Introduction
Lower respiratory tract infections (LRTIs),
including pneumonia, are a primary cause of infection-based
mortalities. The World Health Organization estimates, that
~3,500,000 mortalities are caused by this infection annually
(1). Community-acquired pneumonia
(CAP) is a severe type of pneumonia that typically requires
hospitalization. CAP is a primary factor for adult mortality, and
for those who survive, mortality in the following years remains
high (2,3). Among adolescents, it is estimated that
pneumonia accounts for 15% of mortalities worldwide (4). Although the occurrence of childhood
pneumonia has been reduced, the decline has been small. Reportedly,
~950,000 individuals <5 years old succumbed to pneumonia in 2013
(5).
Severe pneumonia requires treatment in an intensive
care unit. Viral infection is a primary factor for the mortality of
patients with severe pneumonia (6)
and a number of viruses, such as the respiratory syncytial virus,
have been identified in infants and children with severe pneumonia
(7). Although vaccines against
viruses, including Haemophilus influenzae type b and
Streptococcus pneumoniae, have been introduced in numerous
locations, achieving sufficient coverage in developed countries
remains a challenge (5). Therefore,
it is important to develop novel and improved methods of diagnosing
and treating patients with pneumonia.
Targeted therapy has been widely used for the
management of numerous types of cancer and diseases. A previous
study reported that the plasminogen activator inhibitor-1 gene
4G/5G polymorphism may be used as a marker for the diagnosis of
severe pneumonia (8). Elevated
levels of procalcitonin and C-reactive protein are positively
correlated with the severity of CAP (9). MicroRNAs (miRNAs or miRs) are
non-coding RNAs of ≤200 base pairs in length that can regulate gene
expression at transcriptional or post-transcriptional levels,
influencing cellular activities and disease progression (10). Few miRNAs have been detected in the
pathogenesis of pneumonia. Abd-El-Fattah et al (11) demonstrated that miR-155, miR-21 and
miR-197 were markedly increased in patients with lung cancer or
pneumonia. Another previous study, using miRNA microarray data from
patients with severe pneumonia infected with the influenza A
subtype H1N1, identified a number of differentially expressed
miRNAs (DE-miRs; upregulated, hsa-miR-374a, hsa-miR-875-5p,
hsa-miR-342-3p, hsa-miR-150 and hsa-miR-15b; downregulated,
hsa-miR-29c, hsa-miR-1247 and hsa-miR-1233) in patients with severe
pneumonia, compared with patients with non-severe pneumonia or
healthy controls. In addition, upregulated miRNAs that associated
with severe pneumonia primarily affect transforming growth factor-,
apoptosis and Wnt/β-catenin signaling pathways (12). Despite these discoveries, the targets
of these DE-miRs, and their target's function and potential
interactions, remain unknown.
RNA-sequencing (RNA-seq) has been successfully used
to detect gene alterations, gene-fusions and somatic mutations, and
is a markedly more precise method for the detection of the
transcripts compared with other methods (13,14). The
present study utilized RNA-seq to detect miRNA expression in
patients with severe pneumonia, patients with non-severe pneumonia
and healthy controls. The results of these groups were compared to
identify DE-miRs. In addition, target genes of these DE-miRs were
predicted and put through enrichment analysis. Furthermore, a
protein-protein interaction (PPI) network of the predicted targets
was constructed to investigate the potential regulatory
relationships of them at the protein level. Through these
comprehensive bioinformatics analyses, the present study aimed to
explore the pathogenesis of pneumonia and identify important miRNA
therapeutic targets.
Materials and methods
Sample collection
Three types of samples were collected in the present
study: Patients with severe pneumonia (severe sample: n=9; mean
age, 65.2±11.9 years; all male), non-severe pneumonia (non-severe
sample: n=9; mean age, 21.3±2.7 years; all male) and healthy
individuals (controls: n=9; mean age, 19.6±2.2 years; all male).
The patients were hospitalized at the People's Liberation Army
General Hospital (PLA; Beijing, China) between June and December
2013. The average duration of stay in the hospital was 18.2 days in
the severe group and 14.6 days in the non-severe group. Severe
pneumonia was diagnosed if one or more of the following conditions,
based on the previously described CAP guidelines (15), was present: i) Disturbance of
consciousness; ii) respiratory rate ≥30 breaths/min; iii)
PaO2 <60 mmHg, PaO2/FiO2
<300 mmHg, with mechanical ventilation required; vi) systolic
arterial pressure <90 mmHg; v) complications related to septic
shock; vi) chest X-ray showing bilateral lung involvement or lung
lesion expansion of ≥50% within 48 h following hospitalization; and
vii) oliguria, urine volume <20 or >80 ml/h, or acute renal
failure requiring dialysis. The present study was approved by the
Medical Ethics Committee of the PLA General Hospital and all
participants gave informed consent.
RNA isolation and sequencing
Total RNA in the peripheral blood plasma was
isolated using the mirVan miRNA Isolation kit (Ambion; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and purified using the
miRNeasy Mini kit (Qiagen, GmbH, Hilden, Germany), according to the
manufacturer's instructions. Then, the isolated RNA was quantified
using a spectrophotometer (NanoDrop, Thermo Fisher Scientific,
Inc., Wilmington, DE, USA). Purified RNA was frozen with dry ice
and stored at −80°C until required.
As the quantity of RNA extracted was relatively low,
the nine patients within each group (severe pneumonia, non-severe
pneumonia and control) were randomly divided into three sub-groups.
Within each subgroup, the three patients' RNAs were mixed equally
and used as one sample for sequencing. Therefore, there were three
samples from each group [severe pneumonia (WLL1, WLL2 and WLL3),
non-severe pneumonia (WLL4, WLL5 and WLL6) and the control (WLL7,
WLL8 and WLL9)].
Following extraction and purification, an RNA
library was constructed using the NEBNext Ultra RNA Library Prep
kit for Illumina (New England Biolabs, Inc., Ipswich, MA, USA)
based on the manufacturer's instructions. In brief, four steps were
performed: i) Total RNA (1 µg) was sheared into fragments (200–500
nucleotides) in the NEBNext First Strand Synthesis Reaction Buffer
and the fragments were utilized to generate the double-stranded
cDNA; ii) the cDNA was end-repaired and ligated with
Illumina-specific adaptors; iii) size selection of the library was
performed using 200 bp inserts to select suitable fragments for
polymerase chain reaction (PCR) amplification; iv) PCR was
performed using Phusion High-Fidelity DNA polymerase (New England
Biolabs, cat. no. F-530S) and the products were purified with a
QIAquick Nucleotide Removal kit (Qiagen, no. 28304). The
constructed RNA library was sequenced on an Illumina HiSeq 4000
system using 2×50 base pairs paired-end sequencing.
Processing of the sequencing data
FASTX-Toolkit software (ver. 0.0.13, http://hannonlab.cshl.edu/fastx_toolkit)
was used to remove the 3′ adaptor primer from the raw read. In
addition, bases with a quality score <20 were eliminated. If 30%
of bases in a sequence were eliminated, the entire sequence was
discarded and did not undergo further analysis.
