Introduction
Developmental dysplasia of the hip (DDH; Online
Mendelian Inheritance in Man entry no. 142700) is a spectrum of
disorders affecting the proximal femur and acetabulum that leads to
hip subluxation or dislocation and suboptimal joint function. One
to five of 1,000 newborns are affected by DDH in China (1). Early diagnosis and treatment are
important, as failure to diagnose DDH in neonates and young infants
may result in significant morbidity, such as early accelerated wear
of the articular cartilage resulting in 40% osteoarthritis of the
hip at age 20–40 (2). At present, a
sensitive and specific genetic test to accurately identify newborns
susceptible for DDH is an elusive goal for pediatric orthopedic
surgeons.
DDH is a common, complex, osteologic condition with
environmental as well as genetic factors contributing to the risk.
Epidemiologic studies have highlighted the significant influence of
the orientation of the fetus in the womb on DDH, and Breech
presentation (specifically if delivered naturally), primiparity and
high birth weight are significant environmental risk factors
(3). Genetic contributions are
apparent from studies on pedigrees with a 12-fold increase of DDH
among first-degree offspring of those affected (4). Evidence for a genetic cause of DDH has
been provided by studies indicating a genetic predisposition to DDH
based on polygenic inheritance (5,6).
Furthermore, monozygotic twins have a 41% concordance rate as
compared to the 2.8% seen in dizygotic twins (7). At present, few loci and genes have been
identified to be associated with DDH. Genome-wide scans of affected
families have suggested linkage to chromosomes 4q35 (8), 16p (9),
13q22 (10) and 17q21 (11). To date, no mutations have been found
to be definitely linked to DDH in large canine pedigree studies, in
spite of certain dog pedigrees being commonly affected by DDH
(12). Linkage to chromosome 12q13,
where the genes encoding collagen type II, alpha 1 (COL2A1) and
vitamin D receptor are located, were recently excluded, as were two
polymorphic sites within the COL1A1 gene (13). A recent linkage analysis and a
whole-exome sequencing (WES) study in a 71-member family pedigree
revealed that a rs3732378 variant in CX3 chemokine receptor on
chromosome 3p22.2 was shared by all affected family members and by
15% of sporadic DDH cases, but it was also carried by some
unrelated, ‘married-in’ individuals in this family (14,15). To
date, no gene mutations in these reported regions have been found
to be definitely linked to DDH. Indeed, it is the complex genetic
heterogeneity that has previously posed a significant barrier to
the investigation and molecular diagnosis of DDH.
Through WES, the present study identified for the
first time, to the best of our knowledge, that recurrent germline
mutations and somatic deletion or insertion in bone morphogenetic
proteins-2-inducible kinase (BMP2K) correlated with the phenotype
of DDH in a 37-member, three-generation pedigree. The present study
provided further evidence that DDH is a genetically heterogeneous
disease and that BMP2K mutations account for a subset of the
cases.
Materials and methods
Subjects and clinical analysis
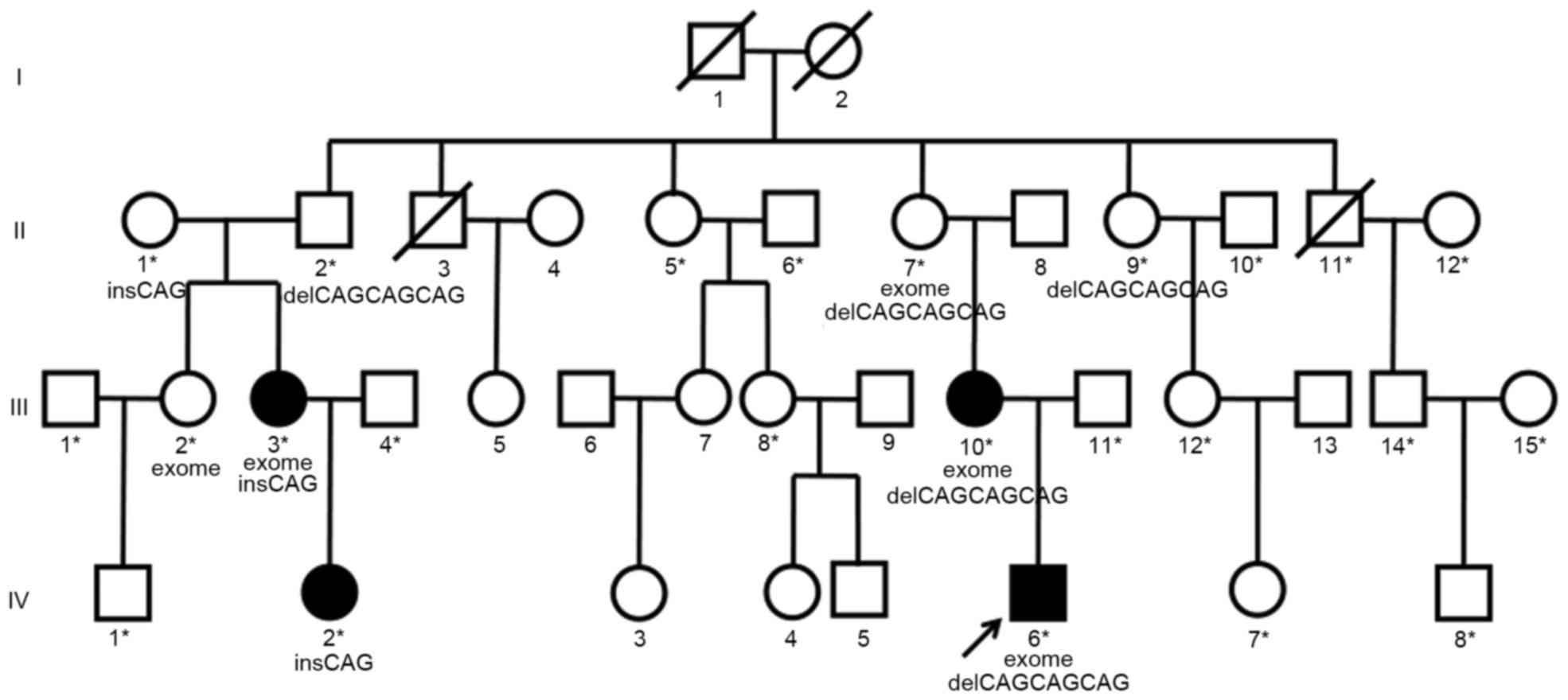
A four-generation, 37-member family from Southern
Suzhou (Anhui, China) of Han Chinese origin, in which DDH was
present in three generations, and 37 sporadic DDH patients from
China were recruited for the present study. The Institutional
Ethical Review Board of the Shanghai Children's Hospital (Shanghai,
China) approved the protocol of the present study. After obtaining
written informed consent from all participants or their legal
guardians, family members and sporadic DDH patients were diagnosed
using detailed clinical exams and supine anterior posterior
radiographs of the pelvis. Results of clinical exams and radiograph
