Introduction
Adoptive immunotherapy is considered a promising
treatment for cancer patients (1).
Patients’ own immune cells are collected and induced ex vivo
to proliferate and differentiate into effector cells with increased
quantity and antitumor effects, and then re-administrated to the
patients via infusion. Effector cells prepared ex vivo for
infusion include non-specifically activated lymphocytes, including
natural killer (NK) cells (2),
cytokine-induced killer (CIK) cells (3), NKT cells, tumor antigen-specific T
cells, including chimeric antigen receptor-engineered T cells
(CAR-T) (4) and T cell receptor
engineered T cells (5). Although a
recent study has demonstrated the efficacy of CAR-T therapy in
treating hematologic malignancies, their effects on solid tumors
are far less known (6).
Adoptive non-specific immune effector cell infusion
has an important role in the treatment of a variety of solid tumor
types. NK cells (CD3−CD56+) are effectors of
innate immunity in peripheral blood, spleen, bone marrow,
intestine, liver and uterus (7).
They migrate to lymph nodes and secondary lymphoid organs to build
the first line of defense against invading pathogens as well as to
provide antitumor immune responses (8). Receptors on the NK cell surface
interact with ligands on tumor cells without restriction by the
major histocompatibility complex (MHC). NK cells recognize and kill
tumor cells, targeting them based on a reduced or absent expression
of human leukocyte antigen class I molecules (9).
CIK cells are generated ex vivo from
peripheral blood mononuclear cells (PBMCs) using anti-CD3
antibodies (OKT3) and various cytokines. Expanded CIK cells are a
heterogeneous lymphocyte population of
CD3+CD56+ NKT cells,
CD3+CD56− T lymphocytes, and a minority of
CD3−CD56+ NK cells (10). Under CIK culture conditions, expanded
CD3+CD56+ cells are derived from
CD3+CD56− T cells rather than
CD3−CD56+ NK cells. The majority of the
CD3+CD56+ cells co-express CD8 but not CD4,
which is consistent with the high level of effector CD8+
T cell cytotoxic activity (11). CIK
cells differ from NK cells in that they do not mediate
antibody-dependent cell-mediated cytotoxicity (ADCC). Alternating
infusions of CIK and NK cells provide an enhanced synergistic
antitumor immunity compared to adoptive immunotherapy with CIK
cells alone (12). Innate immune
cells function to support adaptive immune responses by enhanced
direct tumor cell cytolysis and optimal antitumor T-cell activity
(13).
Within the current regulatory paradigm, clinical
translation of adoptive immunotherapy requires good manufacturing
practice (GMP)-compliant processes to produce clinically relevant
quantities of antitumor immune effectors. In this respect,
clinical-grade CIK cells may be expanded ex vivo under
relatively simple and low-cost GMP-compliant culture conditions,
which offer important advantages over other cell therapy products,
including NK cells, tumor-infiltrating lymphocytes and CAR-T. The
major challenge with NK cell immunotherapy has been to obtain large
quantities of NK cells with high purity. At present, the source of
precursor cells, the collection methods, quality control and
evaluation of treatment outcomes vary among laboratories (14). Certain protocols rely on the use of
feeder cells to promote the proliferation of NK cells (15–18).
However, these methods may be restricted by GMP guidelines, which
hinder the clinical application of NK cells in immunotherapy
(19).
Trastuzumab (TTZ, Herceptin®) is a human
anti-HER-2 monoclonal antibody used for treating breast cancer,
metastatic gastric adenocarcinoma and adenocarcinoma of the
gastroesophageal junction (20). TTZ
bears two antigen-specific sites that bind to the extracellular
domain of the HER2 receptor and that prevent the activation of its
intracellular tyrosine kinase (21).
The remainder of the antibody is human immunoglobulin (Ig)G with a
conserved fragment crystallizable (Fc) portion. Preclinical studies
using models suggested the contribution of ADCC to the therapeutic
benefit of TTZ. ADCC occurs when antibodies bind to antigens on
tumor cells and the Fc domains of the antibody recruit Fc
receptor-bearing effector cells, including NK cells and macrophages
(22). Manipulations of the Fc
domain structure may optimize antibody clearance and the
interaction of Fc domains with cellular Fc receptors (23). Furthermore, immobilization of TTZ
increased the number of PBMCs from 5 healthy donors after 48 h of
culture, which indicated that this technique is effective for
culturing cells for immunotherapy (24).