Identification of DE-miRs
Following processing, the remaining reads were
aligned to the Genome Reference Consortium human genome assembly 38
(GRCH38) reference genome using Bowtie2 software (version 2.1.0,
www.sourceforge.net/projects/bowtie-bio/files/bowtie2).
In the alignment, each read was allowed ≤8 base mismatches and only
reads with a quality score >60 were retained. In addition,
considering RNA splicing, the length of reads was not taken into
account as a restriction when performing the alignment.
The latest (June 2016) human miRNA sequences and
their locations were downloaded from the miRbase database (16) and the aligned sequences of each miRNA
counted using perl programing, written by the present authors. The
total quantity of each miRNA in each sample was used to calculate
the number of mapped reads per kilobase per million reads (RPKM),
which were normalized using DESeq software (version 1.24.0;
www.bioconductor.org/packages/release/bioc/html/DESeq.html).
Then, DE-miRs were selected based on un-paired t-tests in
limma (version 3.22.7; www.bioconductor.org/packages/3.0/bioc/html/limma.html)
(17), followed by adjustment using
the Benjamini-Hochberg method (18).
The adjusted P-value was deemed to be the false discovery rate
(FDR). The cut-off for DE-miRs between different types of samples
(non-severe vs. control; severe vs. non-severe) was a log 2
fold-change (FC) >1 and FDR <0.5. Afterwards, a Venn diagram
was delineated to identify the overlapped DE-miRs in the two
comparisons and their expression trends in the three different
types of samples were portrayed. Two-way hierarchical cluster
analysis was performed to present the expression of DE-miRs in the
three types of samples.
DE-miR target prediction
Gene sequences of GRCH38 reference genome
corresponding to the DE-miRs were downloaded. miRanda software
(ver. 1.0, www.microrna.org) (19) was used to predict target genes of
these DE-miRs. Efficiency of the microRNA target sites predicted by
this software was scored and ranked by the mirSVR algorithm. The
mirSVR score reflects the empirical probability that a gene is
downregulated by a specific microRNA (20). As each DE-miR had multiple targets,
only genes with a mirSVR score <-1.2 were considered to be
targets.
Enrichment analysis of the predicted
targets
Gene ontology (GO; www.geneontology.org) (21) and Kyoto Encyclopedia of Genes and
Genomes (KEGG; www.genome.jp/kegg/pathway.html) (22) enrichment analyses were performed
using the Database for Annotation, Visualization and Integration
Discovery (DAVID; version 6.7; https://david.ncifcrf.gov) (23). Pathway enrichment analysis of targets
