Introduction
Matrix metalloproteinases (MMPs) are a large family
of zinc-dependent enzymes, which are able to collectively degrade
collagen and the majority of the extracellular matrix (ECM)
components (1). ECM provides
structural support to the heart, and ECM quantity and quality are
the major determinants of myocardial passive stiffness (2). Recent evidence demonstrated that
excessive breakdown of the ECM by MMPs serves a pathogenic role in
the atherosclerotic plaque, which is the major cause of mortality
as a result of acute coronary syndrome and stroke (3). Therefore, MMPs are critical for
vascular remodeling by regulating the degradation of the ECM in
atherosclerotic plaques.
Atherosclerosis is a chronic inflammatory disease
that is the basis of atherothrombosis and the development of acute
coronary syndrome (4–6). Increasing evidence indicates that the
concentrations of MMPs are elevated not only in certain patients
with cancer, liver cirrhosis or rheumatoid arthritis (7,8), but is
also involved in the pathogenesis and progression of
atherosclerotic plaque formation and rupture (9). Among the MMP family, MMP-9 (also known
as gelatinase B) is the predominant MMP and can degrade type IV
collagen, which is the major component of the basement membrane
(10). In addition, MMP-9 is
indispensable for collagen-cleaving and degrading of the
extracellular plaque matrix, leading to plaque instability and
rupture (11). MMP-9 can be secreted
by smooth muscle cells (SMCs) and macrophages (12). Furthermore, the macrophage-rich
region of atherosclerotic plaque is the major source of MMP-9
(13). The secretion of specific
proinflammatory cytokines can activate pro-MMP-9, therefore
inducing the degradation of the ECM, which leads to the migration
and proliferation of SMCs (14). In
addition, activated endothelial cells express adhesion molecules
that can promote the infiltration of monocytes and their adhesion
to the endothelial cells, which enhances the MMP-9 production,
consequently enhancing endothelial cell permeability and
acceleration of plaque progression. A previous study has
demonstrated that MMP-9 was highly expressed in humans with
atherosclerotic lesions and animal models (15), and participated in mediating plaque
instability, which is a major cause of acute coronary syndrome and
stroke (12). MMP-9 appears to serve
a central role in the loss of atherosclerotic plaque stability.
Therefore, it is crucial to clarify the regulatory mechanism of
MMP-9 in atherosclerosis and to determine whether inhibiting the
MMP-9 activity may be an effective therapeutic strategy for the
treatment of atherosclerosis.
The expression of MMP-9 appears to be regulated by a
range of different signaling pathways. Several studies have
demonstrated that mitogen-activated protein kinases (MAPKs) are
involved in the regulation of MMPs by various cell types (16,17). The
MAPK pathway is one of the important mediators of signal
transduction, which participates in multiple fundamental cellular
processes, including cell growth, proliferation, differentiation
and death (18). However, tumor
necrosis factor-α (TNF-α) can activate three MAPK cascades,
including the extracellular signal-regulated kinases (ERKs), the
c-Jun N-terminal kinase (JNK)/stress-activated protein kinases and
p38 (19,20). The ERK pathway has been linked to
cell proliferation, cell growth and differentiation, whereas JNK
and p38 MAPK pathways have been linked to apoptosis, cell survival,
transformation, development, cell migration and immune activation
(21). Holvoet et al also
proved that increased expression of MMP-9 induced by TNF-α was
reduced by the specific inhibitors of MAPK signaling pathway in
human keratinocytes (22). Nuclear
factor-κB (NF-κB) binds to the proximal promoter region of the
MMP-9 gene and regulates MMP-9 transcription in response to
distinct extracellular stimulation of TNF-α (23,24),
which is one of the strongest physiological inducers of MMP-9
expression (25).
Aspirin, a conventional nonselective non-steroidal
anti-inflammation drug, is widely used in the primary prevention
against cardiac-cerebral vascular diseases, such as myocardial
infarction and stroke, and 20–25% of patients with various vascular
diseases who were treated with aspirin presented decreased
development of vascular events (26). The anti-platelet function of aspirin
is known to contribute to the therapy of atherosclerotic
cardiovascular disease. However, the anti-inflammatory effect of
aspirin in atherosclerosis is not widely reported. Previous studies
(3–5)
have demonstrated that atherosclerosis is a complex vascular
inflammation disease. A clinical study has shown that patients
receiving treatment with aspirin exhibited lower macrophage density
of the carotid atherosclerotic plaque, suggesting that aspirin is
involved in the suppression of the vascular inflammation process
(27). Hua et al (28) also reported that aspirin prevented
against atherosclerotic plaque rupture by inhibiting MMP-9
expression by upregulating peroxisome proliferator-activated
receptor α/γ (PPARα/γ) expression in oxidized low-density
lipoprotein-stimulated macrophages and by inducing TIMP
metallopeptidase inhibitor 1 (TIMP1) and TIMP2 expression. However,
whether aspirin inhibits the expression of MMP-9 via the MAPK and
NF-κB signaling pathways in TNF-α-stimulated RAW264.7 cells remains
unknown. Therefore, the present study investigated the effects and
mechanisms of aspirin on MMP-9 expression in TNF-α-stimulated
RAW264.7 cells.
Materials and methods
Materials
Antibodies against JNK (1:500 dilution, BS6448), p38
(1:500 dilution, BS3566), ERK (1:1,000 dilution, AP0485),
phospho-JNK (1:500 dilution, BS4763), phospho-p38 (1:500 dilution,
BS4635) and phospho-ERK (1:1,000 dilution, BS4759) were purchased
from Bioworld Technology (Beijing, China). SB203580 (p38MAPK
inhibitor, 5633S), SP600125 (JNK inhibitor, 8177S) and PD98059
(ERK1/2 inhibitor, 9900S) and PDTC (NF-κB inhibitor) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). An
antibody against the p65 subunit of NF-κB was also purchased from
Cell Signaling Technology, Inc. (1:500 dilution, 8242). An antibody
against MMP-9 was purchased from EMD Millipore (Chemicon;
Billerica, MA, USA, 1:500 dilution, AB19016). Recombinant murine
TNF-α was purchased from Thermo Fisher Scientific, Inc. (Biosource;
MA, USA), and aspirin was purchased from Langtze Biomedical
Technology (Nanjing, China).
