Introduction
Metoprolol succinate (MS) is a highly selective
β1-adrenergic receptor blocker typically used for the
treatment of hypertension, coronary heart disease, chronic heart
failure and arrhythmia (1). However,
at higher plasma concentrations, MS may also inhibit
β2-adrenergic receptors located in the vascular and
bronchial musculature. Additionally, when plasma MS concentrations
are greater than required, anesthetic membrane-stabilizing activity
is detected (2,3). Therefore, it is necessary to control
the plasma MS concentration to maximize therapeutic effects and
minimize side effects. Due to the short half-life of metoprolol
(3–4 h) MS must be administered several times a day, leading to
fluctuations in plasma MS concentration (4). Sustained-release (SR) formulations are
able to minimize fluctuations in plasma concentration, hence
minimizing the adverse effects associated with excessively high
plasma concentrations and providing an effective stable dose
(5). This may be achieved through
various formulations, including hydrophilic matrix tablets, SR
pellets, osmotic pump tablets and drug-polymer conjugates. Novel
platforms, such as nano/microcarriers with SR, improved adhesion
and tissue penetration, may be utilized for oral drug delivery
(6).
Microcapsules provide several advantages compared
with conventional dosage forms, including modulated drug release,
enhanced drug stability and reduced gastrointestinal irritation
(7). Microspheres have been
previously used in an SR formulation of MS to provide robust and
consistent control of hypertension and heart rate (8). Recently, metoprolol tartrate
sustained-release capsules, which are polymer-coated metoprolol
tartrate matrix granules, have become commercially available.
Hydrophilic hydroxypropyl methyl cellulose (HPMC) and hydrophobic
ethyl cellulose polymers may also be employed as matrix builders,
and Eudragit® RL/RS as a coating polymer. Microparticles
coated with a film composed of these water-insoluble polymers
exhibit extended release periods of up to 12 h in vitro
(9).
Various encapsulation methods may improve
immobilization, isolation and protection of commercial products and
facilitate control of the transfer rate of pharmacological agents,
nutrients and perfumes (10). The
emulsification-solvent diffusion method is an established method
for the preparation of microcapsules based on an emulsion
technique. This method involves adding capsule material dissolved
in an organic solution to an aqueous solution saturated with
organic solvent to form an emulsion. Removal of organic solvents
induces the formation of microcapsules as a result of phase
separation due to the decreased solubility of the capsule material
in solution. These formulations produce microcapsules with an
evenly rounded shape, a smooth surface and good SR characteristics
(11,12). Ethylcellulose (EC) is a non-ionic,
pH-insensitive cellulose ether, which is insoluble in water but
soluble in numerous polar organic solvents (13), and exhibits SR properties. Dash et
al (14) previously confirmed
that aspirin-loaded EC microcapsules, made using the emulsion
solvent evaporation method, were able to effectively reduce the
drug release rate. Polyethylene glycol (PEG) is a hydrophilic
polymer material and plasticizer. PEG enhances the flexibility and
plasticity of microcapsules, decreases the tendency for aggregation
and adhesion, and improves dispersion. It may also be used as a
porogen for membrane-controlled drug release, and, with a suitable
ratio of polymer semi-permeable membrane and porogenic materials,
is able to reduce the rate of drug release (15).
The objective of the present study was to develop a
method of MS encapsulation to provide microcapsules with high
entrapment efficiency and optimal SR profiles in vitro and
in vivo. The emulsification-solvent diffusion method
(14) was used to prepare
microcapsules of ethyl cellulose and PEG 6,000 and the release of
MS from these capsules was assessed in vitro and in
vivo.
Materials and methods
Preparation of SR microcapsules of
MS
MS (AstraZeneca Pharmaceuticals Co., Ltd., Wuxi
China) was dissolved in water (the internal aqueous phase,
W1) and added to 2 ml ethyl acetate (EA) solution (O)
and ultrasonically emulsified, forming a primary emulsion
(W1/O). The indicated concentration of PEG 6,000
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) was dissolved
in EA-saturated water (containing 0.5% SDS; Tianjin Global Chemical
Technology Co., Ltd., Tianjin, China) to constitute the external
aqueous phase (W2). W1/O was slowly added
drop-wise to W2, under continuous stirring until the
emulsion droplets solidified to form a stable
W1/O/W2 complex emulsion. Water was used to
dilute the emulsion 20-fold. The ethyl cellulose phase separated
and condensed into a capsule, and following sedimentation of the
microcapsules, the supernatant was removed and the retentate
centrifuged and filtered to obtain the microcapsules, which were
subsequently washed with water and dried in a vacuum at 40°C.
Study of microcapsule morphology and
size distribution
Microcapsules were re-suspended in double-distilled
water via ultrasonic dispersion for 20 min. The sample was then
observed under a microscope (Olympus X 51, Olympus Corporation,
Tokyo, Japan; magnification, ×400) and particle diameter was
measured.
Determination of MS drug loading
capacity and encapsulation efficiency
The maximum absorption of MS was previously recorded
at 274 nm when scanned at a wavelength of 200–350 nm (16). Therefore, 274 nm was chosen as the
optimum wavelength for measuring the MS content. A total of 50 mg
of microcapsules were placed in a 100-ml volumetric flask and
dissolved in ethanol. The solution was ultrasonically treated for
30 min and filtered through a 0.8-µm microporous membrane filter.