imaging of the hips were evaluated by three pediatric orthopedic
surgeons, with clinical opinions of two additional surgeons
elicited in any case of disagreement. Radiographic measurements of
the hip were taken and affected individuals were identified
according to the following criteria: Perkin quadrant (the femoral
head is not in the inner lower Perkin quadrant), Acetabular index
(>25 degrees), Shenton's line (disrupted) and center edge angle
(<20 degrees). A pedigree chart was constructed based on the
family information (Fig. 1). Four
patients had hip dislocation with all the four radiographic signs
and were considered to be unequivocally affected by DDH
(individuals III3, III10, IV2 and IV6 in Fig. 1). Patient IV6 was the main proband of
the present study. A total of 18 individuals had no clinical
features or radiographic signs of DDH and were deemed as
unaffected, and their blood samples were obtained (individuals
marked with asterisks in Fig. 1).
Among these, one subject succumbed to lung cancer during the study
period (subject II11 in Fig. 1).
Three individuals died before the present study was initiated
(subjects I1, I2 and II3 in Fig. 1)
and 11 refused to donate DNA samples for the present study and were
therefore excluded from the analysis. None of the subjects assessed
had any systemic syndrome. Peripheral blood samples from
affected/unaffected family members and sporadic DDH patients were
collected. Genomic DNA was then extracted from the 22 available
family members' and 37 sporadic DDH patients' blood samples using
the QIAamp DNA Blood Mini kit (cat. no. 51,104; Qiagen, Hilden,
Germany) according to manufacturer's protocol. Finally, genetic
syndromes involving dysplasia, subluxation or dislocation of the
hip and cardiovascular disease were ruled out and confirmed in this
family and in the sporadic DDH patients.
Exome and Sanger sequencing
Three affected (III3, III10 and IV6 in Fig. 1) and two unaffected (II7 and III2 in
Fig. 1) family members (one was the
grandmother of IV6 and was deemed to be a carrier of mutations, and
the other one was the aunt of IV6 who did not carry any BMP2K
mutation and was used as a control) were selected for WES and for
analysis of exonic variants and are shown in the pedigree (Fig. 1). Exome capture on each individual
was performed using a SureSelect Human All Exon V5+UTRs (Agilent
Technologies, New Castle, DE, USA), guided by the manufacturer's
protocols. Sequencing was performed using an Illumina HiSeq 2500
(Illumina, San Diego, CA, USA) following the manufacturer's
instructions. A control reference sequence was derived from the
1,000 Genomes project (http://www.1000genomes.org) and from the reference
human genome (GRCh37) assembly of the National Center for
Biotechnology Information. Exome reads were analyzed in a standard
bioinformatics pipeline based on Burrows-Wheeler Aligner for
sequence alignment on GRCh37 reference, Broad Institute Genome
Analysis Tool Kit (GATKv2.6) for genotyping, ANNOVAR 11 and SnpEff
for variant annotation and ExomeDepth for copy number variation
detection (16–19). Variants were detected using the GATK
Unified Genotyper (software.broadinstitute.org/gatk) in conjunction with
Southern Han Chinese and Han Chinese in the Beijing exome BAM
files, which is a compressed binary version of a SAM file that is
used to represent aligned sequences, from the 1,000 Genomes
Project. Potentially deleterious variants were analyzed, which were
detected in subjects II7, III3, III10 and IV6 (Fig. 1) but absent in subject III2 (Fig. 1). The single nucleotide
polymorphisms, version 137 (dbSNP137) (ncbi.nlm.nih.gov/projects/SNP/), the 1,000 Genomes
Project (internationalgenome.org/) and the National Heart, Lung
and Blood Institute (NHLBI) databases (nhlbi.nih.gov)
were selected for further scrutiny and segregation analysis.