Cell cultures
Murine macrophage RAW264.7 cells, purchased from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China), were cultured in plastic dishes containing
Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis,
MO, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich),
100 U/ml penicillin and 100 µg/ml streptomycin at 37°C and 5%
CO2. For all experiments, cells were grown to 60–80%
confluence in culture flasks. Then, the medium was replaced with
fresh DMEM and cells were transferred into multiple flasks for
further expansion. The control groups were treated with medium
only. In order to study the expression of MMP-9, TNF-α (10 ng/ml)
was added in the presence or absence of aspirin (75, 150, 300 and
600 µM) for 24 h. For the inhibitory study, PDTC, an inhibitor of
NF-κB, can significantly inhibit NF-κB activity, and further reduce
the production of inflammatory cytokines, alleviating the systemic
inflammatory response (29). In
order to determine the effect of PDTC on TNF-α-induced expression
of MMP-9 in RAW264.7 cells, the cells were divided into six groups
and incubated with either TNF-α or TNF-α plus PDTC, PDTC and
aspirin, aspirin or PDTC only group, respectively. The cells were
treated with or without aspirin and PDTC for 1 h, then stimulated
with TNF-α for 24 h. And for the MAPK inhibitors, the cells were
divided into six groups and incubated with TNF-α or TNF-α plus
PD98059, SB203580, SP600125 or aspirin. Cells were pre-incubated
with or without 10 µM PD98059 (p-ERK inhibitor) (30), 10 µM SB203580 (p-p38 inhibitor)
(30), SP600125 (p-JNK inhibitor)
(31) and aspirin (600 µM) for 1 h
with TNF-α (10 ng/ml). These cultured cells and supernatants were
then collected for measurement of the following design parameters
after treatment with TNF-α for 24 h.
Cell viability
MTT was used to evaluate the cytotoxicity of aspirin
in RAW264.7 cells. Briefly, the cells were seed at a density of
4×104 cells/ml in a 96-well plate. The cells were
pretreated with various concentrations of aspirin (75, 150, 300 and
600 µM) for 1 h, and then stimulated with or without TNF-α (10
ng/ml) for 24 h at 37°C in an atmosphere with 5% CO2.
Subsequently, MTT solution (5 mg/ml in a phosphate-buffered saline)
was added to each well and the cells were incubated for a further 4
h. The medium was then discarded and 100 ml dimethyl sulfoxide was
added. The absorbance was recorded at 490 nm with a microplate
reader in order to determine the cell viability.
Determination of MMP-9 mRNA levels by
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR)
Total RNA was abstracted from the TNF-α-stimulated
RAW264.7 cells with TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Equal amounts of RNA (1 µg) were reverse transcribed using a
First-Strand cDNA synthesis kit (Takara Biotechnology Co., Ltd.,
Dalian, China, 639504). qPCR was then performed using SYBR-Green
(Takara Biotechnology Co., Ltd., 639655) on a Real-Time
Quantitative Thermal Block (Biometra, Göttingen, Germany). The
following specific primers were used: MMP-9 forward,
5′-TTCACCCGGTTGTGGAAACT-3′, and reverse,
5′-AAATGTGGGTGTACACAGGC-3′; GAPDH forward,
5′-TGGAATCCTGTGGCATCCATGAAA-3′, and reverse,
5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. The entire amplification course
was initiated at 95°C for 5 min, followed by 40 cycles of 95°C for
10 sec and 60°C for 30 sec, and a final step at 60°C for 30 sec.
The specificity of the amplified products was analyzed through
dissociation curves generated by the equipment yielding single
peaks. GAPDH was used as an internal control to normalize samples.
PCR reactions of each sample were conducted in triplicate. Data
were analyzed through the comparative cycle threshold (Cq) method,
obtained from the iQ5 Optical System software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Gelatin zymography assay
To analyze MMP-9 enzyme expression, RAW264.7 cells
were seeded in 6-well culture plates (2×106 cells/well).
After the medium was changed with serum-free medium, the cells were
pretreated with aspirin for 1 h and then stimulated with TNF-α for
24 h. Next, the samples were collected and separating by passing
throughout 10% zymography gels. Following electrophoresis, sodium
dodecyl sulfate (SDS) was removed by washing the gels three times
with buffer (50 mM Tris/HCl, pH 7.6, 150 mM NaCl, 5 mM
CaCl2, 2 mM ZnCl2 and 0.1% Triton X-100) for
30 min at room temperature with gentle agitation to renature
enzymes. The gels were subsequently incubated in zymogen
development buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM
CaCl2, 0.02% NaN3, and 1 µM ZnCl2]
at 37°C for 24–48 h. After briefly washing in water, gels were
stained with Coomassie blue R-250 (Bio-Rad Laboratories, Inc.) for
1 h. Gels were destained with 40% methanol and 5% acetic acid until
clear white bands against a blue background were visible.
Western blot analysis
Total cells were washed with ice-cold phosphate
buffer saline and then harvested using RIPA buffer containing 1 mM
phenylmethylsulfonyl fluoride. In order to obtain the nuclear
extracts of NF-κB, the nuclear proteins were prepared using a
nuclear protein extraction kit (Beyotime Institute of Institute of
Biotechnology, Haimen, China, p0027) according to the
manufacturer's protocol. Protein concentrations were then measured
using a bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology, p0012s). Following incubation on ice for 30 min, the
supernatant was collected by centrifugation at 12,000 × g for 10
min at 4°C, and the amount of protein was measured using a Bradford
assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA,
5000202EDU). Subsequently, proteins were denatured in sample buffer
containing 2-mercaptoethanol and bromophenol blue for 10 min at
100°C. Equal quantities of total cell lysates were size
fractionated by 10% SDS-polyacrylamide gel electrophoresis and
transferred to polyvinylidene difluoride membranes using the Hoefer
electrotransfer system (GE Healthcare, Chicago, IL, USA).
Subsequent to blocking, the membrane was incubated with primary
antibodies against NF-κB p65, β-actin (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA; 1:1,000 dilution, sc-7210), phospho-ERK,
ERK, phospho-p38, p38, phospho-JNK, JNK and MMP-9 overnight at 4°C.
The membrane was then washed with Tris-buffered saline/Tween 20 and
incubated with anti-mouse or anti-rabbit horseradish peroxidase
(HRP)-conjugated immunoglobulin G secondary antibodies (Santa Cruz
Biotechnology, Inc.; 1:10,000 dilution, sc-2030) for 1 h at room
temperature. The specific proteins were detected using enhanced
chemiluminescence, and images were captured with a Fluorochem Gel
Image Analyzer (ProteinSimple, San Jose, CA, USA).