Of the resulting filtrate, 5 ml was diluted with 20 ml water and
the ultraviolet (UV) absorbance at 274 nm was measured using an
ultraviolet spectrophotometer (Shimadzu UV2550; Shimadzu
Corporation, Kyoto, Japan). MS drug loading capacity was calculated
as: Quantity of agent in microcapsules/microcapsule weight ×100.
Encapsulation efficiency was calculated as follows: Quantity of
agent in the microcapsules/total quantity of agent used ×100.
A linear correlation was obtained with
A=0.0042C+0.0014 (r=0.9997), Where A is the absorbance intensity, C
is the concentration of drug and r is the linearly dependent
coefficient. The intra-day relative standard deviation (RSD) was
0.41% (n=6), and the inter-day RSD was 0.85% (n=6), with the
average recovery being 99.2 and the average RSD being 0.37%
(n=9).
MS microcapsule in vitro release
Artificial gastric fluid was prepared by diluting
16.4 ml hydrochloric acid in 800 ml water and 10 g pepsin (3,800
U/mg; Sichuan Deyang Biochemical Products Co., Ltd., Deyong, China)
and diluting again with water to 1,000 ml. Artificial intestinal
fluid was prepared by dissolving 6.8 g potassium dihydrogen
phosphate in 500 ml water, which was adjusted to pH 6.8 with sodium
hydroxide, to which 10 g trypsin (2,500 U/mg; Sichuan Deyang
Biochemical Products Co., Ltd.) was added, followed by further
dilution with water to 1,000 ml.
Prepared microcapsules were placed in a basket with
1,000 ml of release medium (water, artificial gastric fluid or
artificial intestinal fluid) and the rotation speed was fixed at
100 rpm at 37±0.5°C. A 5-ml sample was taken from the release
solution at predetermined time intervals (0.5, 2, 4, 6, 8, 10, 12,
15 and 18 h) and 5 ml of release medium (water, artificial gastric
fluid or artificial intestinal fluid) was added at 37±0.5°C to
compensate for the volume loss. Following filtration through a
0.8-µm microporous membrane, the UV absorbance of release medium
and microcapsule release medium was measured at 274 nm.
MS release from microcapsule SR tablet
and regular SR tablet in vitro
Excipients, including HPMC (viscosity, 5 cPs),
METHOCEL DC2 K4M (all donated by Shanghai Colorcon Coating
Technology Co., Ltd., Shanghai, China) and microcrystalline
cellulose (MCC; Shanghai Chineway Pharma Tech Co., Ltd., Shanghai,
China) were crushed and passed through a 100-mesh sieve. Weighed MS
microcapsules and excipients were uniformly mixed in a mortar,
passed through a 40-mesh sieve three times and mixed with 85%
ethanol to prepare a soft material. Granules were produced by
passing material through a 16-mesh nylon screen and dried at
50–60°C for 1 h. Dry granules were forced through a 16-mesh sieve
and talcum powder (Shanghai Ju Qian Chemical Co., Ltd., Shanghai,
China) was added, prior to being mixed uniformly and
compressed.
In vitro release of MS microcapsule
SR-tablets was compared with the release from conventional SR
tablets (metoprolol tartrate tablets; AstraZeneca Pharmaceutical
Co., Ltd.) in water. At the indicated time intervals (0.5, 2, 4, 6,
8, 10, 12, 15 and 18 h), samples of release liquid were drawn and,
following filtration through a 0.8-µm microporous membrane, the UV
absorbance of conventional release medium and microcapsule release
medium was measured at 274 nm.
Pharmacokinetic studies of MS
microcapsules in dogs
Six male beagle dogs (age, 8 months; weight, 8–10
kg) were obtained from the Guangzhou General Pharmaceutical
Research Institute Co., Ltd. (Guangzhou, China), housed in
air-conditioned chambers at ambient temperature and humidity, fed a
standard laboratory diet and a 12-h light/dark cycle. Dogs had free
access to food and water. All animal experiments were performed in
full compliance with local, national, ethical and regulatory
principles with the approval of the Institutional Animal Care and
Use Committee of China Pharmaceutical University (Nanjing,
China).
Dogs were fasted overnight and randomly divided into
two groups (n=3 in each). Each dog was orally administered 47.5 mg
MS microcapsules or 50 mg metoprolol tartrate tablets (AstraZeneca
Pharmaceutical Co., Ltd.). Prior to administration, 2 ml venous
blood was collected from the dogs, and further samples were drawn
at 0.5, 1, 1.5, 2, 2.5, 3, 4, 8 and 16 h post-administration. Dogs
had free access to food and water after 4 h administration. Samples
were mixed with 1% sodium heparin solution anti-coagulant (Nanjing
King-Friend Biochemical Pharmaceutical Co., Ltd., Nanjing, China),
centrifuged for 10 min at 4,500 × g, siphoned and stored at
−20°C.
The concentration of MS in blood samples was
determined using a Shimadzu 10-Avp high performance liquid
chromatography system (Shimadzu, Kyoto, Japan) with a C18 column
(Inertsil® ODS-SP, 4.6×250 mm; particle size, 5 µm, GL
Sciences, Inc., Tokyo, Japan) and a mobile phase of methanol/water
equal to 6/4 (v/v) (containing 960 mg sodium heptane sulfonate and
82 mg anhydrous sodium acetate, adjusted to pH 4.7 with glacial
acetic acid) at 30°C; Absorbance was measured with an excitation
wavelength of 285 nm and an emission wavelength of 316 nm, with a
sample injection volume of 50 µl.