Candidate variants were validated in the four affected family
members, other available family members as well as in-laws
(unrelated individuals married into the family). Whole exons and
adjacent splicing site of the candidate gene were tested using
Sanger sequencing in 37 sporadic DDH cases. Polymerase chain
reaction products were amplified using the primers shown in
Table I, which were synthesized by
Shanghai Majorbio Co., Ltd. (Shanghai, China). The reaction mixture
(25 µl) contained 2 µl DNA template (200 ng), 1 µl of each primer
(10 µM) and 2X Premix master mix (cat. no. RR820A; Takara
Biotechnology Co., Ltd., Dalian, China). PCR thermal cycling was
performed as follows: Initial denaturation at 95°C for 1 min,
followed by 45 cycles of 94°C for 10 sec, 60°C for 20 sec and 72°C
for 25 sec. The reaction products were analyzed using the ABI
3730XL Genetic Analyzer equipment (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
 | Table I.Primers used to amplify the sequences
harboring the variants in the present study. |
Table I.
Primers used to amplify the sequences
harboring the variants in the present study.
| Primer name | Sequence (5′-3′) |
|---|
| Chr4:exon 1-F |
TTTCGCTCTCTTCCTCTACCC |
| Chr4:exon 1-R |
GCAAGAAAAGCCAGCTTAAGAG |
| Chr4:exon 2-F |
GAGAAGCGTCTTCATTCGAGA |
| Chr4:exon 2-R |
AGCCAGGTATGGTGATGGAC |
| Chr4:exon 3-F |
TCGCATGCAGGTAAACTTTTT |
| Chr4:exon 3-R |
TGCGTTTACATGCTCATATCATAA |
| Chr4:exon 4-F |
GATGATCTTTTGAGATGTAAAATGTG |
| Chr4:exon 4-R |
CTCCACAGCCATGCTAGAAA |
| Chr4:exon 5-F |
TTTGACCTTCCTCATAATTTGG |
| Chr4:exon 5-R |
AAAATCTGCATCTATGGCAAAA |
| Chr4:exon 6-F |
TTCAAGGAAAACTGTTTCCAA |
| Chr4:exon 6-R |
AAGCTTTCCATCTCAGGATTG |
| Chr4:exon 7-F |
TTCAGCCAACTAGCCACAGA |
| Chr4:exon 7-R |
TCAATAATTAGGTTGCTTTCATTCTT |
| Chr4:exon 8-F |
CCTTAGCTTGGGATAATGCTT |
| Chr4:exon 8-R |
TCCCCAAGGTATCTCAGTGC |
| Chr4:exon 9-F |
TGGCACATGAATATGAAATTGAC |
| Chr4:exon 9-R |
TCCAGTGAGATGATGCCAGA |
| Chr4:exon 10-F |
TGGTTGTGTACTGTTTTAAGGAAA |
| Chr4:exon 10-R |
TGCCAGTAACTTCCCTTTGC |
| Chr4:exon 11-F |
CGACAAAATTAAGCTTCTTCGGTA |
| Chr4:exon 11-R |
TTTCACTGATTGCAAATTAAATACG |
| Chr4:exon 12-F |
TTTGAAACCAGTTCTGATAAGCAT |
| Chr4:exon 12-R |
TGCCATATTCAGGTGCCATA |
| Chr4:exon 13-F |
TTTAAAAAGATTTATTTCATCTTGCTG |
| Chr4:exon 13-R |
GCAGTTAGCAGAATTAACTGAGGA |
| Chr4:exon 14-F |
TCTCCCTCTCCATCTCATGC |
| Chr4:exon 14-R |
GCATACTATCAGCAATATTGGGTTT |
| Chr4:exon 15-F |
TTTTGCAGCAACAAATATGTATTAAA |
| Chr4:exon 15-R |
TGTTGGCAAAGATACAACTGAA |
| Chr4:exon 16-F |
CCTGAGGTCAAATGCATTCTCT |
| Chr4:exon 16-R |
GGGAGTCTCCACTGCAACAT |
Results
Whole-exome analysis
Whole-exome analysis of the three affected and two
unaffected family members (subjects II7, III2, III3, III10 and IV6
in Fig. 1) was performed. An average
of 159.74 million 100-bp paired-end sequencing reads were generated
for each individual, 98.8% of which aligned to the targeted region.