Statistical analysis
Statistical analysis was performed using SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). The results are expressed
as the mean ± standard deviation, and differences between the means
of two groups were determined by unpaired Student's t-test. The
minimum significance level was set at P<0.05 for all analyses.
All experiments were performed at least three times.
Results
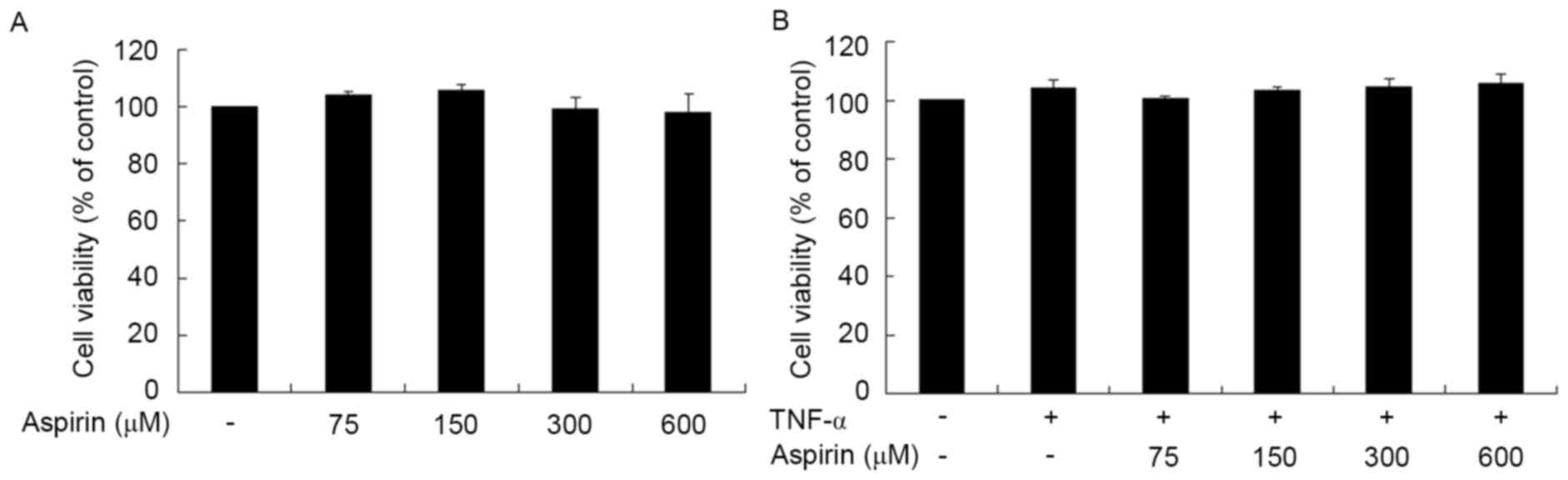
Cytotoxicity of aspirin on RAW264.7
cells
The cytotoxic effect of aspirin on RAW264.7 cells
was evaluated using MTT assay (Fig.
1A). Treatment of aspirin for 24 h did not have a significant
cytotoxic effect on the cells at the concentrations of 75, 150, 300
and 600 µM, when compared with the untreated cells. Next, the
cytotoxic effects of aspirin on TNF-α-treated RAW264.7 cells were
determined. Cells were incubated in the presence of aspirin (0–600
µM) in serum-depleted medium for 1 h and then stimulated with TNF-α
(10 ng/ml) for 24 h. The results indicated that aspirin had no
evident cytotoxic effect on TNF-α-stimulated RAW264.7 cells at a
concentration of up to 600 µM (Fig.
1B). Therefore, an aspirin dose of up to 600 µM was used in
subsequent experiments.
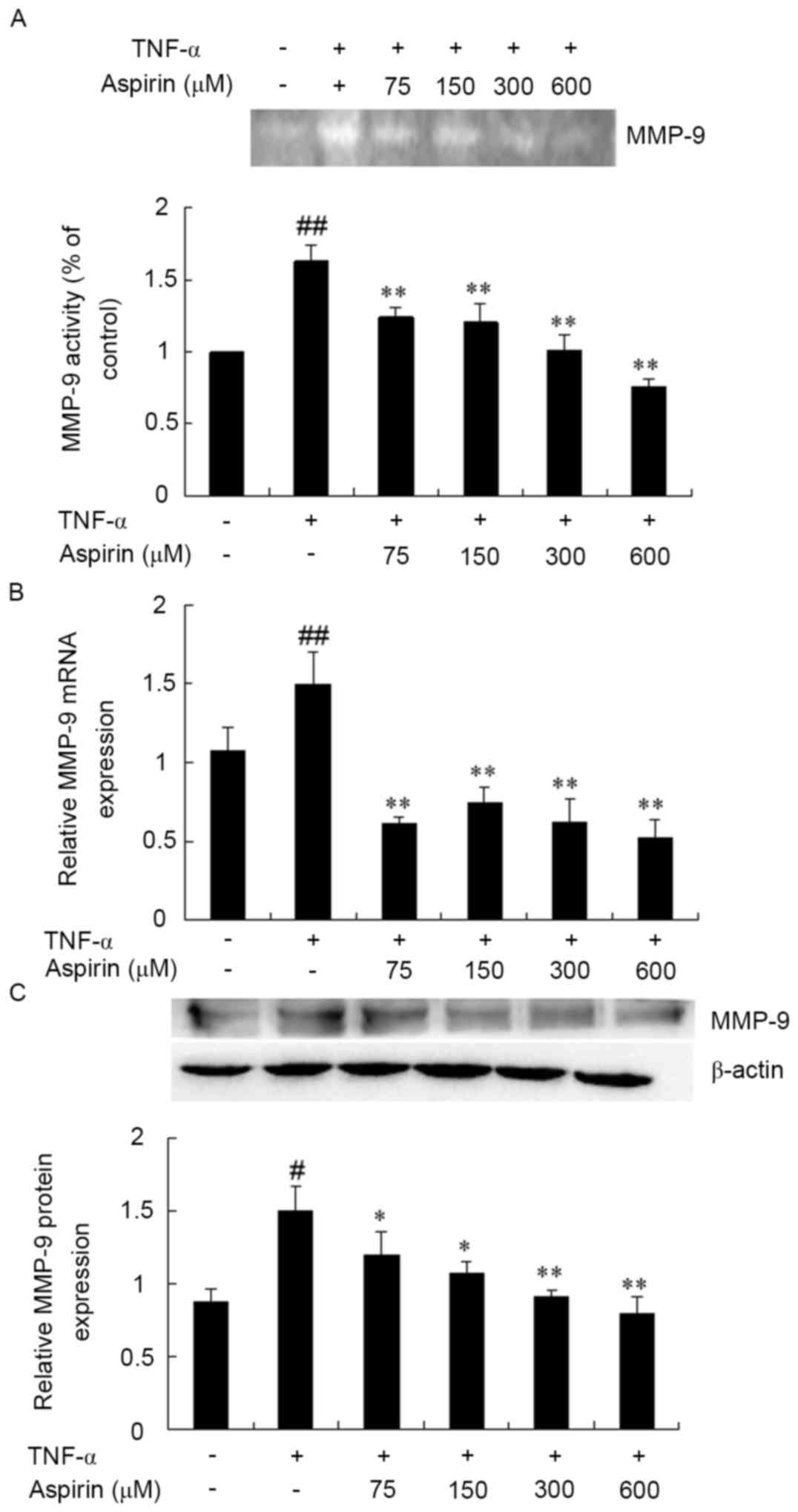
Inhibitory effects of aspirin on MMP-9
expression in TNF-α-treated RAW264.7 cells
The next experiment attempted to determine the
effect of aspirin on TNF-α-induced MMP-9 expression. RAW264.7 cells
were pretreated without or with aspirin (75–600 µm/l) for 1 h and