Statistical analysis
Statistical analysis was performed with SPSS 13.0
software (SPSS Inc., Chicago, IL, USA). Values are expressed as the
mean ± standard deviation. Multiple regression was used to
determine the optimal conditions for preparation of MS
microcapsules. Statistical significance was determined using a
two-tailed Student's t-test. Fisher's Least Significant Difference,
Sidak and Tukey's post hoc analysis were applied as the post hoc
analysis following one-way analysis of variance for the homogeneity
variance data. Statistical significance was set at P<0.05.
Results
Optimization of microcapsule
preparation
Microcapsules were prepared using the
W1/O/W2 double emulsification-solvent
diffusion method, with W1 being an MS solution, O being
EC dissolved in EA and W2 being PEG 6,000. The impact of
the following factors on the encapsulating efficiency was analyzed:
Volume ratio of W1:O, MS content in W1, EC
concentration, power and duration of ultrasonic emulsification, PEG
6,000 concentration, W1/O:W2 volume ratio,
stirring speed and time, and diffusion time (Table I).
 | Table I.Levels of single factor analysis. |
Table I.
Levels of single factor analysis.
|
| Level |
|---|
|
|
|
|---|
| Factor | 1 | 2 | 3 | 4 | 5 |
|---|
| W1:O | 1:20 | 1:15 | 1:10 | 1:5 | 1:1 |
| MS content (mg) | 1 | 2 | 3 | 4 | 5 |
| EC concentration
(mg/ml) | 5 | 10 | 20 | 40 | 80 |
| Power of ultrasonic
emulsification (W) | 40 | 80 | 120 | 160 | 200 |
| Duration of
ultrasonic emulsification (sec) | 30 | 60 | 90 | 120 | 180 |
| PEG 6,000
concentration (%) | 1 | 2 | 3 | 4 | 5 |
|
W1/O:W2 (v/v) | 1:5 | 1:10 | 1:15 | 1:20 | 1:25 |
| Mixing speed of
multiple emulsion (rcf, × g) | 450 | 750 | 1,050 | 1,350 | 1,650 |
| Multiple emulsion
mixing time (min) | 0.5 | 1 | 2 | 3 | 4 |
| Diffusion time
(h) | 0.5 | 1 | 2 | 3 | 4 |
As demonstrated in Table
II, single factor analysis indicated that microcapsule loading
capacity was influenced by W1:O, MS content in
W1, EC, W1/O:W2 volume ratio and
multiple emulsion mixing time.
| Table II.Analysis of single factors affecting
microcapsule loading capacity. |
Table II.
Analysis of single factors affecting
microcapsule loading capacity.
|
| Drug loading amount
(%) |
|---|
|
|
|
|---|
| Factor | Level 1 | Level 2 | Level 3 | Level 4 | Level 5 |
|---|
|
W1:O | 8.45±0.36 | 8.57±0.71 | 8.69±0.45 | 8.73±0.38 | 8.76±0.32 |
| MS content |
7.14±0.28a |
7.52±0.17b |
8.68±0.31c |
9.31±0.34d |
9.72±0.51e |
| EC content | 7.89±0.21 | 8.53±0.19 | 8.71±0.47 | 7.84±0.51 | 7.16±0.15 |
| Power of ultrasonic
emulsification | 7.99±0.36 | 8.65±0.43 | 8.71±0.25 | 8.91±0.78 | 8.72±0.13 |
| Time of ultrasonic
emulsification | 8.40±0.82 | 8.68±0.20 | 8.69±0.27 | 8.72±0.71 | 8.62±0.50 |
| Concentration of
PEG 6,000 | 8.35±0.67 | 8.39±0.58 | 8.43±0.17 | 8.41±0.13 | 8.36±0.41 |
|
W1/O:W2 | 7.87±0.52 | 7.91±1.16 | 8.06±0.39 | 8.01±0.36 | 7.95±0.27 |
| Mixing speed of
multiple emulsion | 8.42±0.34 | 8.67±0.28 | 8.69±0.23 | 8.75±0.51 | 8.71±0.39 |
| Multiple emulsion
mixing time |
8.39±0.36f | 8.78±0.47 | 9.02±0.68 | 9.06±1.08 | 9.14±0.38 |
| Time of
diffuser | 8.56±0.43 | 8.59±0.30 | 8.61±0.77 | 8.58±0.50 | 8.55±0.12 |
Uniform design is an algorithm used to test the
influence of multiple factors with fewer experiments by evenly
distributing the tested factors in the experimental design
(17). Uniform design was applied to
determine the optimal conditions for the preparation of
microcapsules. Each factor was tested at five levels, using the
principle of the intended level (repeating the same level multiple
times without adding new levels), so that the number of levels
reached more than twice the number of factors. A total of 15 level
experiments were designed, using the U15
(155) uniform design table (18) and the resultant drug loading is
illustrated in Table III.
| Table III.Assessment of the influence of
multiple factors on drug loading capacity using the uniform design
algorithm. |
Table III.