An average of 96.6% of targeted regions were covered at a read
depth of at least 20x. On average, 38,810 SNPs were identified per
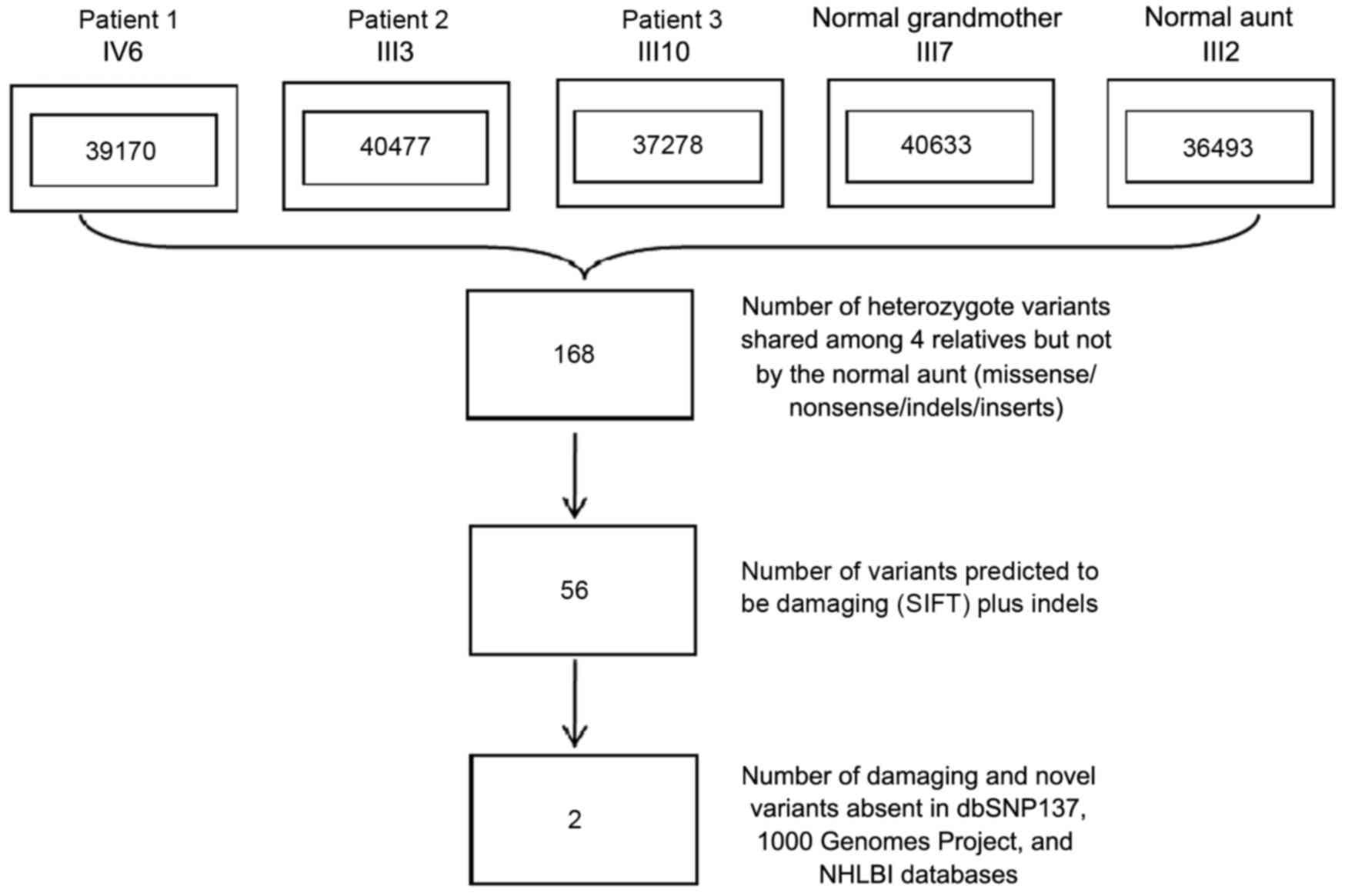
individual (range, 36,493–40,633). A total of 168 heterozygote
variants (missense/nonsense/indels/inserts), of which 92.8% were
already annotated in a public database (dbSNP v137), found in all
three affected individuals and the main proband's grandmother, who
was deemed to be the carrier of mutations (individuals II7, III3,
III10 and IV6 in Fig. 1), but not
found in the aunt of one family member with DDH who was used as a
control (subject III2 in Fig. 1). In
order to refine the list of variants, a filtering strategy was
applied with only those variants included that were indels, those
predicted to be loss of function (damaging) mutations by SIFT
(sift.jcvi.org/) and ANNOVAR programs (annovar.openbioinformatics.org/) and
those that were not included in the dbSNP137, 1,000 Genomes Project
and NHLBI databases, and as shown in Fig. 2 (20–22).