was conducted using clusterProfiler software (ver. 2.8, www.bioconductor.org/packages/2.8/bioc/html/clusterProfiler.html)
(24). Function and pathways of the
reference species were deposited in the GO and KEGG databases.
Fisher's exact test was used for statistical analysis and the
threshold for significant function and pathway categories of the
target genes was P<0.05, compared with those of the reference
species.
Construction of PPI networks
To identify potential regulatory relationships of
targets of the DE-miRs at protein level, the Search Tool for the
Retrieval of Interacting Genes (STRING; ver. 10.0; www.string-db.org) database (25) was used to build PPI networks of the
targets. DEGs with a combined score >0.9 were entered into the
STRING database. PPI networks were visualized using the Cytoscape
software (ver. 3.1.0; www.cytoscape.org) (26). A node in the network is defined as an
encoded protein of a target gene and the nodal degree represents
the linked gene numbers of this node. In addition, a literature
search was conducted to identify reports of interactions between
these targets. The key search terms used were the name of the
specific target gene and the gene that linked to it. These were:
‘CTNNB1’ AND ‘CRBE’, or ‘CTNNB1’ AND ‘FOXO1’, OR ‘CTNNB1’ AND
‘XPO1’.
Results
Identification of DE-miRs
Under the inclusion criteria of log 2 FC >1 and
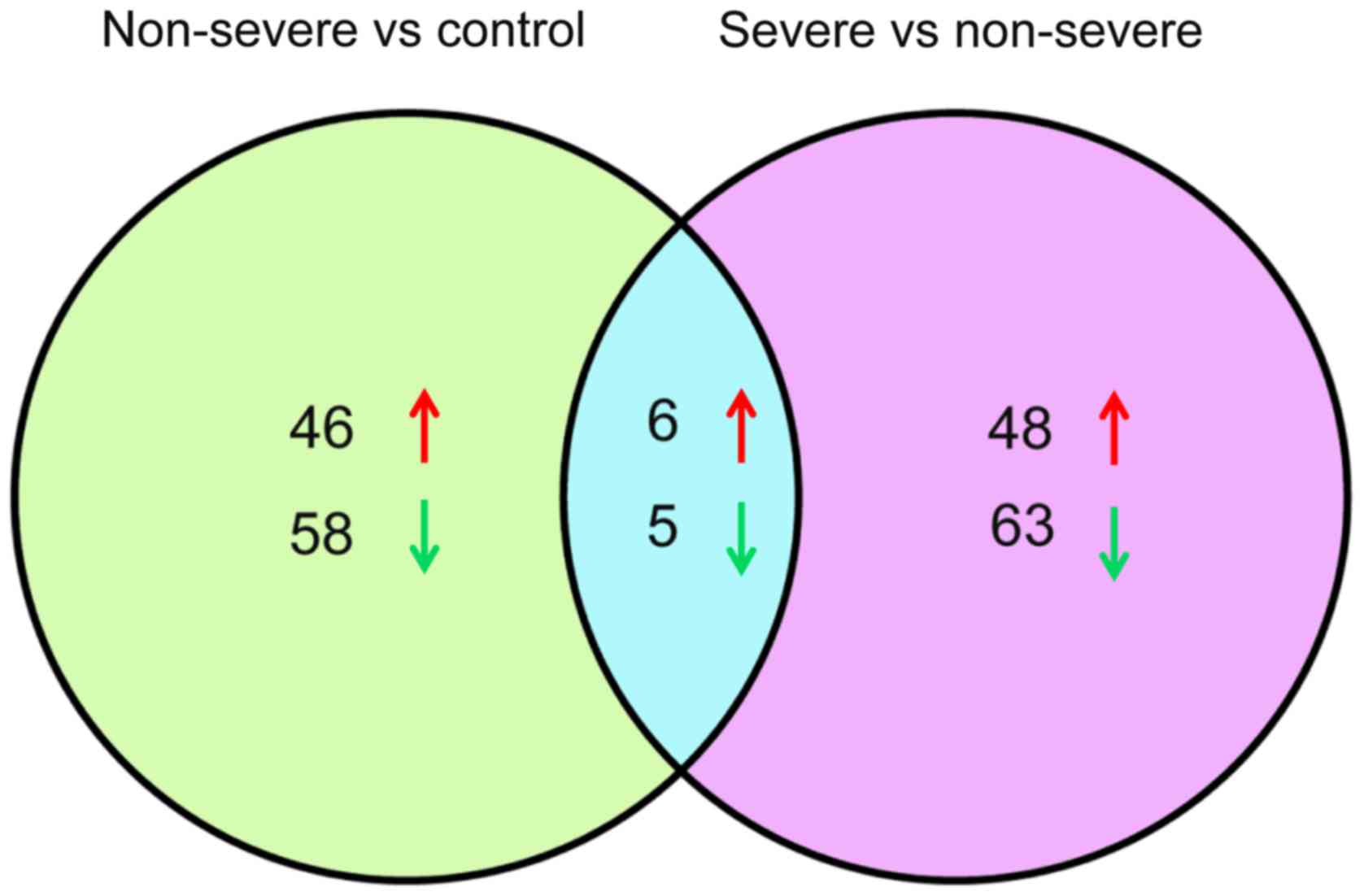
FDR <0.05, a total of 237 DE-miRs were identified. From the
comparison of the non-severe and control groups, there were 52
upregulated and 63 downregulated miRNAs, whereas following the
comparison of the severe and non-severe groups, there were 54
upregulated and 68 downregulated miRNAs (Fig. 1). Venn analysis identified 11
overlapping DE-miRs from both comparisons, of which 6 were
upregulated (hsa-miR-200b, hsa-miR-483, hsa-miR-34a, hsa-miR-193b,
hsa-miR-455 and hsa-miR-95), and 5 were downregulated
(hsa-miR-3617, hsa-miR-664b, hsa-miR-4485, hsa-miR-3161 and
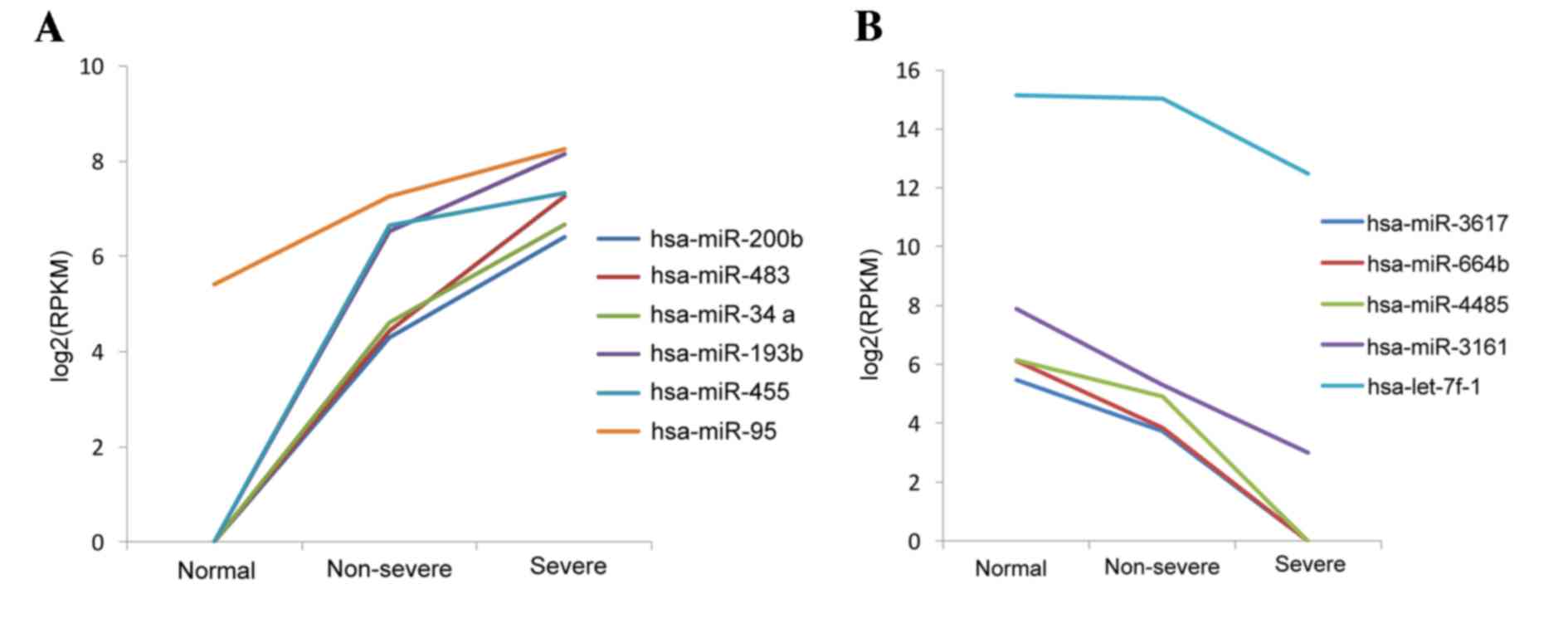
hsa-let-7f-1; Fig. 1). Based on this
result, the levels of expression of the 11 DE-miRs in the three
types of samples were assessed. It was observed that the 6 miRNAs
were upregulated and 5 were downregulated. These miRNAs were up and
downregulated, respectively, in the pneumonia groups compared with
the controls, which may reflect the three phases of pneumonia
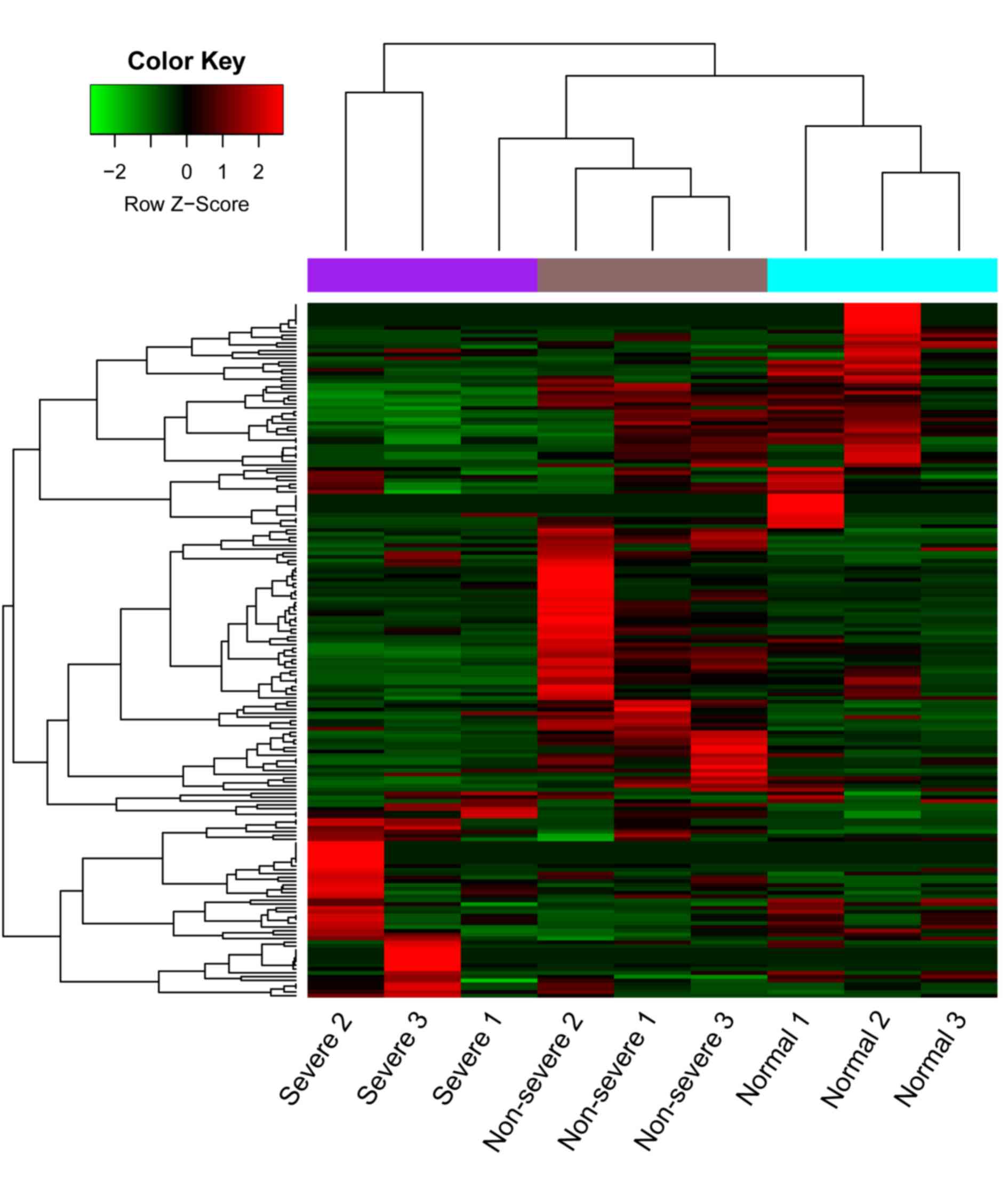
progression (Fig. 2). A heat map of
the cluster analysis is presented in Fig. 3, which indicated that the three types
of samples could be identified by their differing expression of
DE-miRs.