stimulated with TNF-α (10 ng/ml) for 24 h. Thereafter, the cultured
medium was harvested for analysis of MMP-9 enzymatic activity, mRNA
levels and protein expression by gelatin zymography, RT-qPCR and
western blot analysis, respectively. As shown in Fig. 2A, MMP-9 secretion was significantly
induced by TNF-α. However, the induction of MMP-9 activity by TNF-α
was significantly inhibited by aspirin pre-treatment in a
dose-dependent manner. Similarly, aspirin exhibited an inhibitory
effect on TNF-α-induced MMP-9 expression at the mRNA and protein
levels in a dose-dependent manner (Fig.
2B and C). Therefore, these findings suggest that aspirin
effectively inhibits the TNF-α-stimulated MMP-9 expression and
activity without any cytotoxicity observed at the dosage tested in
RAW264.7 cells.
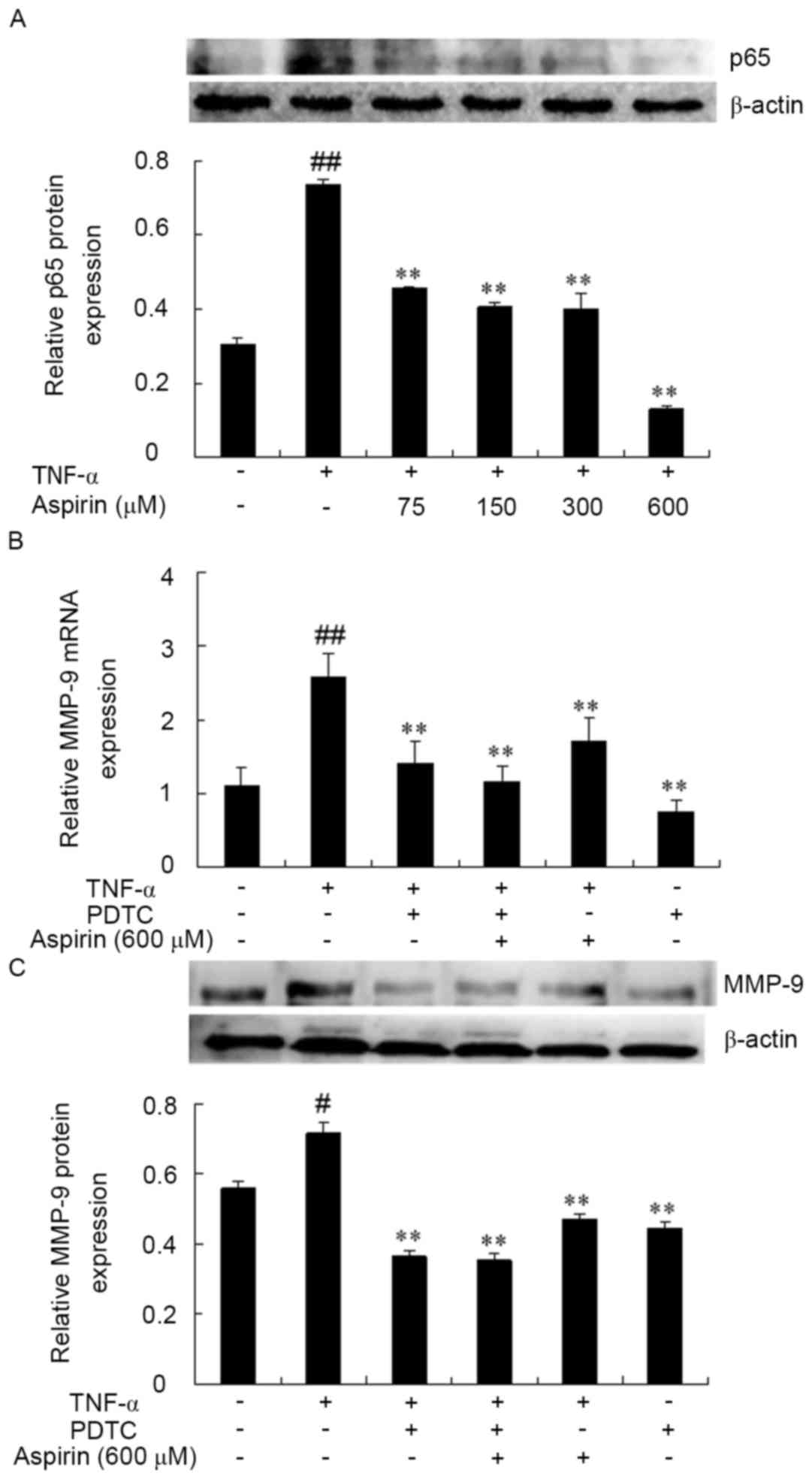
Effect of aspirin on TNF-α-induced
activation of NF-κB in RAW264.7 cells
NF-κB, which has a binding site located in the MMP-9
promoter region, has been implicated in the TNF-α-induced
expression of MMP-9 in several cell lines (32). In order to determine whether the
inhibitory effect of aspirin on the TNF-α-induced expression of
MMP-9 is mediated by NF-κB, the effect of the nuclear translocation
of p65 was investigated, which is a major subunit of NF-κB that has
been shown to be induced by TNF-α. The cells were treated with
aspirin in the presence of TNF-α for 1 h and then assessed by
western blotting. As shown in Fig.