After these three filtering steps shown in Fig. 2, two novel heterozygous, inframe
mutations causing multi-nucleotide substitution polymorphisms
(MNPs) (c.1432_1440delCAGCAGCAG corresponding with p.Gln478_480del
and c.1440_1441insCAG corresponding with p.Gln480ins) in exon 11 of
chromosome 4q21.21 in the BMP2K gene remained, which were possibly
correlated with DDH (Fig. 1). These
two variants shared the same genomic coordinate but with different
types of mutation in BMP2K. The family members affected by DDH and
the unaffected grandmother of subject IV6 had either one of these
two variants, while the unaffected aunt of another family member
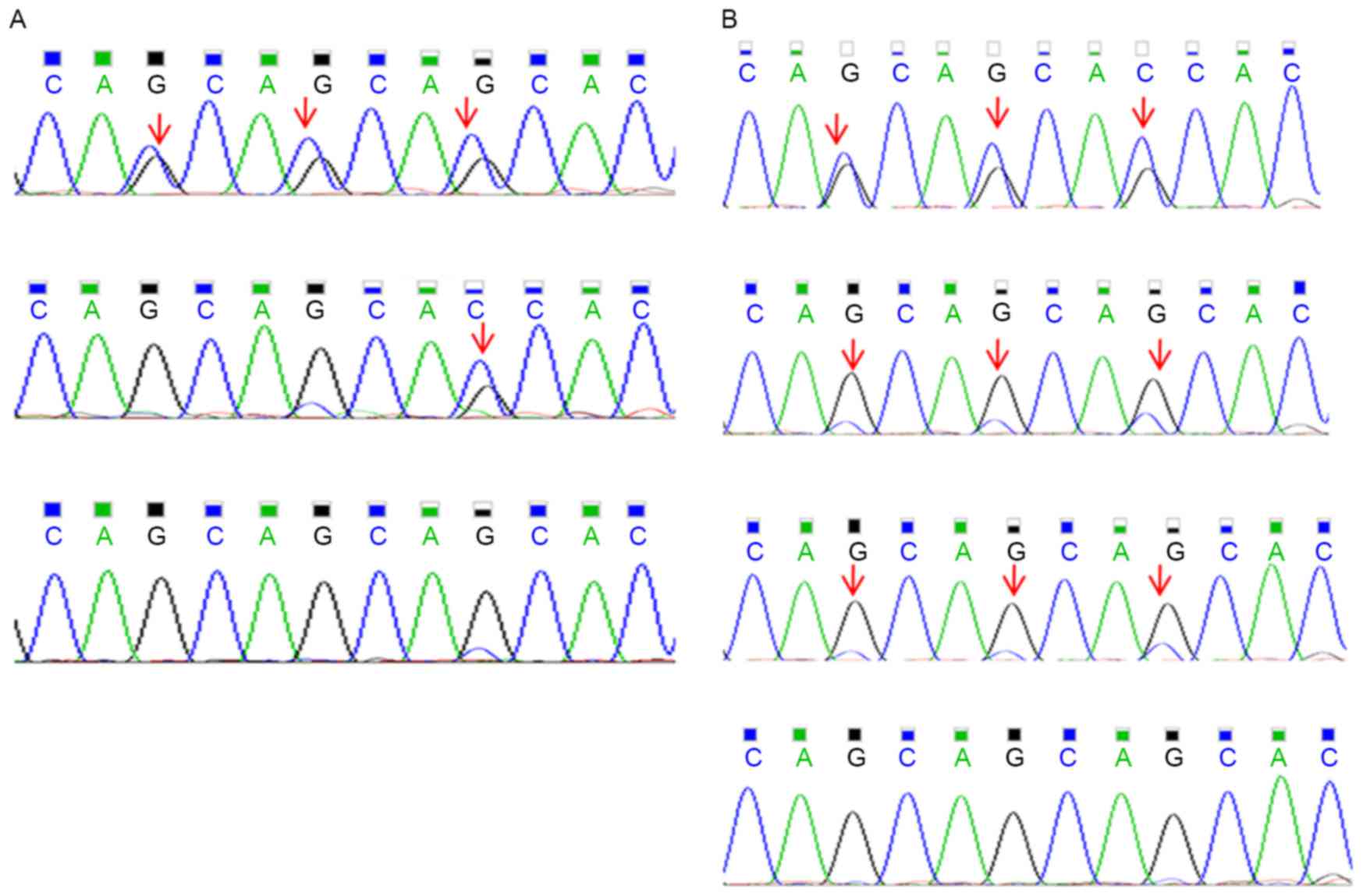
had no mutation in BMP2K (Fig. 3A).
No shared splice-site mutations or variants were found. The BMP2K
variants encode a mutation that caused the deletion of three amino
acids or insertion of one amino acid in the BMP2K transcript
(NM_017593). This indel was found to affect a evolutionarily
conserved glutamine residue within the BMP2K gene.