Enrichment analysis of the predicted
targets of the DE-miRs
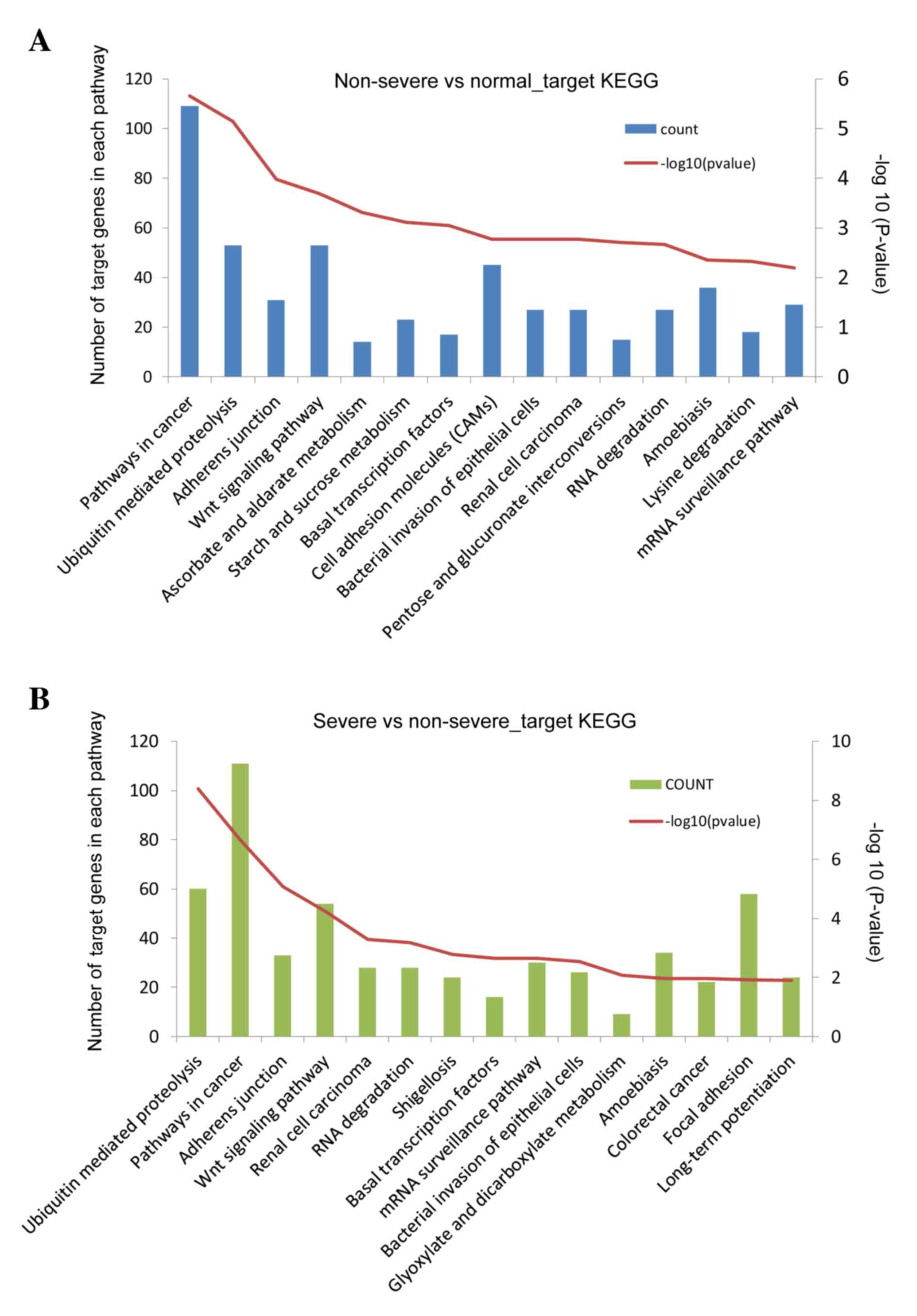
A total of 9,026 targets of the DE-miRs were
identified (non-severe pneumonia vs. the control group, 4,498;
severe pneumonia vs. non-severe pneumonia, 4,528). Targets of
DE-miRs from the comparison of non-severe pneumonia and the control
groups were primarily enriched in anatomical structure
morphogenesis and metabolism regulation-associated functions
(Table I), and pathways involved in
cancer, ubiquitin mediated proteolysis, adherens junction and Wnt
signaling (Fig. 4A). Target genes of
DE-miRs from the comparison of the severe pneumonia and non-severe
pneumonia groups produced similar results in the enrichment
analysis (Table II; Fig. 4B).
 | Table I.Top 10 enriched functions of the
target genes of DE-miRs from the comparison of the non-severe
pneumonia and control groups. |
Table I.
Top 10 enriched functions of the
target genes of DE-miRs from the comparison of the non-severe
pneumonia and control groups.
| GO ID no. | Function | Count | P-value |
|---|
| GO:0009653 | Anatomical
structure morphogenesis | 757 |
<1.0E−16 |
| GO:0009893 | Positive regulation
of metabolic process | 938 |
<1.0E−16 |
| GO:0010604 | Positive regulation
of macromolecule metabolic process | 770 |
<1.0E−16 |
| GO:0019222 | Regulation of
metabolic process | 1,637 |
<1.0E−16 |
| GO:0032502 | Developmental
process | 1,439 |
<1.0E−16 |
| GO:0044767 | Single-organism
developmental process | 1,424 |
<1.0E−16 |
| GO:0048518 | Positive regulation
of biological process | 1,355 |
<1.0E−16 |
| GO:0048522 | Positive regulation
of cellular process | 1,188 |
<1.0E−16 |
| GO:0007275 | Multicellular
organismal development | 1,231 |
2.22E−16 |
| GO:0045893 | Positive regulation
of transcription, DNA-templated | 433 |
2.22E−16 |
| Table II.Top 10 enriched functions of the
target genes of DE-miRs from the comparison of the severe and
non-severe pneumonia groups. |
Table II.