3A, the nuclear translocation of p65 as a result of TNF-α
stimulation was strongly inhibited in the presence of aspirin at a
concentration of 600 µM. This inhibitory effect was increased in a
dose-dependent manner.
The association between NF-κB activation and MMP-9
expression was then further examined by exposure of cells to a
specific inhibitor of NF-κB, pyrrolidine dithiocarbamate (PDTC),
prior to TNF-α stimulation. PDTC can inhibit NF-κB activity and
further reduce the production of inflammatory cytokines,
alleviating the systemic inflammatory response (29). The results demonstrated that the
combination of aspirin and PDTC also reduced TNF-α-induced MMP-9
expression (P<0.01; Fig. 3B and
C). Therefore, these results indicated that the inhibitory
effect of aspirin on MMP-9 expression and activity are associated
with the suppression of NF-κB activation in TNF-α-stimulated
RAW264.7 cells.
Effect of aspirin on the inhibition of
ERK1/2, JNK and p38 phosphorylation
Several studies have indicated that MAPK pathways
are involved in the expression of MMP-9 (33,34). To
explore whether the inhibitory effect of aspirin on the expression
of MMP-9 was mediated through the MAPK pathway, the phosphorylated
protein levels of ERK1/2, JNK and p38 were examined by western blot
in RAW264.7 cells pre-treated with aspirin and then with TNF-α for
various times (0, 10, 20, 30 and 60 min). As shown in Fig. 4A, the protein expression levels of
non-phosphorylated ERK, JNK and p38 were not evidently altered in
the TNF-α alone and the TNF-α plus aspirin stimulation groups. By
contrast, the expression of phosphorylated (p)-p38, p-ERK1/2 and
p-JNK significantly increased by TNF-α stimulation, whereas aspirin
inhibited the increase of p-p38, p-ERK1/2 and p-JNK induced by
TNF-α at each time point.
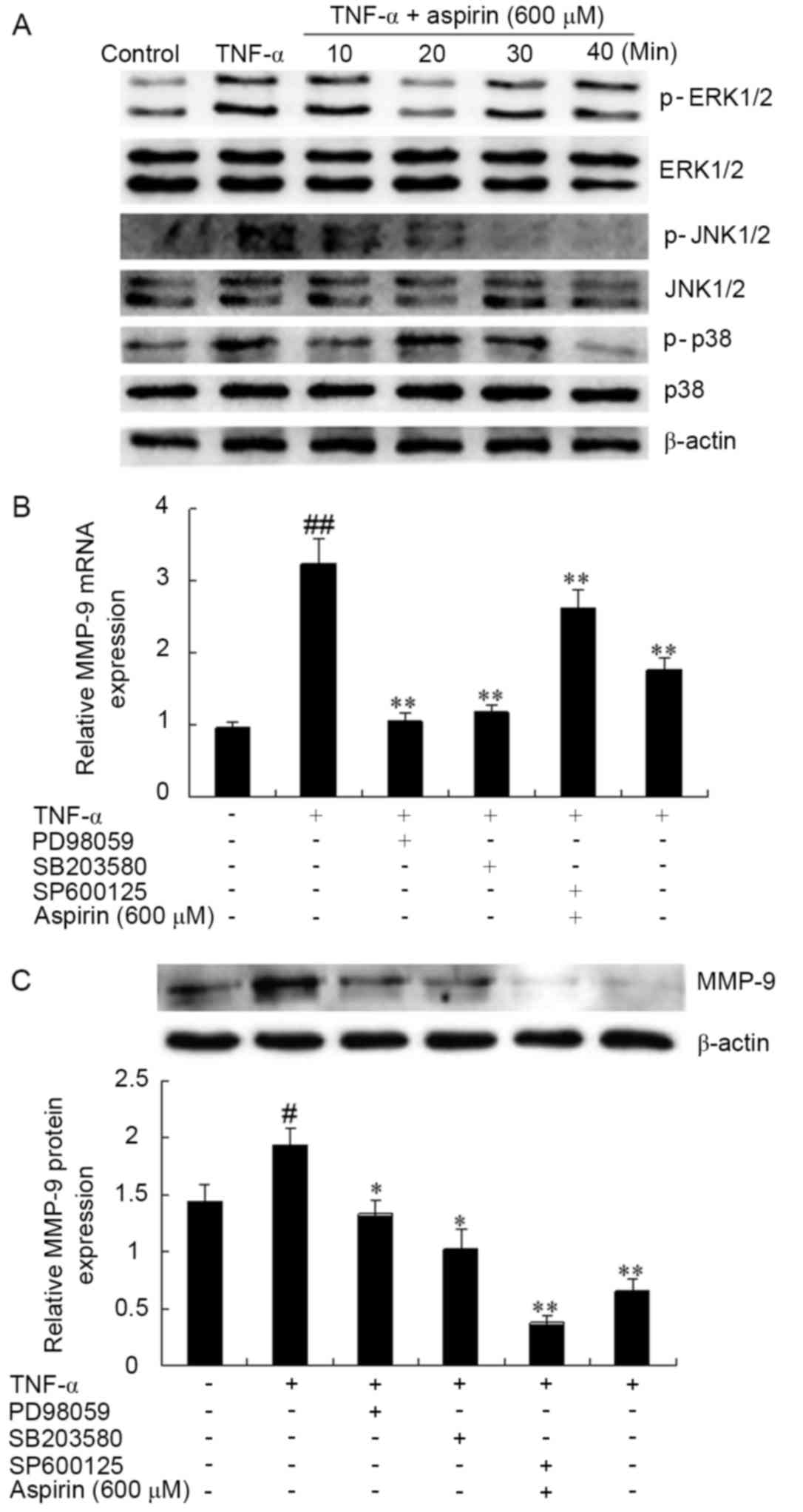 | Figure 4.Effects of aspirin on
TNF-α-stimulated activation of MAPK signaling pathway in RAW264.7
cells. (A) Aspirin inhibited the TNF-α-stimulated phosphorylation
levels of ERK1/2, p38 MAPK and JNK, as determined using western
blot analysis. Cells were incubated for 1 h in the absence or
present of aspirin (600 µM) and then stimulated for 10, 20, 30 and
60 min with 10 ng/ml of TNF-α. (B) Reverse
transcription-quantitative polymerase chain reaction and (C)
western blot analysis were performed to examine the effect of MAPK
inhibitors on the mRNA and protein expression levels of MMP-9,
respectively. Cells were pre-incubated with or without 10 µM
PD98059 (p-ERK inhibitor), 10 µM SB203580 (p-p38 inhibitor),
SP600125 (p-JNK inhibitor) and aspirin (600 µM) for 1 h and then
with TNF-α (10 ng/ml) for 24 h. Densitometric results are
represented the mean ± standard deviation of three independent
measurements. #P<0.05 and ##P<0.01 vs.