Validation of the presence of the
mutations in BMP2K by Sanger sequencing
The presence of CAGCAGCAG/− or -/CAG indels in BMP2K
was validated by Sanger sequencing in the four affected
individuals, the unaffected related as well as the unaffected
unrelated family members. Of the affected individuals, a total of
4/4 (100%) were positive for the BMP2K variants. The unaffected
family members with DNA available (Fig.
1) were also genotyped and 4/15 (26.7%) of the unaffected
related family members and 0/7 (0%) of the unaffected unrelated
(married-in) family members were positive for the variants
(Fig. 1). Subsequently, 37 sporadic
DDH patients were assessed, three of which were positive for the
BMP2K variants (8.12%) (Fig.
3B).
Discussion
DDH has a complex etiology with environmental as
well as genetic causes, and appears to be inherited in an autosomal
dominant fashion and exhibits incomplete penetrance as described by
Feldman et al (11). Previous
studies have revealed five loci and a few genes associated with
DDH; however, the exact genetic factors associated with a risk of
DDH have remained elusive. The present study performed WES and
mutational analyses in a large family showing intergenerational
inheritance of DDH to identify the genetic basis of the disease.
WES analysis, which makes no assumptions regarding the location of
a mutation, identified two novel inframe mutations in BMP2K to be
the probable disease-causing mutations in a family with DDH in the
present study. Sanger sequencing confirmed this result and the
mutations were found to be associated with the clinical findings in
the three generations with regard to DDH. DDH is a complex disorder
with environmental as well as genetic causes and shows incomplete
penetrance, and not all individuals who appear unaffected by DDH
are free mutations associated with the disease, as was shown in the
inheritance of the BMP2K variant in the pedigree. As expected, all
affected individuals in the family assessed had either one of these
two variants. These two mutations were also present in the DNA of
obligate heterozygotes (II2, II7 and II9) even though these
individuals did not have any signs of DDH. The validation of these
two MNP mutations in 37 unrelated sporadic cases of DDH in the
present study appears to show the overrepresentation of the
c.1432_1440delCAGCAGCAG mutation. While the present study indicated
that the mutations identified represent a considerable genetic risk
factor associated with DDH, more sporadic DDH cases and DDH
pedigrees require to be tested to determine the prevalence of BMP2K
mutations in the overall DDH patient population.
The BMP2K gene is located on chromosome 4q21.21 and
was originally identified by Kearns et al (23) as a BMP2-inducible gene. BMPs are
multifunctional cytokines belonging to the transforming growth
factor-β superfamily, comprised of ~50 genes (24), and were originally identified to have
a central role in the normal development of skeletal and osteoblast
differentiation during embryonic, fetal and infantile growth
periods (25). BMP2 is the most
clinically evaluated BMP and has a critical role in early
embryogenesis, skeletal development and the differentiation of
osteoblasts (25,26). Signaling by BMP2 requires the binding
of the BMP2 molecule to the BMP receptors, a set of
serine/threonine kinase receptors located on the cell surface of
osteoblasts (27,28). The protein encoded by BMP2K is a
126-kDa serine/threonine protein kinase containing a nuclear
localization signal (23). Protein
levels of BMP2K were shown to increase during BMP2-induced
differentiation of a mouse osteoblastic cell line, and BMP2K has
been identified as an important regulator of the cell
differentiation process, suggesting that it is critical for
skeletal development and osteoblast differentiation (23). It is therefore plausible that the
BMP2K mutations affect the development of the hip.
BMP2K contains a nuclear localization signal to
direct protein to nuclei and may affect transcription activities of
target genes. It has been identified as a clathrin-coated,
vesicle-associated protein, suggesting it may also function to
regulate endocytic complexes (29).
BMP2K was identified to be an interacting protein of Numb and to
participate in the endocytic functions of Numb, which is essential
for mammalian development and has a role in regulating the
proliferation and differentiation of neural progenitor cell
populations during embryogenesis (30,31). It
has been reported that signaling pathways associated with Akt, a
serine/threonine protein kinase, are involved in osteogenic
processes and suppress osteoblast apoptosis (32). Mukherjee and Rotwein (33) found that an intact insulin-like
growth factor-induced phosphoinositide-3 kinase/Akt signaling
cascade is essential for BMP2-activated osteoblast differentiation
and maturation, bone development and growth, and demonstrated that
activation of Akt promotes BMP2-mediated osteoblast
differentiation. Tseng et al (34) demonstrated that hypoxia induced BMP2
expression via integrin-linked kinase/Akt/mammalian target of
rapamycin and hypoxia-inducible factor-1α pathways in osteoblasts
in a time-dependent manner. BMP2K, a serine/threonine kinase
protein, may regulate osteoblast differentiation through via
Akt-associated signaling pathways. It remains elusive why mutations
in BMP2K result in the phenotype of DDH. In addition, whether the
BMP2K variants are associated with the risk for DDH remains to be
determined. In vitro and in vivo experiments are
required to examine the genotype-phenotype correlation.