The present study investigated the proliferation of
PBMCs derived from 22 healthy donors, their expression of
activating and inhibitory receptors and the cytotoxicity in
effector cell culture using large-scale ex vivo
GMP-compliant culture systems based on anti-HER-2 antibody in order
to optimize the culture conditions for clinical-grade effector
cells.
Materials and methods
Study design
Following the attainment of informed consent, fresh
heparinized peripheral blood samples were collected from 22 healthy
donors (13 males and 9 females; median age, 34.5 years; age range,
22–68 years). PBMCs were isolated by density gradient
centrifugation in lymphocyte separation medium (GE Healthcare,
Little Chalfont, UK) according to the manufacturer's protocol. The
PBMCs from all 22 healthy donors were cultured in one of the
expansion systems listed below. Cell proliferation, phenotype,
expression of NK cell receptors and cytotoxicity against target
cells were compared between the two systems.
Effector cell expansion system
In brief, PBMCs were cultured in effector cell
expansion systems (system A or system B). Cells were suspended in
20 ml ALyS505NK-AC culture media (Center for Stem Cell Therapeutics
and Imaging, Inc., Sendai, Japan) containing 1,000 IU/ml
interleukin 2 (IL-2; Sansheng, Jiangyin, China) and 1% autologous
heat-inactivated plasma. All cells were then inoculated into
75-cm2 culture flasks coated with 0.48 mg/ml anti-HER2
monoclonal antibody (GTIN: 07640149610680; Genentech Inc., Roche,
San Francisco, CA, USA) at a concentration of 2×106
cells/ml and incubated for 5 days. The modification of system B was
the addition of anti-CD3 antibody (cat. no. 170-076-116; Miltenyi
Biotec, Bergisch Gladbach, Germany) to the media at a concentration
of 50 ng/ml on day 1. In the two systems, cells and media were
transferred to a gas-permeable cell culture bag (Nipro Corp.,
Osaka, Japan) with 200 ml ALyS505NK-EX culture media containing
1,000 IU/ml IL-2 on day 5 and cultured for another 8 days. The
media were changed every 2–3 days.
Flow cytometry
Aliquots of cells were incubated for 15 min at room
temperature (20–25°C) in the dark with fluorochrome-conjugated mAb
for phenotypic analysis. CD4 fluorescein isothiocyanate (FITC)/CD8
phycoerythrin (PE)/CD3 peridinin chlorophyll (PerCp) (cat. no.
340298; 1:5), CD56 allophycocyanin (APC) (cat. no. 341025; 1:20),
CD3 FITC (cat. no. 555339; 1:5), CD8 PerCP-Cy™5.5 (cat. no. 560662;
1:20), CD94/NKG2C PE (cat. no. 555889; 1:5), CD158a PE (cat. no.
556063; 1:5), CD158b PE (cat. no. 559785; 1:5) and CD335/NKP46 PE
(cat. no. 557991; 1:5) were obtained from BD Biosciences (San Jose,
CA, USA), and CD314/NK group 2, member D (NKG2D) APC (cat. no.
FAB139P; 1:10) from R&D Systems (Minneapolis, MN, USA).
Isotypes of the equivalent antibodies were used as negative
controls and incubated for 15 min at room temperature in the dark.
FastImmune™ Control γ1/γ1/CD3 (cat. no. 340369; 1:5), PE Mouse IgM,
κ Isotype Control (cat. no. 555584; 1:5), PerCP-Cy™ 5.5 Mouse IgG1,
κ Isotype Control (cat. no. 550795; 1:5), PE Mouse IgG1, κ Isotype
Control (cat. no. 555749; 1:5), FITC Mouse IgG2a, κ Isotype Control
(cat. no. 555573; 1:5), PE Mouse IgG2b, κ Isotype Control (cat. no.
555743; 1:5), APC Mouse IgG2b, κ Isotype Control (cat. no. 555745;
1:5) were obtained from BD Biosciences with the exception of mouse
IgG1 PE-conjugated antibody (cat. no. IC002P; 1:10; R&D
Systems). Samples were measured with a flow cytometer (FACSCanto
II; BD Biosciences). Analysis of flow cytometric data was performed
using of BD FACSDiva software version 6.1.2 (BD Biosciences).