untreated control; *P<0.05 and **P<0.01 vs. TNF-α treatment
alone. TNF-α, tumor necrosis factor-α; MMP-9, matrix
metalloproteinase-9; MAPK, mitogen-activated protein kinase; ERK,
extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase; p-, phosphorylated. |
The present study also examined whether MAPK
pathways are involved in the MMP-9 expression in the
TNF-α-stimulated RAW264.7 cells using inhibitors of ERK1/2
inhibitor (PD98059), p38 (SB203580) and JNK (SP600125). As shown in
Fig. 4B and C, the mRNA and protein
levels of TNF-α-induced MMP-9 expression levels were significantly
downregulated in the presence of each of the MAPK inhibitors. These
results suggest that the specific inhibition of the MAPK signaling
pathway may be involved in the regulation of TNF-α-induced MMP-9
expression by aspirin in RAW264.7 cells.
Discussion
Cardiovascular disease, in particular
atherosclerosis, is regarded as a type of inflammatory disease
(35). Macrophages serve a major
role during atherosclerotic lesion development in atherosclerotic
plaque at various stages of development, partially through
participation in the inflammatory response (4). Therefore, understanding the regulatory
mechanisms of inflammation and finding pharmacological agents that
can inhibit the inflammatory disease may have a potential effect in
the prevention and treatment of atherosclerosis.
Aspirin, a platelet-inhibitory drug, is used to
prevent complications of atherosclerotic cardiovascular disease,
such as myocardial infarction and stroke (36). A previous study has demonstrated that
aspirin, together with its anti-platelet activity, suppressed
vascular inflammation and increased the stability of
atherosclerotic plaques in murine atherosclerosis, thus exhibiting
an anti-atherogenic effect (37).
Aspirin can inhibit the expression and release of MMP-2/9 by
upregulation of PPARα/γ gene expression, and also inhibit the
activity of MMP-2/9 by induction of TIMP1 expression, which may be
beneficial for the stabilization of atherosclerotic plaques
(38). However, to the best of our
knowledge, no studies have reported the potential effects of
aspirin on MMP-9 expression in TNF-α-treated RAW264.7 cells. In the
present study, the mechanism underlying the MMP-9 inhibition by
aspirin treatment in TNF-α-stimulated RAW264.7 cells was
investigated. It was demonstrated that aspirin has potent
inhibitory effects on MMP-9 expression, possibly together with an
anti-inflammatory and anti-platelet function.
MMPs are a major family of endopeptidases that can
selectively degrade various components of the ECM and serve crucial
roles in various physiological and pathological process, including
wound healing, vascular remodeling, rheumatoid arthritis,
angiogenesis and invasion (39–41).
MMP-9 is a member of the MMPs family and a marker for coronary
atherosclerosis. Plasma MMP-9 concentration has been identified as
a predictor of cardiovascular mortality in patients with coronary
artery disease (42), and the
atherosclerotic lesion development is initiated by infiltrated
macrophages that mainly produce MMP-9. In turn, the expression and
activity of MMP-9 were increased in advanced atherosclerotic
lesions, followed by macrophage infiltration (43). Additionally, accumulating evidence
demonstrates that the activity of MMP-9 is induced by TNF-α in a
variety of cell types (44,45). The results of the present study
consistently identified that TNF-α enhanced MMP-9 expression and
activity in cultured RAW264.7 cells. Notably, these data indicated
that the elevated mRNA expression, protein level and activity of
MMP-9 by TNF-α stimulation were inhibited by aspirin pre-treatment
in a dose-dependent manner in RAW264.7 cells. Therefore, the
present results suggested that the inhibition of MMP-9 expression
and activity may be responsible for the inhibitory effects of
aspirin on TNF-α-treated RAW264.7 cells.
The binding site for NF-κB in the promoter region of
MMP-9 serves a key function in the upregulation of MMP-9 expression
by TNF-α induction. (46,47). NF-κB serves an important role in
regulating the inflammatory and immune responses to extracellular
stimuli. NF-κB is normally sequestered in the cytoplasm by
inhibitory IκB proteins. Once activated, the NF-κB subunit p65
dissociates from its inhibitory protein IκB and translocates from
the cytoplasm to the nucleus. Western blot analysis in the current
study revealed that aspirin inhibited the nuclear translocation of
NF-κB p65 in TNF-α-stimulated RAW264.7 cells in a dose-dependent
manner. Furthermore, the NF-κB signaling specific inhibitor PDTC
was used to determine whether NF-κB signaling is involved in the
regulation of MMP-9 expression. The present data demonstrated that
PDTC suppressed the MMP-9 expression, which agrees with a previous
study (48) showing that aspirin
inhibited MMP-9 mRNA expression and the nuclear translocation of
NF-κB p65 subunit, thus suppressing the activity of this
inflammatory molecule and maintaining the plaque stability.
Collectively, the results of the present study imply that the
inhibitory effects of aspirin on TNF-α-induced MMP-9 expression
were mediated, at least partially, by suppression of the NF-κB
transcription factor.
MMP-9 has been demonstrated to be stimulated by
TNF-α via activating MAPKs (49).
Considerable evidence indicated that numerous natural products
inhibit the expression of pro-inflammatory genes by modulating the
activation of MAPK pathways (50,51).
Therefore, the present study aimed to reveal the inhibitory
mechanism of aspirin on MMP-9 transcription through MAPK pathways.
The data demonstrated that aspirin downregulated TNF-α-stimulated
the phosphorylation of ERK1/2, p38 and JNK upon treatment for
different time points. In addition, exposure to PD98059 (an
inhibitor of ERK1/2 phosphorylation), SB203580 (an inhibitor of p38
phosphorylation) and SP600125 (an inhibitor JNK phosphorylation)
also suppressed TNF-α-induced MMP-9 expression. According on the
aforementioned results, it is strongly suggested that aspirin
inhibits TNF-α-induced MMP-9 expression possibly by blocking the
NF-κB and MAPK signaling pathways in RAW264.7 cells. However, the
MMP-9 promoter has several transcription factor-binding motifs,
including the AP-1, Sp-1 and NF-κB binding sites (52). Therefore, other possible
transcription factors and signaling pathways may be involved in the
regulation of MMP-9 expression, which requires further
investigation.