In recent years, functional studies have shown the
importance of BMP2K in the regulation of human osteoblast
proliferation and differentiation. Shock wave-stimulated cell
proliferation and differentiation, along with induced upregulation
of BMP2K gene expression, have been reported in human primary
osteoblast cultures (35). Stable
expression of BMP2K in MC3T3-E1 osteoprogenitor cells suppressed
mature osteoblast function, suggesting the important role of BMP2K
in attenuating the program of osteoblast differentiation in
mineralized tissue. Furthermore, treatment with
1,25-dihydroxyvitamin D, which is the active form of vitamin D and
has a pivotal role in bone homeostasis and is a potent regulator of
osteoblast transcription contributing to fetal and neonatal bone
development (36), decreased the
levels of BMP2K protein expression in human osteoblasts, indicating
that BMP2K negatively regulates human osteoblast proliferation and
differentiation (37). These studies
suggest that BMP2K is closely associated with the normal
development of bone during embryonic, fetal and infantile growth
periods. This MNP of c.1432_1440delCAGCAGCAG or c.1440_1441insCAG,
corresponding with the Gln478_480del or Gln480ins variations, may
cause changes of BMP2K protein activity, affecting bone development
and leading to DDH.
In conclusion, the present study proposed two novel
polymorphisms in the BMP2K gene, which is known to be associated
with skeletal development, and suggested an association of BMP2K
with DDH susceptibility in a Han Chinese pedigree and in sporadic
DDH cases. While DDH has been previously shown to be associated
with genetic factors, the mechanisms have remained largely elusive,
and the present study provided a possible mechanism, suggesting
that novel BMP2K mutations are implicated in the pathogenesis of
DDH. Furthermore, the present study provided important insight into
the BMP family as potential signaling molecules in the pathogenesis
of DDH.
Acknowledgements
The authors are grateful for the participation of
the family involved, to whom the present study is dedicated. The
present study was supported by the National Natural Science
Foundation of China (no. 81401763) the Health and Family Planning
Committee of Shanghai Science Foundation (no. 20144Y0176), the
Shanghai Science and Technology Committee (no. 12DZ2295006) and
‘985 Project’ Funds from Shanghai Jiaotong University School of
Medicine (no. YBKL2013008).
Glossary
Abbreviations
Abbreviations:
|
BMP2K
|
bone morphogenetic
proteins-2-inducible kinase
|
|
DDH
|
developmental dysplasia of the hip
|
|
WES
|
whole-exome sequencing
|
|
NHLBI
|
National Heart, Lung and Blood
Institute
|
|
MNP
|
multinucleotide substitution
polymorphism
|
References
|
1
|
Kokavec M and Bialik V: Developmental
dysplasia of the hip: Prevention and real incidence. Bratisl Lek
Listy. 108:251–254. 2007.PubMed/NCBI
|
|
2
|
Jacobsen S, Sonne-Holm S, Søballe K,
Gebuhr P and Lund B: Hip dysplasia and osteoarthrosis: A survey of
4151 subjects from the osteoarthrosis substudy of the copenhagen
city heart study. Acta Orthop. 76:149–158. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stein-Zamir C, Volovik I, Rishpon S and
Sabi R: Developmental dysplasia of the hip: Risk markers, clinical
screening and outcome. Pediatr Int. 50:341–345. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stevenson DA, Mineau G, Kerber RA,
Viskochil DH, Schaefer C and Roach JW: Familial predisposition to
developmental dysplasia of the hip. J Pediatr Orthop. 29:463–466.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Czeizel A, Szentpétery J, Tusnády G and
Vizkelety T: Two family studies on congenital dislocation of the
hip after early orthopaedic screening Hungary. J Med Genet.
12:125–130. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wynne-Davies R: Acetabular dysplasia and
familial joint laxity: Two etiological factors in congenital
dislocation of the hip. A review of 589 patients and their
families. J Bone Joint Surg Br. 52:704–716. 1970.PubMed/NCBI
|
|
7
|
Wilkinson JA: Etiologic factors in
congenital displacement of the hip and myelodysplasia. Clin Orthop
Relat Res. 75–83. 1992.PubMed/NCBI
|
|
8
|
Cilliers HJ and Beighton P: Beukes
familial hip dysplasia: An autosomal dominant entity. Am J Med
Genet. 36:386–390. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Loughlin J, Mustafa Z, Irven C, Smith A,
Carr AJ, Sykes B and Chapman K: Stratification analysis of an
osteoarthritis genome screen-suggestive linkage to chromosomes 4,
6, and 16. Am J Hum Genet. 65:1795–1798. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mabuchi A, Nakamura S, Takatori Y and
Ikegawa S: Familial osteoarthritis of the hip joint associated with
acetabular dysplasia maps to chromosome 13q. Am J Hum Genet.