Cytotoxicity assay
Cytotoxicity was determined using a
CytoTox96® Non-Radioactive Cytotoxicity Assay (Promega
Corp., Madison, WI, USA) based on the exocytic release of lactate
dehydrogenase (LDH), according to the manufacturer's protocol. On
day 14, the cells were harvested, washed, counted and added to
target cells (K562 cells, ATCC® CCL-243™; American Type
Culture Collection, Manassas, VA, USA) at a ratio of 20:1 or 10:1
in 96-well, round-bottomed plates (Falcon; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in triplicates and incubated
for 4 h at 37°C in a humidified atmosphere containing 5%
CO2. Plates were then centrifuged for 3 min at 200 × g
and the supernatant from each well was transferred to a 96-well
flat-bottomed plate. The LDH levels in the supernatant were
quantified according to the absorbance at 492 nm (Thermo Multiskan
MK3; Thermo Fisher Scientific, Inc.). The percentage of LDH
released reflected the antitumor activities of the effector cells.
The cytotoxicity percentage was calculated as follows: Cytotoxicity
(%) = [(experimental release-spontaneous release of effector
cells-spontaneous release of target cells)/(maximal release of
target cells-spontaneous release of target cells)] ×100%.
Re-stimulation of CD8+ T
cells and NK cells with soluble anti-CD3 and IL-2
CD8+ T cells and NK cells were purified
from the cultured cells harvested on day 9 from system B using a
human CD8+ T cell enrichment kit (cat. no. 19053;
Stemcell Technologies, Inc., Vancouver, BC, Canada) and a human NK
cell enrichment kit (cat. no. 19055; Stemcell Technologies, Inc.),
respectively. Purified CD8+ T cells and NK cells were
either cultured with medium alone or re-stimulated with IL-2 (1,000
IU/ml), or with anti-CD3 (50 ng/ml) and IL-2 (1,000 IU/ml) for 24 h
prior to being harvested for western blot analysis.
Western blot analysis
Cells were harvested and washed twice with PBS. The
cell pellets were lysed with sample buffer (62.5 mM Tris-HCl, 10%
glycerol, 10% (w/v) SDS, 1 mM phenylmethylsulfonyl fluoride and 50
mM dithiothreitol) and sonicated for 6 min on ice to shear DNA and
reduce sample viscosity. Cell lysates were cleared by
centrifugation at 9600 × g for 5 min at 4°C. The protein
concentration was measured by means of a bicinchoninic acid protein
assay reagent kit (Pierce; Thermo Fisher Scientific, Inc.) to
ensure equal loading. Lysates (30 µg/lane) were subjected to 6–12%
SDS-PAGE gel and transferred to a nitrocellulose membrane. Blots
were blocked with 5% non-fat milk in Tris-buffered saline
containing 0.1% Tween-20 (TBST) for 2 h at room temperature. The
membranes were incubated overnight at 4°C with different primary
antibodies. Akt (cat. no. 4691; 1:1,000), p-Akt (Ser473; cat. no.
9271; 1:1,000), CDK2 (cat. no. 2546S; 1:1,000), p-CDK2 (Thr160;
cat. no. 2561S; 1:1,000), JAK (cat. no. 3344; 1:1,000), PI3K p85
(cat. no. 4257; 1:1,000), p-PI3K [p85 (Tyr458)/p55 (Tyr199); cat.
no. 4228; 1:1,000], p-P70S6K (Thr421/Ser424; cat. no. 9204;
1:1,000), S6 (cat. no. 2217; 1:1,000), P-S6 (Ser235/236; cat. no.
2211; 1:1,000), p-S6 (Ser240/244; cat. no. 2215; 1:1,000), STAT3
(cat. no. 4904; 1:1,000), p-STAT3 (Tyr705; cat. no. 9131; 1:1,000),
STAT5 (cat. no. 9358; 1:1,000) and p-STAT5 (Tyr694; cat. no. 9351;
1:1,000) were obtained from Cell Signaling Technologies, Inc.