In conclusion, the results of the present study
suggested that aspirin effectively inhibited the expression of
MMP-9 in TNF-α-stimulated RAW264.7 cells, possibly by inhibiting
the activation of NF-κB and MAPK pathways. It was, thus,
demonstrated that aspirin may contribute to the stabilization of
atherosclerotic plaque and prevention of atherosclerosis.
Acknowledgements
The present study was supported by the National
Natural Science Fund of China (grant no. 81173133).
References
|
1
|
Welgus HG, Campbell EJ, Cury JD, Eisen AZ,
Senior RM, Wilhelm SM and Goldberg GI: Neutral metalloproteinases
produced by human mononuclear phagocytes. Enzyme profile,
regulation, and expression during cellular development. J Clin
Invest. 86:1496–1502. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo
SM, Dai Q, Zhang J, Jin YF and Lindsey ML: Matrix
metalloproteinase-9 deletion attenuates myocardial fibrosis and
diastolic dysfunction in ageing mice. Cardiovasc Res. 96:444–455.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arroyo-Espliguero R, Avanzas P,
Cosín-Sales J, Aldama G, Pizzi C and Kaski JC: C-reactive protein
elevation and disease activity in patients with coronary artery
disease. Eur Heart J. 25:401–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fujimoto N, Mouri N, Iwata K, Ohuchi E,
Okada Y and Hayakawa T: A one-step sandwich enzyme immunoassay for
human matrix metalloproteinase 2 (72-kDa gelatinase/type IV
collagenase) using monoclonal antibodies. Clin Chim Acta.
221:91–103. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fujimoto N, Hosokawa N, Iwata K, Okada Y
and Hayakawa T: A one-step sandwich enzyme immunoassay for human
matrix metalloproteinase 9 using monoclonal antibodies. Ann N Y
Acad Sci. 732:359–361. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sundström J and Vasan RS: Circulating
biomarkers of extracellular matrix remodeling and risk of
atherosclerotic events. Curr Opin Lipidol. 17:45–53. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim HS, Kim HJ, Park KG, Kim YN, Kwon TK,
Park JY, Lee KU, Kim JG and Lee IK: Alpha-lipoic acid inhibits
matrix metalloproteinase-9 expression by inhibiting NF-kappaB
transcriptional activity. Exp Mol Med. 39:106–113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gross J and Lapiere CM: Collagenolytic
activity in amphibian tissues: A tissue culture assay. Proc Natl
Acad Sci USA. 48:pp. 1014–1022. 1962, View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galis ZS, Sukhova GK, Lark MW and Libby P:
Increased expression of matrix metalloproteinases and matrix
degrading activity in vulnerable regions of human atherosclerotic
plaques. J Clin Invest. 94:2493–2503. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Major TC, Liang L, Lu X, Rosebury W and
Bocan TM: Extracellular matrix metalloproteinase inducer (EMMPRIN)
is induced upon monocyte differentiation and is expressed in human
atheroma. Arterioscler Thromb Vasc Biol. 22:1200–1207. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chandrasekar B, Mummidi S, Mahimainathan
L, Patel DN, Bailey SR, Imam SZ, Greene WC and Valente AJ:
Interleukin-18-induced human coronary artery smooth muscle cell
migration is dependent on NF-kappaB- and AP-1-mediated matrix
metalloproteinase-9 expression and is inhibited by atorvastatin. J
Biol Chem. 281:15099–15109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johnson JL, George SJ, Newby AC and
Jackson CL: Divergent effects of matrix metalloproteinases 3, 7, 9
and 12 on atherosclerotic plaque stability in mouse brachiocephalic
arteries. Proc Natl Acad Sci USA. 102:pp. 15575–15580. 2005,
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reunanen N, Westermarck J, Häkkinen L,
Holmström TH, Elo I, Eriksson JE and Kähäri VM: Enhancement of
fibroblast collagenase (matrix metalloproteinase-1) gene expression
by ceramide is mediated by extracellular signal-regulated and
stress-activated protein kinase pathways. J Biol Chem.
273:5137–5145. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McCawley LJ, Li S, Wattenberg EV and
Hudson LG: Sustained activation of the mitogen-activated protein
kinase pathway. A mechanism underlying receptor tyrosine kinase
specificity for matrix metalloproteinase-9 induction and cell
migration. J Biol Chem. 274:4347–4353. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lai CS, Lee JH, Ho CT, Liu CB, Wang JM,
Wang YJ and Pan MH: Rosmanol potently inhibits
lipopolysaccharide-induced iNOS and COX-2 expression through
downregulating MAPK, NF-kappaB, STAT3 and C/EBP signaling pathways.
J Agric Food Chem. 57:10990–10998. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biol.
11:372–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wajant H, Pfizenmaier K and Scheurich P:
Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holvoet S, Vincent C, Schmitt D and Serres
M: The inhibition of MAPK pathway is correlated with
down-regulation of MMP-9 secretion induced by TNF-alpha in human
keratinocytes. Exp Cell Res. 290:108–119. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rhee JW, Lee KW, Kim D, Lee Y, Jeon OH,
Kwon HJ and Kim DS: NF-kappaB-dependent regulation of matrix
metalloproteinase-9 gene expression by lipopolysaccharidein a
macrophage cell line RAW 264.7. J Biochem Mol Biol. 40:88–94.
2007.PubMed/NCBI
|
|
24
|
Westermarck J and Kähäri VM: Regulation of
matrix metalloproteinase expression in tumor invasion. FASEB J.
13:781–792. 1999.PubMed/NCBI
|
|
25
|
Meisser A, Chardonnens D, Campana A and
Bischof P: Effects of tumour necrosis factor-alpha, interleukin-1
alpha, macrophage colony stimulating factor and transforming growth
factor beta on trophoblastic matrix metalloproteinases. Mol Hum
Reprod. 5:252–260. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Antithrombotic Trialists' Collaboration:
Collaborative meta-analysis of randomised trials of antiplatelet
therapy for prevention of death, myocardial infarction, and stroke
in high risk patients. BMJ. 324:71–86. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mazzone A, Venneri L and Berti S: Aortic
valve stenosis and coronary artery disease: Pathophysiological and
clinical links. J Cardiovasc Med (Hagerstown). 8:983–989. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hua Y, Xue J, Sun F, Zhu L and Xie M:
Aspirin inhibits MMP-2 and MMP-9 expressions and activities through
upregulation of PPARalpha/gamma and TIMP gene expressions in
ox-LDL-stimulated macrophages derived from human monocytes.