Assessment of the influence of
multiple factors on drug loading capacity using the uniform design
algorithm.
| Level,
W1:O, (v/v) | Level, MS content
(mg) | Level, EC
concentration (mg/ml) | Level,
W1/O:W2 (v/v) | Level, multiple
emulsion mixing time (min) | Drug loading
(%) |
|---|
| 1
(1:20) | 4
(2) | 7
(20) | 11 (1:20) | 13 (4) |
6.27±1.46 |
| 2
(1:20) | 8
(3) | 14 (80) | 7
(1:15) | 11 (3) |
7.18±1.39 |
| 3
(1:20) | 12 (4) | 6
(10) | 3 (1:5) | 9
(2) |
8.61±1.08 |
| 4
(1:15) | 1
(1) | 13 (80) | 14 (1:25) | 7
(2) |
4.31±0.73 |
| 5
(1:15) | 5
(2) | 5
(10) | 10 (1:20) | 5
(1) |
5.74±0.97 |
| 6
(1:15) | 9
(3) | 12 (40) | 6
(1:10) |
3 (0.5) |
6.86±1.33 |
| 7
(1:10) | 13 (5) | 4
(10) | 2 (1:5) |
1 (0.5) |
9.69±1.55 |
| 8
(1:10) | 2
(1) | 11 (40) | 13 (1:25) | 14 (4) |
4.88±0.89 |
| 9
(1:10) | 6
(2) | 3 (5) | 9
(1:15) | 12 (3) |
6.17±1.29 |
| 10 (1:5) | 10 (4) | 10 (40) | 5
(1:10) | 10 (3) |
8.77±1.76 |
| 11 (1:5) | 14 (5) | 2 (5) | 1 (1:5) | 8
(2) | 10.06±2.21 |
| 12 (1:5) | 3
(1) | 9
(20) | 12 (1:20) | 6
(1) |
4.45±0.62 |
| 13 (1:1) | 7
(3) | 1 (5) | 8
(1:15) | 4
(1) |
7.66±1.92 |
| 14 (1:1) | 11 (4) | 8
(20) | 4
(1:10) |
2 (0.5) |
8.84±1.57 |
| 15 (1:1) | 15 (5) | 15 (80) | 15 (1:25) | 15 (4) | 10.63±1.86 |
Using mean drug loading as an index, multiple
regression analysis was performed for each factor. At α=0.05, the
regression equation revealed that it was possible to calculate mean
drug loading according to the formula 2.865+0.597
(W1:O)+1.341 (MS content of W1) + 0.194
(multiple emulsion mixing time) (F=328.265; P<0.001;
r2=0.990). Regression analysis indicated that
W1:O, MS content of W1 and multiple emulsion
mixing time were significantly correlated with drug loading
(P<0.05), whereas EC or the W1/O:W2 volume
ratio were not. The optimal microcapsule preparation conditions
were determined as follows: Internal W1:O, 1:1; MS
content of W1, 5 mg; EC, 20 mg/ml;
W1/O:W2 volume ratio, 1:15; and multiple
emulsion stirring time, 4 min. The microencapsulated drug loading