79:163–168. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Feldman GJ, Dalsey C, Fertala K, Azimi D,
Fortina P, Devoto M, Pacifici M and Parvizi J: The otto aufranc
award: Identification of a 4 Mb region on chromosome 17q21 linked
to developmental dysplasia of the hip in one 18-member,
multigeneration family. Clin Orthop Relat Res. 468:337–344. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marschall Y and Distl O: Mapping
quantitative trait loci for canine hip dysplasia in German Shepherd
dogs. Mamm Genome. 18:861–870. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rubini M, Cavallaro A, Calzolari E,
Bighetti G and Sollazzo V: Exclusion of COL2A1 and VDR as
developmental dysplasia of the hip genes. Clin Orthop Relat Res.
466:878–883. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feldman GJ, Parvizi J, Levenstien M, Scott
K, Erickson JA, Fortina P, Devoto M and Peters CL: Developmental
dysplasia of the hip: Linkage mapping and whole exome sequencing
identify a shared variant in CX3CR1 in all affected members of a
large multigeneration family. J Bone Miner Res. 28:2540–2549. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feldman GJ, Parvizi J, Sawan H, Erickson
JA and Peters CL: Linkage mapping and whole exome sequencing
identify a shared variant in CX3CR1 in a large multi-generation
family. J Arthroplasty. 29:(9 Suppl). S238–S241. 2014. View Article : Google Scholar
|
|
16
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Plagnol V, Curtis J, Epstein M, Mok KY,
Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO,
Thrasher AJ, et al: A robust model for read count data in exome
sequencing experiments and implications for copy number variant
calling. Bioinformatics. 28:2747–2754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cinqolani P, Platts A, le Wang L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: Mutation taster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kearns AE, Donohue MM, Sanyal B and Demay
MB: Cloning and characterization of a novel protein kinase that
impairs osteoblast differentiation in vitro. J Biol Chem.
276:42213–42218. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen G, Deng C and Li YP: TGF-β and BMP
signaling in osteoblast differentiation and bone formation. Int J
Biol Sci. 8:272–288. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li X and Cao X: BMP signaling and
skeletogenesis. Ann N Y Acad Sci. 1068:26–40. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanzler B, Foreman RK, Labosky PA and
Mallo M: BMP signaling is essential for development of skeletogenic
and neurogenic cranial neural crest. Development. 127:1095–1104.
2000.PubMed/NCBI
|
|
27
|
Nakamura Y, Wakitani S, Nakayama J,
Wakabayashi S, Horiuchi H and Takaoka K: Temporal and spatial
expression profiles of BMP receptors and noggin during
BMP-2-induced ectopic bone formation. J Bone Miner Res.
18:1854–1862. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ishidou Y, Kitajima I, Obama H, Maruyama
I, Murata F, Imamura T, Yamada N, ten Dijke P, Miyazono K and Sakou
T: Enhanced expression of type I receptors for bone morphogenetic
proteins during bone formation. J Bone Miner Res. 10:1651–1659.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Borner GH, Antrobus R, Hirst J, Bhumbra
GS, Kozik P, Jackson LP, Sahlender DA and Robinson MS: Multivariate
proteomic profiling identifies novel accessory proteins of coated
vesicles. J Cell Biol. 197:141–160. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Krieger JR, Taylor P, Gajadhar AS, Guha A,
Moran MF and McGlade CJ: Identification and selected reaction
monitoring (SRM) quantification of endocytosis factors associated
with Numb. Mol Cell Proteomics. 12:499–514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Petersen PH, Zou K, Hwang JK, Jan YN and
Zhong W: Progenitor cell maintenance requires numb and numblike
during mouse neurogenesis. Nature. 419:929–934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choi YH, Jeong HM, Jin YH, Li H, Yeo CY
and Lee KY: Akt phosphorylates and regulates the osteogenic
activity of Osterix. Biochem Biophys Res Commun. 411:637–641. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mukherjee A and Rotwein P: Akt promotes
BMP2-mediated osteoblast differentiation and bone development. J
Cell Sci1. 22:716–726. 2009. View Article : Google Scholar
|
|
34
|
Tseng WP, Yang SN, Lai CH and Tang CH:
Hypoxia induces BMP-2 expression via ILK, Akt, mTOR, and HIF-1
pathways in osteoblasts. J Cell Physiol. 223:810–818.
2010.PubMed/NCBI
|
|
35
|
Hofmann A, Ritz U, Hessmann MH, Alini M,
Rommens PM and Rompe JD: Extracorporeal shock wave-mediated changes
in proliferation, differentiation, and gene expression of human
osteoblasts. J Trauma. 65:1402–1410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goltzman D, Hendy GN and White JH: Vitamin
D and its receptor during late development. Biochim Biophys Acta.
1849:171–180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lisse TS, Chun RF, Rieger S, Adams JS and
Hewison M: Vitamin D activation of functionally distinct regulatory
miRNAs in primary human osteoblasts. J Bone Miner Res.
28:1478–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|