Top 10 enriched functions of the
target genes of DE-miRs from the comparison of the severe and
non-severe pneumonia groups.
| GO ID no. | Function | Count | P-value |
|---|
| GO:0007275 | Multicellular
organismal development | 1,252 |
<1.00E−16 |
| GO:0009653 | Anatomical
structure morphogenesis | 777 |
<1.00E−16 |
| GO:0009893 | Positive regulation
of metabolic process | 943 |
<1.00E−16 |
| GO:0019222 | Regulation of
metabolic process | 1,646 |
<1.00E−16 |
| GO:0032502 | Developmental
process | 1,447 |
<1.00E−16 |
| GO:0044767 | Single-organism
developmental process | 1,433 |
<1.00E−16 |
| GO:0048518 | Positive regulation
of biological process | 1,348 |
<1.00E−16 |
| GO:0048856 | Anatomical
structure development | 1,300 |
<1.00E−16 |
| GO:0010604 | Positive regulation
of macromolecule metabolic process | 768 |
1.11E−16 |
| GO:0016043 | Cellular component
organization | 1,484 |
4.44E−16 |
Pathway enrichment of the 11
overlapping DE-miRs
Following the integration of KEGG pathway analysis
of targets of the 11 DE-miRs and their miRNA-target interactions,
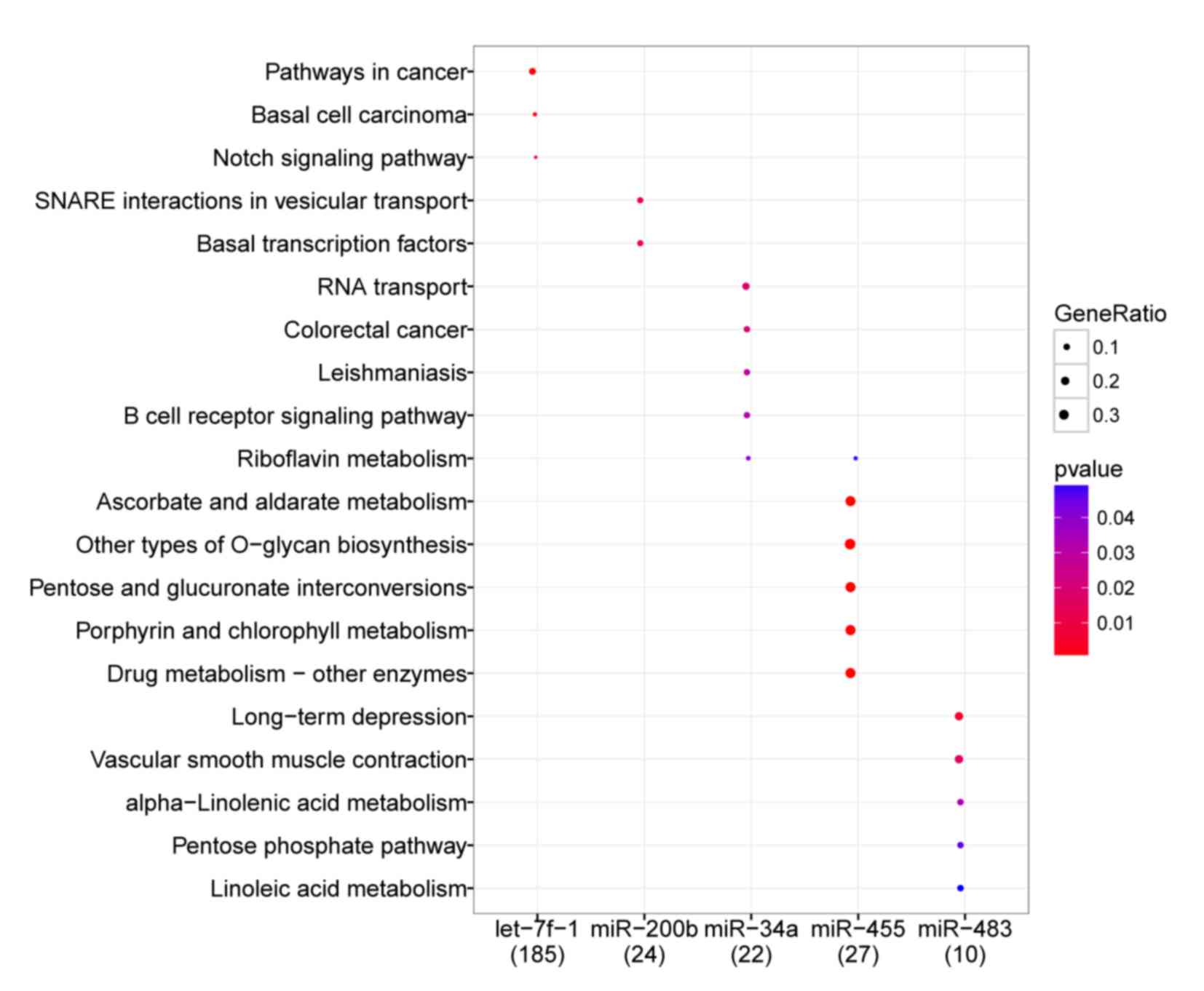
the enriched pathways of 5 DE-miRs were obtained (Fig. 5). The miRNA hsa-let-7f-1 was
significantly enriched in a number of pathways compared with the
reference species in the GO and KEGG databases. These included
‘pathways in cancer’ (P=0.001), ‘basal cell carcinoma’ (P=0.002)
and ‘Notch signaling pathways’ (P=0.003); hsa-miR-200b was enriched
in ‘SNARE interactions in vesicular transport’ (P=0.009) and ‘basal
transcription factors’ (P=0.010); hsa-miR-34a was enriched in ‘RNA
transport’ (P=0.018), ‘colorectal cancer’ (P=0.022),
‘leishmaniasis’ (P=0.030), ‘B cell receptor signaling pathway’
(P=0.031) and ‘riboflavin metabolism’ (P=0.040); hsa-miR-455 was
enriched in ‘riboflavin metabolism’ (P<0.001), ‘ascorbate and
aldarate metabolism’ (P<0.001) and ‘drug metabolism-other
enzymes’ (P<0.001); and hsa-miR-483 was enriched in pathways
associated with ‘long term depression’ (P=0.006), the ‘pentose
phosphate pathway’ (P=0.045) and ‘linoleic acid metabolism’
(P=0.050).