capacity predicted using a regression equation under optimal
conditions was 10.94%. Consistent with this predicted value, the
three batches of microcapsules prepared were determined to have an
average drug loading capacity of 10.91%, as well as an
encapsulation efficiency of 83.16%.
Microcapsule morphology and size
distribution
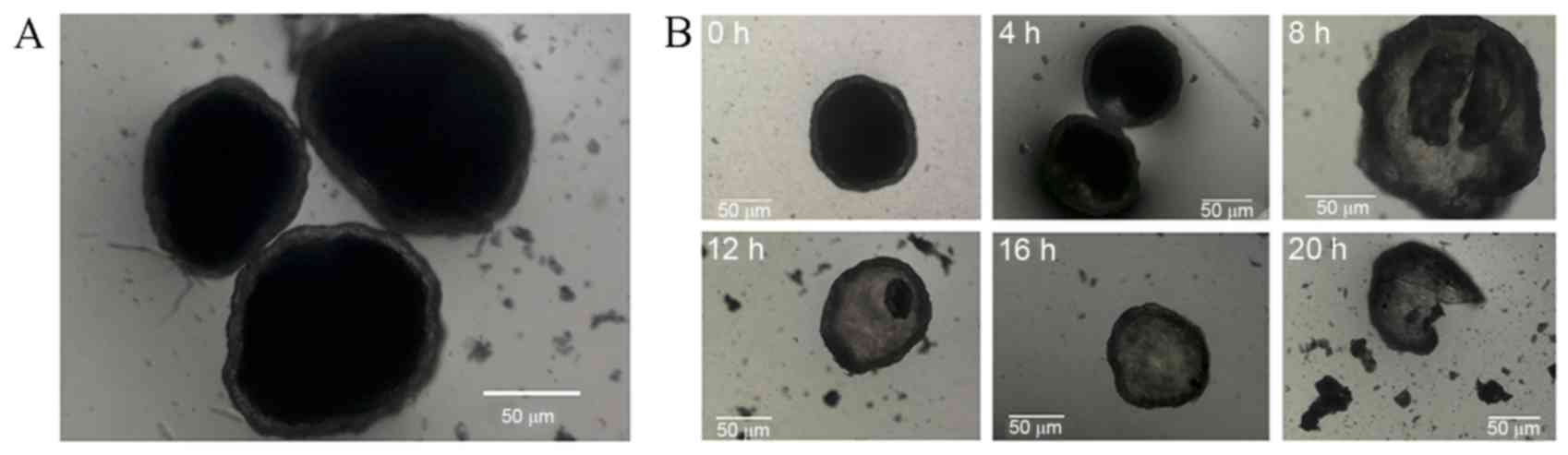
Prepared microcapsules were spherical, with
relatively uniform size, and did not adhere to one another
(Fig. 1). Microcapsule wall
integrity was maintained for 20 h prior to rupture when suspended
in distilled water, indicating a 20-h period of
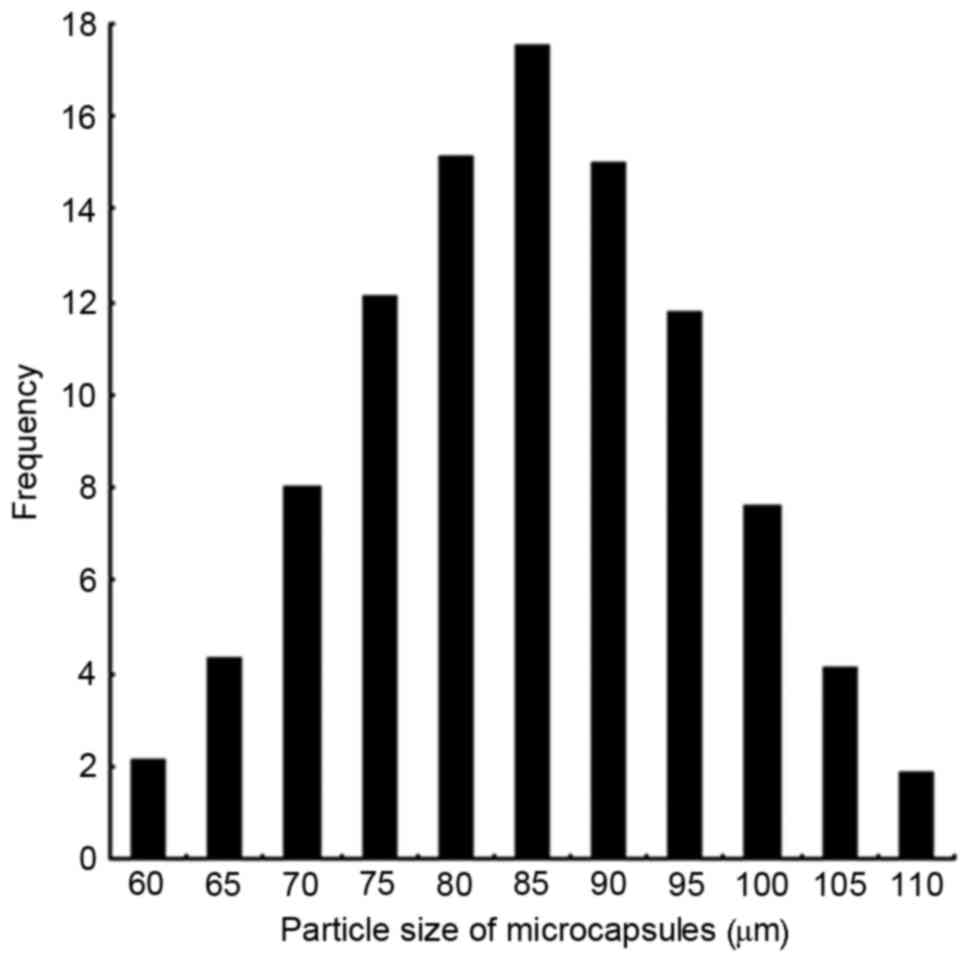
diffusion-controlled drug release. Microcapsule size was relatively
narrowly distributed, with 86% of capsules being 70–100 µm in
diameter, and a mean particle size of 85 µm (Fig. 2).
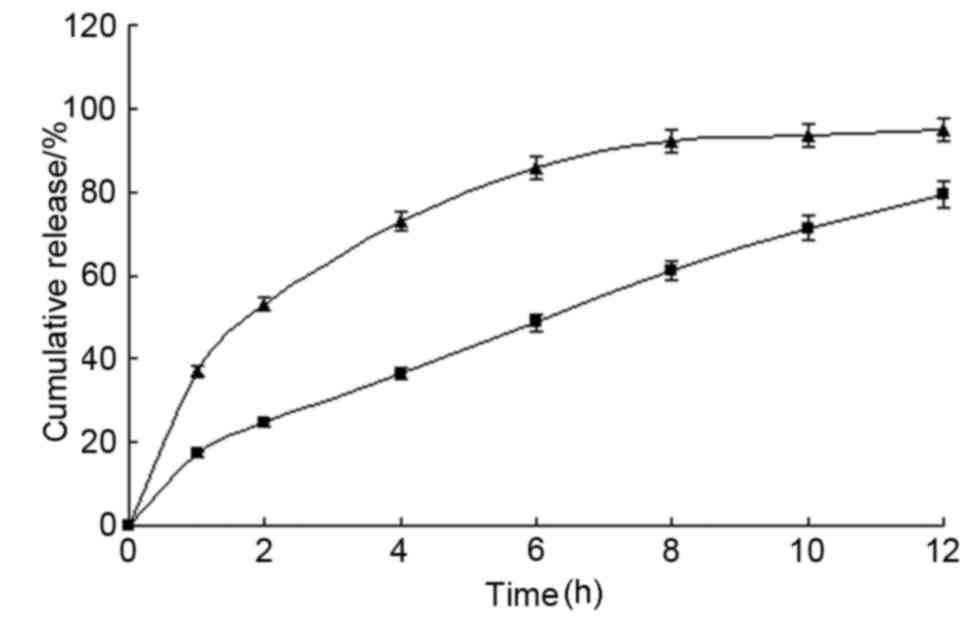
In vitro release
The in vitro drug release profiles of the MS
microcapsules in various dissolution media are presented in
Fig. 3. The release equations of MS
microcapsules in water, artificial gastric fluid, and in artificial
intestinal fluid were determined to be
Q=22.346t1/2+1.6068 (r=0.992);
Q=26.438t1/2+2.5376 (r=0.990); and
Q=22.553t1/2-1.3337 (r=0.997), respectively. The
cumulative release was found to fit the Higuchi equation (19), indicating that MS was released from
microcapsules via diffusion. The MS microcapsules demonstrated a
good SR profile in all three media, with a mean release of 96.1%
within 18 h.
 | Figure 3.Drug release from MS microcapsules
in vitro. Prepared microcapsules were placed in a basket
with 1,000 ml of release medium (water, artificial gastric fluid or
artificial intestinal fluid). Samples were taken from the release
solution at pre-determined time intervals (0.5, 2, 4, 6, 8, 10, 12,
15 and 18 h) to detect the released drug concentration. The total
percentages of MS released at different time-points are plotted.