Pharmacology. 83:18–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tan JX, Shi W, Liao AJ, Peng AB and Zeng
B: Effect of Pyrrolidine Dithiocarbamate (PDTC) on NF-κB activity
of pancreatic acinar cells and blood inflammatory cytokines in
murine. Chin J Clin Gastroenterol. 19:147–149. 2007.
|
|
30
|
Cho A, Graves J and Reidy MA:
Mitogen-activated protein kinases mediate matrix
metalloproteinase-9 expression in vascular smooth muscle cells.
Arteriosclerosis, Thrombosis and Vascular Biology. 20:2527–2532.
2000. View Article : Google Scholar
|
|
31
|
Kim HH, Lee Y, Eun HC and Chung JH:
Eicosapentaenoic acid inhibits TNF-alpha-induced matrix
metalloproteinase-9 expression in human keratinocytes, HaCaT cells.
Biochem Biophys Res Commun. 368:343–349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moon SK, Cha BY and Kim CH: ERK1/2
mediates TNF-alpha-induced matrix metalloproteinase-9 expression in
human vascular smooth muscle cells via the regulation of NF-kappaB
and AP-1: Involvement of the ras dependent pathway. J Cell Physiol.
198:417–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cho A, Graves J and Reidy MA:
Mitogen-activated protein kinases mediate matrix
metalloproteinase-9 expression in vascular smooth muscle cells.
Arterioscler Thromb Vasc Biol. 20:2527–2532. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim KC and Lee CH: MAP kinase activation
is required for the MMP-9 induction by TNF-stimulation. Arch Pharm
Res. 28:1257–1262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ley K, Miller YI and Hedrick CC: Monocyte
and macrophage dynamics during atherogenesis. Arterioscler Thromb
Vasc Biol. 31:1506–1516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peters KD, Kochanek KD and Murphy SL:
Deaths: Final data for 1996. Natl Vital Stat Rep. 47:1–100.
1998.PubMed/NCBI
|
|
37
|
Cyrus T, Sung S, Zhao L, Funk CD, Tang S
and Praticò D: Effect of low-dose aspirin on vascular inflammation,
plaque stability, and atherogenesis in low-density lipoprotein
receptor-deficient mice. Circulation. 106:1282–1287. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yiqin Y, Meilin X, Jie X and Keping Z:
Aspirin inhibits MMP-2 and MMP-9 expression and activity through
PPARalpha/gamma and TIMP-1-mediated mechanisms in cultured mouse
celiac macrophages. Inflammation. 32:233–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Visse R and Nagase H: Matrix
metalloproteinases and tissue inhibitors of metalloproteinases:
Structure, function, and biochemistry. Circ Res. 92:827–839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chambers AF and Matrisian LM: Changing
views of the role of matrix metalloproteinases in metastasis. J
Natl Cancer Inst. 89:1260–1270. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Blankenberg S, Rupprecht HJ, Poirier O,
Bickel C, Smieja M, Hafner G, Meyer J, Cambien F and Tiret L:
AtheroGene Investigators: Plasma concentrations and genetic
variation of matrix metalloproteinase 9 and prognosis of patients
with cardiovascular disease. Circulation. 107:1579–1585. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wågsäter D, Zhu C, Björkegren J, Skogsberg
J and Eriksson P: MMP-2 and MMP-9 are prominent matrix
metalloproteinases during atherosclerosis development in the
Ldlr(−/-)Apob(100/100) mouse. Int J Mol Med. 28:247–253.
2011.PubMed/NCBI
|
|
44
|
Birkedal-Hansen H, Moore WG, Bodden MK,
Windsor LJ, Birkedal-Hansen B, DeCarlo A and Engler JA: Matrix
metalloproteinases: A review. Crit Rev Oral Biol Med. 4:197–250.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fabunmi RP, Baker AH, Murray EJ, Booth RF
and Newby AC: Divergent regulation by growth factors and cytokines
of 95 kDa and 72 kDa gelatinases and tissue inhibitors or
metalloproteinases-1, −2 and −3 in rabbit aortic smooth muscle
cells. Biochem J. 315:335–342. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Borden P and Heller RA: Transcriptional
control of matrix metalloproteinases and the tissue inhibitors of
matrix metalloproteinases. Crit Rev Eukaryot Gene Expr. 7:159–178.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bond M, Fabunmi RP, Baker AH and Newby AC:
Synergistic upregulation of metalloproteinase-9 by growth factors
and inflammatory cytokines: An absolute requirement for
transcription factor NF-kappa B. FEBS Lett. 435:29–34. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu L, Liu H, Peng J, Gan L, Shen L, Zhang
Q, Li L, Zhang L, Su C and Jiang Y: Regulations of the key
mediators in inflammation and atherosclerosis by aspirin in human
macrophages. Lipids Health Dis. 9:162010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Suh SJ, Cho KJ, Moon TC, Chang HW, Park YG
and Kim CH: 3,4,5-trihydroxybenzaldehyde from Geum japonicum has
dual inhibitory effect on matrix metalloproteinase 9; inhibition of
gelatinoytic activity as well as MMP-9 expression in TNF-alpha
induced HASMC. J Cell Biochem. 105:524–533. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cheng YW, Chang CY, Lin KL, Hu CM, Lin CH
and Kang JJ: Shikonin derivatives inhibited LPS-induced NOS in RAW
264.7 cells via downregulation of MAPK/NF-kappaB signaling. J
Ethnopharmacol. 120:264–271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu MJ, Wang L, Ding HY, Weng CY and Yen
JH: Glossogyne tenuifolia acts to inhibit inflammatory mediator
production in a macrophage cell line by downregulating LPS-induced
NF-kappa B. J Biomed Sci. 11:186–199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Himelstein BP, Lee EJ, Sato H, Seiki M and
Muschel RJ: Tumor cell contact mediated transcriptional activation
of the fibroblast matrix metalloproteinase-9 gene: Involvement of
multiple transcription factors including Ets and an alternating
purine-pyrimidine repeat. Clin Exp Metastasis. 16:169–177. 1998.
View Article : Google Scholar : PubMed/NCBI
|