PPI network of 11 key targets of
DE-miRs
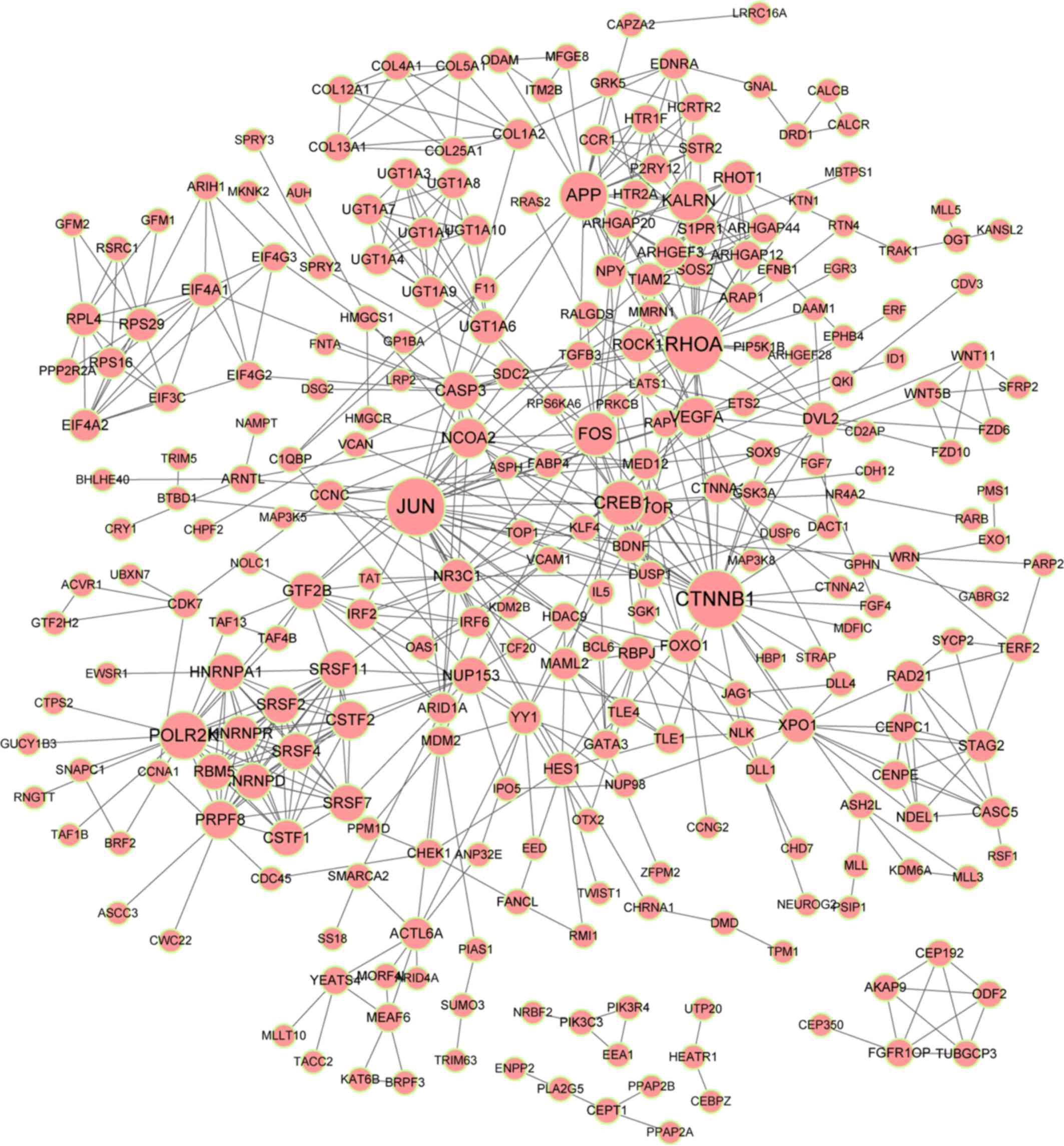
A PPI network of targets of the DE-miRs was
constructed (Fig. 6), as
aforementioned in the methods section. Five predominant protein
interactors were identified: Ras homolog family member A (RhoA;
degree, 28), β-catenin (CTNNB1; degree, 28), polymerase II
subunit K (POLR2K; degree, 20), amyloid precursor protein
(APP; degree, 20) and Kalirin, RhoGEF Kinase (KALRN;
degree, 15). RHOA, CTNNB1, POLR2K and
APP were predicted targets of hsa-let-7f-1.
Discussion
In the present study, 11 key DE-miRs were identified
in pneumonia, including 6 upregulated miRNAs (including
hsa-miR-200b and hsa-miR-455) and 5 downregulated miRNAs (including
hsa-let-7f-1). Notably, these 11 miRNAs kept their
upregulation/downregulation expression pattern in the three types
of samples, which may reflect the three phases of pneumonia
development. Enrichment analysis indicated that the target genes of
DE-miRs identified from comparisons of non-severe pneumonia and the
control, and severe pneumonia and non-severe pneumonia were highly
associated with functions in the ‘adherens junction’ and ‘Wnt
signaling pathway’. In addition, KALRN, RhoA, CTNNB1,
POLR2K and APP were determined to be crucial nodes in
the PPI network of targets of the DE-miRs. KALRN was
predicted as a target of hsa-miR-200b, while RHOA,
CTNNB1, POLR2K and APP were predicted targets
of hsa-let-7f-1.
miRNAs in the let-7 family function as regulators of
cell proliferation. let-7 acts as an inhibitor during the
development of lung cancer, indicating that its downregulation
facilitates cell proliferation (27). In mice, let-7b and let-7c have been
identified as important miRNAs in the lung restoration at the late
stage of influenza pneumonia (27).
In human non-small cell lung cancer (NSCLC), hsa-let-7c inhibits
tumor metastasis and invasion via targeting the transcripts
encoding integrin 3 and mitogen-activated protein kinase 3
(28). Previous studies have
demonstrated that let-7 family members are involved in many
carcinogenic pathways, including the Notch signaling pathway, which
is associated with a number of diseases in humans (29,30). In
the present study, hsa-let-7f-1 was found to be downregulated in
pneumonia. Compared with the reference pathways in the reference
species, this miRNA was significantly enriched in ‘pathways in
cancer’ and the ‘Notch signaling pathway’, suggesting that let-7f-1
could function in these signaling pathways to regulate the
progression of pneumonia. Downregulation of hsa-let-7f-1 may
facilitate the invasion and metastasis of pneumonia-associated
pathogens into lung epithelial cells, or reduce lung damage repair
functions.
RhoA belongs to the Rho family, whose members are
small guanosine-5′-triphosphate phosphatases and serve as molecular
switches in signal transduction cascades. Overexpression of RhoA is
associated with tumor cell proliferation and metastasis (31,32). In
antecedent influenza, Staphylococcus aureus strains are
typically the cause of invasive infections, including pneumonia
(33). A conserved surface protein
of S. aureus, SpA (protein A), is overexpressed in the early
stages of bacterial growth. Previous experiments in models of
pneumonia have indicated that SpA activates the RhoA/Rho-associated
protein kinase/myosin light chain protein signaling cascade, which
results in contraction of the epithelial cytoskeleton that
facilitates translocation via the paracellular junctions of the
mucosal epithelium (34).