Values are expressed as the mean ± standard deviation (n=12). ■,
water; ●, artificial gastric fluid; ▲, artificial intestinal fluid.
MS, metoprolol succinate. |
When submerged in water, conventional SR-tablets
exhibited a burst of release with >50% released in 2 h.
Conversely, the MS microcapsule SR-tablets exhibited an almost
linear SR in a 12-h test period (Fig.
4).
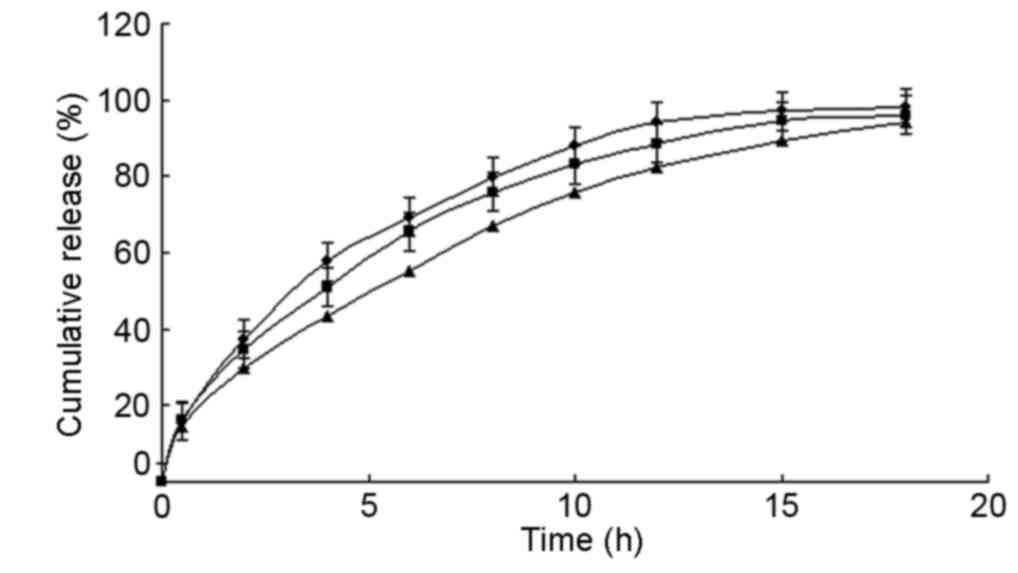
Pharmacokinetic studies in vivo
Pharmacokinetic studies of MS microcapsules and
conventional SR tablets were performed in dogs to evaluate the
in vivo performance of these formulations (Fig. 5). The results indicated that
conventional tablets and microcapsules fitted a single-compartment
model. The pharmacokinetic parameters are presented in Table IV. Both formulations exhibited a
similar area under curve; however, the microcapsules exhibited a
significantly longer half-life and time to peak, and a markedly
lower maximum drug concentration (Cmax).
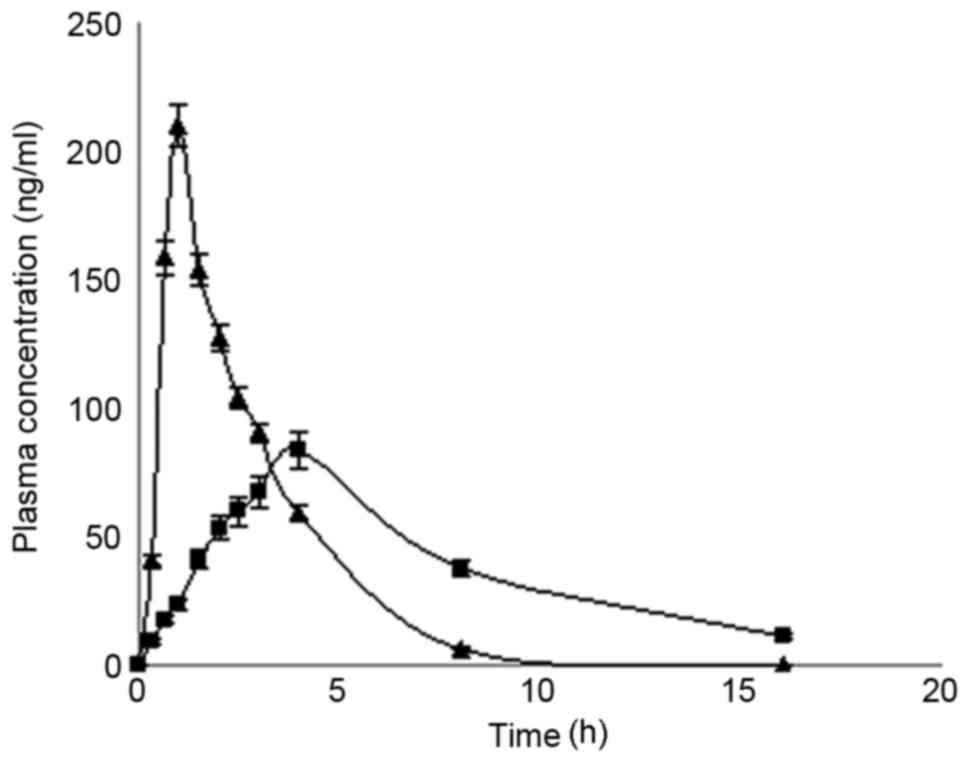 | Figure 5.Microcapsule drug release profile
in vivo. Six dogs were fasted overnight and administered
47.5 mg MS microcapsules or 50 mg metoprolol tartrate tablets
(conventional SR tablets). Venous blood samples harvested prior to
and following administration at 0.5, 1, 1.5, 2, 2.5, 3, 4, 8 and 16
h were used to detect MS concentration by HPLC analysis. Values are
expressed as the mean ± standard deviation (n=3). ■, SR
microcapsules; ▲, conventional SR tablets. MS, metoprolol
succinate; SR, sustained-release; HPLC, high-performance liquid
chromatography. |
| Table IV.Pharmacokinetic parameters. |
Table IV.
