response in acute traumatic brain
injury: A double-edged sword. Curr Opin Crit Care. 8:101–105. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heneka MT, Sharp A, Klockgether T,
Gavrilyuk V and Feinstein DL: The heat shock response inhibits
NF-kappaB activation, nitric oxide synthase type 2 expression, and
macrophage/microglial activation in brain. J Cereb Blood Flow
Metab. 20:800–811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ding XZ, Fernandez-Prada CM, Bhattacharjee
AK and Hoover DL: Overexpression of hsp-70 inhibits bacterial
lipopolysaccharide-induced production of cytokines in human
monocyte-derived macrophages. Cytokine. 16:210–219. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zheng Z, Kim JY, Ma H, Lee JE and Yenari
MA: Anti-inflammatory effects of the 70 kDa heat shock protein in
experimental stroke. J Cereb Blood Flow Metab. 28:53–63. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Palmer AM, Marion DW, Botscheller ML,
Swedlow PE, Styren SD and DeKosky ST: Traumatic brain
injury-induced excitotoxicity assessed in a controlled cortical
impact model. J Neurochem. 61:2015–2024. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tanaka K, Watase K, Manabe T, Yamada K,
Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi
T, et al: Epilepsy and exacerbation of brain injury in mice lacking
the glutamate transporter GLT-1. Science. 276:1699–1702. 1997.
View Article : Google Scholar : PubMed/NCBI
|