Pseudomonas aeruginosa induces severe pneumonia and its type
III secretion system facilitates the spread of this bacteria
(35). A previous study in bovine
pulmonary artery endothelial cells demonstrated that ExoS and ExoT,
type III cytotoxins of P. aeruginosa, are responsible for
increased vascular permeability in the lungs, through the
activation of the RhoA signaling pathway (35). In the present study, RhoA was
predicted as a target of hsa-let-7f-1, which was downregulated in
pneumonia samples. These results suggest that the downregulation of
hsa-let-7f-1 may induce the upregulation of RHO, activating
the RhoA signaling pathway, thus increasing vascular permeability
of the lungs and leading to pneumonia.
POLR2K encodes the smallest subunit of RNA
polymerases II, which is the common subunit of three RNA
polymerases (36). A previous study
speculated that upregulation of POLR2K may facilitate the
assembly of polymerase III, thus contributing to cell proliferation
and cancer development (36).
APP encodes a transmembrane precursor protein that is
cleaved into peptides by secretases. In NSCLC, alternative splicing
events influence the expression of AAP (37). In other types of cancer, increased
expression of APP is associated with cell proliferation and
tumorigenesis (38,39). CTNNB1 encodes a protein that
is a key downstream component of the Wnt signaling pathway
(40). CTNNB1 is associated
with cadherin-mediated cell-cell adhesion systems and has been
suggested as a biomarker for NSCLC prognosis (41). However, to the best of our knowledge,
no studies investigating the association between
POLR2K/CTNNB1/APP and pneumonia have been performed. In the
present study, POLR2K, CTNNB1 and APP were
downregulated in pneumonia and identified as targets of
hsa-let-7f-1. In addition, their resulting proteins were
functionally enriched in adherens junction and Wnt signaling
pathways. These results suggest that POLR2K, CTNNB1 and
APP, regulated by hsa-let-7f-1, regulate pneumonia
development through the adherens junction and Wnt signaling
pathways.
miR-200b has an important role in lung cancer
development; reportedly, aberrant expression of miR-200b in sputum
may be used as a diagnostic marker for lung adenocarcinoma and
squamouscell carcinoma (42). In a
murine influenza pneumonia model, miR-200b-5p was predicted to be
an important miRNA, connecting regulation of gene functions in
repair (43). This suggests that the
dysregulation of miR-200b may be a key mediator in pulmonary
injury. KALRN encodes the protein kalirin, and decreased
kalirin is implicated in the severity of allergic asthma (44). However, to the best of our knowledge,
no current study elucidates the relationship between KALRN
and pneumonia. In the present study, miR-200b was upregulated and
predicted to target KALRN, suggesting that expression of
KALRN may be downregulated by miR-200b, and this
downregulation may subsequently promote pneumonia development. A
previous study reported that miR-455 suppresses NSCLC by targeting
the transcript encoding Zinc finger E-box-binding homeobox 1
(45). This indicates that
upregulation of miR-455 may inhibit the development of pneumonia.
However, the specific mechanisms underlying this process need to be
further investigated.
In conclusion, the present study identified a number
of key DE-miRs, such as hsa-miR-200b, hsa-miR-455 and hsa-let-7f-1
in pneumonia. The results of bioinformatics analysis suggest that
hsa-let-7f-1 contributes to the development of pneumonia by
affecting cancer and Notch signaling pathways, through targeting
and regulating transcripts encoding RhoA, CTNNB1, POLR2K and APP,
while miR-200b may promote pneumonia via targeting KALRN. In
addition, the results of the present study indicate that
hsa-miR-455 may serve as an inhibitor of pneumonia development.
Further studies are required to validate these predictive
results.
Acknowledgements
The present study was supported by grants from
Welfare Industry Research Program of Ministry of Health (no.
201302017, 201502019), the National Natural Science Fund (No.
81272060, 81371561), the Hai Nan Natural Science Fund (20158315),
the Youth Training Program of the PLA (no. 13QNP171), Beijing
Scientific and Technologic Supernova Supportive Project
(Z15111000030000/XXJH2015B100), PLA General Hospital Science and
Technology Innovation Nursery Fund Project (16KMM56) and PLA
Logistic Major Science and Technology Project (14CXZ005, AWS15J004,
BWS14J041).
References
|
1
|
Organization WH: The top 10 causes of
death. July. 2013, simplewhoint/mediacentre/factsheets/fs310/en2014
|
|
2
|
Wunderink RG and Waterer GW: Clinical
practice. Community-acquired pneumonia. N Engl J Med. 370:543–551.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Said MA, Johnson HL, Nonyane BA,
Deloria-Knoll M, O'Brien KL; AGEDD Adult Pneumococcal Burden Study
Team, ; Andreo F, Beovic B, Blanco S, Boersma WG, et al: Estimating
the burden of pneumococcal pneumonia among adults: A systematic
review and meta-analysis of diagnostic techniques. PLoS One.
8:e602732013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rakha MA, Abdelmoneim AN, Farhoud S,
Pièche S, Cousens S, Daelmans B and Bahl R: Does implementation of
the IMCI strategy have an impact on child mortality? A
retrospective analysis of routine data from Egypt. BMJ Open.
3:pii.e0018522013. View Article : Google Scholar
|
|
5
|
Floyd J, Wu L, Hay Burgess D, Izadnegahdar
R, Mukanga D and Ghani AC: Evaluating the impact of pulse oximetry
on childhood pneumonia mortality in resource-poor settings. Nature.
528:S53–S59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Choi SH, Hong SB, Ko GB, Lee Y, Park HJ,
Park SY, Moon SM, Cho OH, Park KH, Chong YP, et al: Viral infection
in patients with severe pneumonia requiring intensive care unit
admission. Am J Respir Crit Care Med. 186:325–332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berkley JA, Munywoki P, Ngama M, Kazungu
S, Abwao J, Bett A, Lassauniére R, Kresfelder T, Cane PA, Venter M,
et al: Viral etiology of severe pneumonia among Kenyan infants and
children. JAMA. 303:2051–2057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sapru A, Hansen H, Ajayi T, Brown R,
Garcia O, Zhuo H, Wiemels J, Matthay MA and Wiener-Kronish J: 4G/5G
polymorphism of plasminogen activator inhibitor-1 gene is
associated with mortality in intensive care unit patients with
severe pneumonia. Anesthesiology. 110:1086–1091. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
España PP, Capelastegui A, Bilbao A, Diez
R, Izquierdo F, de Goicoetxea MJ Lopez, Gamazo J, Medel F, Salgado
J, Gorostiaga I, et al: Utility of two biomarkers for directing
care among patients with non-severe community-acquired pneumonia.
Eur J Clin Microbiol Infect Dis. 31:3397–3405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong KY, Huang X and Chim CS: DNA
methylation of microRNA genes in multiple myeloma. Carcinogenesis.
33:1629–1638. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abd-El-Fattah AA, Sadik NA, Shaker OG and
Aboulftouh ML: Differential microRNAs expression in serum of
patients with lung cancer, pulmonary tuberculosis, and pneumonia.
Cell Biochem Biophys. 67:875–884. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramirez G, Uribe-Boll J, Cruz A, Jimenez
L, Banales J, Romero S, Hidalgo A, Bautista E, Merino E and Moran
J: Circulating microRNA profiles in patients with severe pneumonia
associated to the A/H1N1 virus. Am J Respir Crit Care Med.