Pharmacokinetic parameters.
| Parameter | Conventional
SR-tablets | SR
microcapsules |
|---|
| t1/2
(h) | 0.84±0.09 |
2.84±0.37a |
| Ka (1/h) | 0.88±0.12 |
0.255±0.10a |
| Tmax
(h) | 1.17±0.27 |
4.01±0.53a |
| Cmax
(ng/ml) | 216.13±48.79 |
86.69±27.71a |
| CL (l/kg) | 0.42±0.07 | 0.43±0.09 |
| AUC (ng·h/ml) | 738.50±150.82 | 710.71±131.64 |
Discussion
SR formulations of MS may minimize fluctuations in
plasma concentration, avoiding the adverse effects associated with
excessively high plasma concentrations and providing a stable
effective dose. Microspheres have been previously used in SR
formulations of MS to provide robust and consistent control of
hypertension and heart rate (20).
The objective of the present study was to develop a method of MS
encapsulation to provide microcapsules with high entrapment
efficiency and optimal sustained-release profiles in vitro
and in vivo.
EC was employed as the capsule material and EA as a
solvent to prepare multiple emulsions. At room temperature, the
emulsion was added to distilled water to cause phase separation and
condensation of microcapsules. The solvent was removed with excess
water, avoiding the elevated temperatures usually employed in
solvent evaporation methods, which may have negatively affected the
stability of the encapsulated compound. Water-insoluble EC was
employed as a blocker film coating.
Increased ultrasonic power and time during
emulsification may increase drug loading; however, excessive
emulsification may lead to excess evaporation of EA in the primary
emulsion. In the current study, when agitated too quickly,
excessive foam was produced and the primary emulsion particle size
decreased, reducing the dose loaded per particle. Emulsification at
<160 W with a mixing time of <120 sec and 1,350 × g
using PEG 6,000 as a plasticizer in the W2 phase
produced microcapsules with favorable mechanical properties, as
walls were less prone to rupture and drug leakage was reduced. An
excess of PEG may reduce the amount of drug loading and accelerate
drug release, and may also cause microcapsules to aggregate into
larger clumps, with oil leach induced by mechanical impact
adversely affecting the process of preparation (21). As such, the optimal PEG 6,000
concentration was determined to be 3%. It was also necessary to
determine the optimum diffusion time, as excessively long diffusion
periods may result in loss of the pharmacological agent (20). The optimum diffusion time was
determined to be 2 h.
Given the complexity of the microcapsule preparation
process, numerous variables may be adjusted to optimize the drug
loading capacity. By using a single factor method and applying a
uniform design to assess the contribution of individual parameters,
and subsequently performing multiple regression analysis to select
the optimal conditions, the loading capacity of MS microcapsules
was optimized (22). The MS
microcapsules prepared under optimal conditions exhibited good
morphological characteristics, a high encapsulation efficiency and
good reproducibility.
It is difficult to achieve prolonged SR of
water-soluble pharmacological agents with conventional tablets
(23). Microcapsules formed with EC
and PEG 6,000 exhibited SR of MS in vivo and in
vitro. The 18-h release behavior followed the Higuchi equation
in water (19), and release rates in
artificial gastric and intestinal fluid were similar and
reproducible, with a release of ~80% of the encapsulated MS within
12 h. Furthermore, the novel microcapsule formula developed in the
present study exhibited slower in vitro release of MS than
the conventional tablet formula. These novel microcapsules are
advantageous, as they achieve a more even SR in various
environments, including water, artificial gastric fluid and
artificial intestinal fluid.
When dogs were orally administered microcapsules and
conventional tablets, the plasma half-life of MS with the
microcapsule tablet formulation was longer than with the
conventional tablet and the peak plasma concentration was
significantly lower (P<0.05). MS microcapsule SR tablets may
provide additional advantages by reducing the dose frequency and
minimizing adverse effects associated with Cmax.
In conclusion, the present study reported on the
development of microcapsules with high entrapment efficiency and
desirable SR properties in vitro and in vivo. An
entrapment efficiency of 83.16% was achieved and 96.1% of MS was
released in vitro within 18 h. Pharmacokinetic studies of MS
microcapsules in dogs indicated a superior SR profile compared with
conventional SR tablets. These findings suggested that the use of
microcapsules in tablets may provide therapeutic benefits over
conventional tablets by SR of pharmacological agents.