189:A26942014.
|
|
13
|
Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X,
Cui Z, Zhang J, Yi K, Xu W, et al: RNA-seq analysis of prostate
cancer in the Chinese population identifies recurrent gene fusions,
cancer-associated long noncoding RNAs and aberrant alternative
splicings. Cell Res. 22:806–821. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weiwu D: Guidelines for the diagnosis and
treatment of community acquired pneumonia. Chinese J Tuberculosis
Respiratory Dis. 29:651–655. 2001.
|
|
16
|
Griffiths-Jones S, Saini HK, van Dongen S
and Enright AJ: miRBase: Tools for microRNA genomics. Nucleic Acids
Res. 36:D154–D158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smyth GK: Limma: Linear models for
microarray dataBioinformatics and Computational Biology Solutions
Using R and Bioconductor. Springer; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
18
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Statistical Society Series B
(Methodological). 57:289–300. 1995.
|
|
19
|
John B, Enright AJ, Aravin A, Tuschl T,
Sander C and Marks DS: Human microRNA targets. PLoS Biol.
2:e3632004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Palmieri D, Capponi S, Geroldi A, Mura M,
Mandich P and Palombo D: TNFα induces the expression of genes
associated with endothelial dysfunction through p38MAPK-mediated
down-regulation of miR-149. Biochem Biophys Res Commun.
443:246–251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harris MA, Clark J, Ireland A, Lomax J,
Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C,
et al: The gene ontology (GO) database and informatics resource.
Nucleic Acids Res. 32(Database Issue): D258–D261. 2004.PubMed/NCBI
|
|
22
|
Kanehisa M, Goto S, Sato Y, Furumichi M
and Tanabe M: KEGG for integration and interpretation of
large-scale molecular data sets. Nucleic Acids Rese. 40(Database
Issue): D109–D114. 2012. View Article : Google Scholar
|
|
23
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Methods Mol Biol. 696:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tan KS, Choi H, Jiang X, Yin L, Seet JE,
Patzel V, Engelward BP and Chow VT: Micro-RNAs in regenerating
lungs: An integrative systems biology analysis of murine influenza
pneumonia. BMC Genomics. 15:5872014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao B, Han H, Chen J, Zhang Z, Li S, Fang
F, Zheng Q, Ma Y, Zhang J, Wu N and Yang Y: MicroRNA let-7c
inhibits migration and invasion of human non-small cell lung cancer
by targeting ITGB3 and MAP4K3. Cancer Lett. 342:43–51. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Cao L, Wang Y, Wang X, Liu N and
You Y: Regulation of let-7 and its target oncogenes (Review). Oncol
Lett. 3:955–960. 2012.PubMed/NCBI
|
|
30
|
Liu XS, Chopp M, Zhang RL, Tao T, Wang XL,
Kassis H, Hozeska-Solgot A, Zhang L, Chen C and Zhang ZG: MicroRNA
profiling in subventricular zone after stroke: MiR-124a regulates
proliferation of neural progenitor cells through Notch signaling
pathway. PLoS One. 6:e234612011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pillé JY, Denoyelle C, Varet J, Bertrand
JR, Soria J, Opolon P, Lu H, Pritchard LL, Vannier JP, Malvy C, et
al: Anti-RhoA and Anti-RhoC siRNAs inhibit the proliferation and
invasiveness of MDA-MB-231 breast cancer cells in vitroin vivo. Mol
Ther. 11:267–274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chan CH, Lee SW, Li CF, Wang J, Yang WL,
Wu CY, Wu J, Nakayama KI, Kang HY, Huang HY, et al: Deciphering the
transcriptional complex critical for RhoA gene expression and
cancer metastasis. Nat Cell Biol. 12:457–467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
DeLeo FR and Musser JM: Axis of
coinfection evil. J Infect Dis. 201:488–490. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Soong G, Martin FJ, Chun J, Cohen TS, Ahn
DS and Prince A: Staphylococcus aureus protein A mediates invasion
across airway epithelial cells through activation of RhoA GTPase
signaling and proteolytic activity. J Biol Chem. 286:35891–35898.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ganter MT, Roux J, Su G, Lynch SV,
Deutschman CS, Weiss YG, Christiaans SC, Myazawa B, Kipnis E and
Wiener-Kronish JP: Role of small GTPases and alphavbeta5 integrin
in Pseudomonas aeruginosa-induced increase in lung endothelial
permeability. Am J Respir Cell Mol Biol. 40:108–118. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin Y, Li Z, Ozsolak F, Kim SW,
Arango-Argoty G, Liu TT, Tenenbaum SA, Bailey T, Monaghan AP, Milos
PM and John B: An in-depth map of polyadenylation sites in cancer.
Nucleic Acids Res. 40:8460–8471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Misquitta-Ali CM, Cheng E, O'Hanlon D, Liu
N, McGlade CJ, Tsao MS and Blencowe BJ: Global profiling and
molecular characterization of alternative splicing events
misregulated in lung cancer. Mol Cell Biol. 31:138–150. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Krause K, Karger S, Sheu SY, Aigner T,
Kursawe R, Gimm O, Schmid KW, Dralle H and Fuhrer D: Evidence for a
role of the amyloid precursor protein in thyroid carcinogenesis. J
Endocrinol. 198:291–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Takayama K, Tsutsumi S, Suzuki T,
Horie-Inoue K, Ikeda K, Kaneshiro K, Fujimura T, Kumagai J, Urano
T, Sakaki Y, et al: Amyloid precursor protein is a primary androgen
target gene that promotes prostate cancer growth. Cancer Res.
69:137–142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Polakis P: Casein kinase 1 A Wnt'er of
disconnect. Curr Biol. 12:R499–R501. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Woenckhaus M, Merk J, Stoehr R, Schaeper
F, Gaumann A, Wiebe K, Hartmann A, Hofstaedter F and Dietmaier W:
Prognostic value of FHIT, CTNNB1, and MUC1 expression in non-small
cell lung cancer. Hum Pathol. 39:126–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shen J, Liao J, Guarnera MA, Fang H, Cai
L, Stass SA and Jiang F: Analysis of MicroRNAs in sputum to improve
computed tomography for lung cancer diagnosis. J Thorac. 9:33–40.
2014. View Article : Google Scholar
|
|
43
|
Tan KS, Choi H, Jiang X, Yin L, Ju ES,
Patzel V, Engelward BP and Chow VT: Micro-RNAs in regenerating
lungs: An integrative systems biology analysis of murine influenza
pneumonia. BMC Genomics. 15:1–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dewan AT, Egan KB, Hellenbrand K,
Sorrentino K, Pizzoferrato N, Walsh KM and Bracken MB: Whole-exome
sequencing of a pedigree segregating asthma. BMC Medical Genetics.
13:1–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li YJ, Ping C, Tang J and Zhang W:
MicroRNA-455 suppresses non-small cell lung cancer through
targeting ZEB1. Cell Biol Int. 40:621–628. 2016. View Article : Google Scholar : PubMed/NCBI
|