Acknowledgements
The authors would like to thank Professor Liu at
Guangzhou General Pharmaceutical Research Institute for providing
the animal models used in the present study.
References
|
1
|
Papadopoulos DP and Papademetriou V:
Metoprolol succinate combination in the treatment of hypertension.
Angiology. 60:608–613. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reiter MJ: Cardiovascular drug class
specificity: Beta-blockers. Prog Cardiovasc Dis. 47:11–33. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frishman WH and Saunders E: β-Adrenergic
blockers. J Clin Hypertens (Greenwich). 13:649–653. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bengtsson C, Johnsson G and Regårdh CG:
Plasma levels and effects of metoprolol on blood pressure and heart
rate in hypertensive patients after an acute dose and between two
doses during long-term treatment. Clin Pharmacol Ther. 17:400–408.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guan G and Li Z: Evaluation of the effect
on metoprolol succinate sustained release tablets in reducing blood
pressure. Jian Yan Yi Xue Yu Lin Chuang. 6:1989–1990. 2009.(In
Chinese).
|
|
6
|
Chirra HD and Desai TA: Emerging
microtechnologies for the development of oral drug delivery
devices. Adv Drug Deliv Rev. 64:1569–1578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu B, Wen R, Yang H and He Y:
Sustained-release tablets of indomethacin-loaded microcapsules:
Preparation, in vitro and in vivo characterization. Int J Pharm.
333:87–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frishman WH, Hainer JW and Sugg J; M-FACT
Study Group, : A factorial study of combination hypertension
treatment with metoprolol succinate extended release and felodipine
extended release results of the Metoprolol Succinate-Felodipine
Antihypertension Combination Trial (M-FACT). Am J Hypertens.
19:388–395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Siddique S, Khanam J and Bigoniya P:
Development of sustained release capsules containing ‘coated matrix
granules of metoprolol tartrate’. AAPS PharmSciTech. 11:1306–1314.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gong X, Lu Y, Xiang Z and Luo G:
Preparation of polysulfone microcapsules containing 1-octanol for
the recovery of caprolactam. J Microencapsul. 26:104–110. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Zhu Y, Wang J, Sun Y, Li S and
Pan W: Preparation of sustained-release microspheres of
clarithromycin by quasi-emulsion solvent diffusion method. Zhongguo
Yao Ye. 18:39–40. 2009.(In Chinese).
|
|
12
|
You J, Cui FD, Han X, Wang YS, Yang L, Yu
YW and Li QP: Study of the preparation of sustained-release
microspheres containing zedoary turmeric oil by the
emulsion-solvent-diffusion method and evaluation of the
self-emulsification and bioavailability of the oil. Colloids Surf B
Biointerfaces. 48:35–41. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jelvehgari M and Montazam SH: Comparison
of microencapsulation by emulsion-solvent extraction/evaporation
technique using derivatives cellulose and acrylate-methacrylate
copolymer as carriers. Jundishapur J Nat Pharm Prod. 7:144–152.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dash V, Mishra SK, Singh M, Goyal AK and
Rath G: Release kinetic studies of aspirin microcapsules from ethyl
cellulose, cellulose acetate phthalate and their mixtures by
emulsion solvent evaporation method. Sci Pharm. 78:93–101. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saringat HB, Alfadol KI and Khan GM: The
influence of different plasticizers on some physical and mechanical
properties of hydroxypropyl methylcellulose free films. Pak J Pharm
Sci. 18:25–38. 2005.PubMed/NCBI
|
|
16
|
Huang G, Deng S, Wang R and Xi Y:
Preparation and in vitro-in vivo correlation in dogs of metoprolol
succinate sustained-released tablets. Zhongguo Yi Yao Gong Ye Za
Zhi. 36:412–414. 2005.(In Chinese).
|
|
17
|
Xu W, Li N and Gao C: Preparation of
controlled porosity osmotic pump tablets for salvianolic acid an
doptimization of the formulation using an artificial neural network
method. Acta Pharm Sin B. 1:64–70. 2011. View Article : Google Scholar
|
|
18
|
Shen LN, Zhang YT, Wang Q, Xu L and Feng
NP: Enhanced in vitro and in vivo skin deposition of apigenin
delivered using ethosomes. Int J Pharm. 460:280–288. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saurí J, Millán D, Suñé-Negre JM, Colom H,
Ticó JR, Miñarro M, Pérez-Lozano P and García-Montoya E: Quality by
design approach to understand the physicochemical phenomena
involved in controlled release of captopril SR matrix tablets. Int
J Pharm. 477:431–441. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Albin P, Markus A, Ben-Zvi Z and Pelah Z:
A new slow release formulation of metoprolol: In-vitro and in-vivo
evaluation in dogs. J Control Release. 23:1–11. 1993. View Article : Google Scholar
|
|
21
|
Liu Y, Feng Y and Xu D: Influence of
excipients on the mechanical properties of microcapsules. Chin
Traditional Pat Med. 29:1602–1605. 2007.
|
|
22
|
Zhao W: Uniform design application in
pharmaceutics. Anhui Med Pharm J. 14:610–612. 2010.
|
|
23
|
Pradhan R, Kim YI, Chang SW and Kim JO:
Preparation and evaluation of once-daily sustained-release coated
tablets of tolterodine-L-tartrate. Int J Pharm. 460:205–211. 2014.
View Article : Google Scholar : PubMed/NCBI
|