(Danvers, MA, USA). Cyclin D3 (cat. no. sc-182; 1:2,00), cyclin B1
(cat. no. sc-752; 1:5,00) and p70S6Kα (cat. no. sc-230; 1:5,00)
were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). p-p70S6K (Thr389; cat. no. MA5-15117, 1:1,000) was obtained
from Thermo Fisher Scientific Inc. and GAPDH (cat. no. G9545;
1:60,000) was obtained from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany). Following three washes in TBST, the membranes were
incubated with horseradish peroxidase-conjugated goat anti-rabbit
IgG (heavy and light chains; cat. no. 31460; 1:60,000; Pierce;
Thermo Fisher Scientific, Inc.) at room temperature for 2 h.
Reactive proteins were detected with LumiGLO™ reagent (Cell
Signaling Technologies, Inc.) and immunoblots were imaged on X-ray
film (cat. no. 4741008378; Fujifilm Holdings Corporation, Tokyo,
Japan).
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Differences between groups were assessed for statistical
significance by the Mann-Whitney test or paired Student's t-test
depending on the distribution of the data. One-way analysis of
variance and Tukey-Kramer multiple comparisons post hoc test were
applied when comparing more than two groups. SPSS software (version
22; IBM Corp., Armonk, NY, USA) was used for all statistical
analyses. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expansion and immunophenotypic
characterization of effector cells
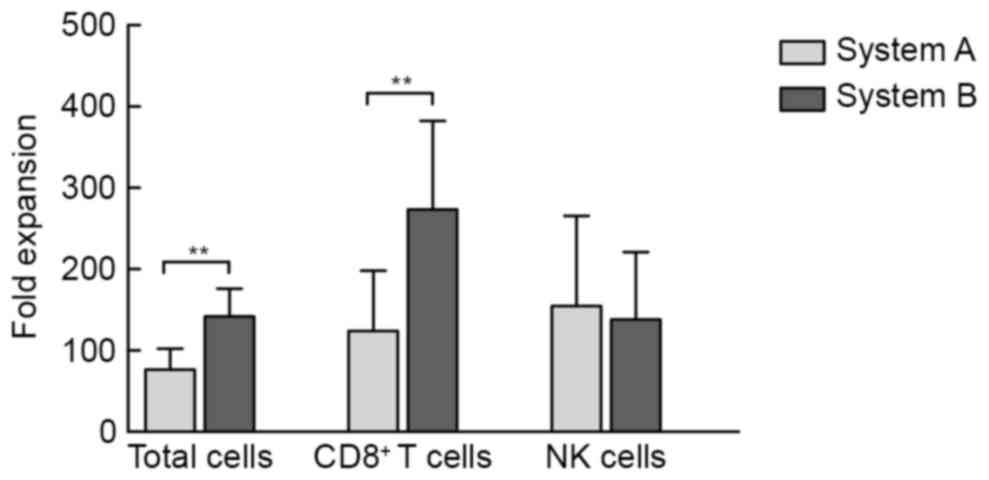
At 14 days after induction, the median expansion of
the cultured products from 22 healthy donors was 80-fold (range,
18.5 to 125-fold) and 147.50-fold (range, 64.0- to 205-fold) in
system A and system B, respectively. The mean expansion fold of
total cells as well as CD8+ T cells in system B was
significantly greater than that in system A (P<0.01; Fig. 1).
Additional phenotypic analysis revealed that the
percentages of CD3+ T cells,
CD3+/CD8+ T cells and
CD3+/CD4+ T cells in the final products of
system B were significantly higher than those of system A. By
contrast, the percentages of NK (CD3−/CD56+)
and NKT (CD3+/CD56+) cells in system B were
significantly lower than those expanded in system A (Table I).
 | Table I.Phenotype analysis (%) of the final
cell products in different culture systems. |
Table I.
Phenotype analysis (%) of the final
cell products in different culture systems.
| System |
CD3+ |
CD3+CD8+ |
CD3+CD4+ |
CD3+CD56+ |
CD3−CD56+ |
|---|
| A | 66.2±20.6 | 46.1±21.6 | 9.6±7.2 | 25.2±19.3 | 31.2±20.5 |
| B |
81.9±13.4a |
52.8±13.0b |
26.9±15.3a |
16.5±11.3a |
16.9±13.3a |
Expression of cell surface receptors
on NK and NKT cells
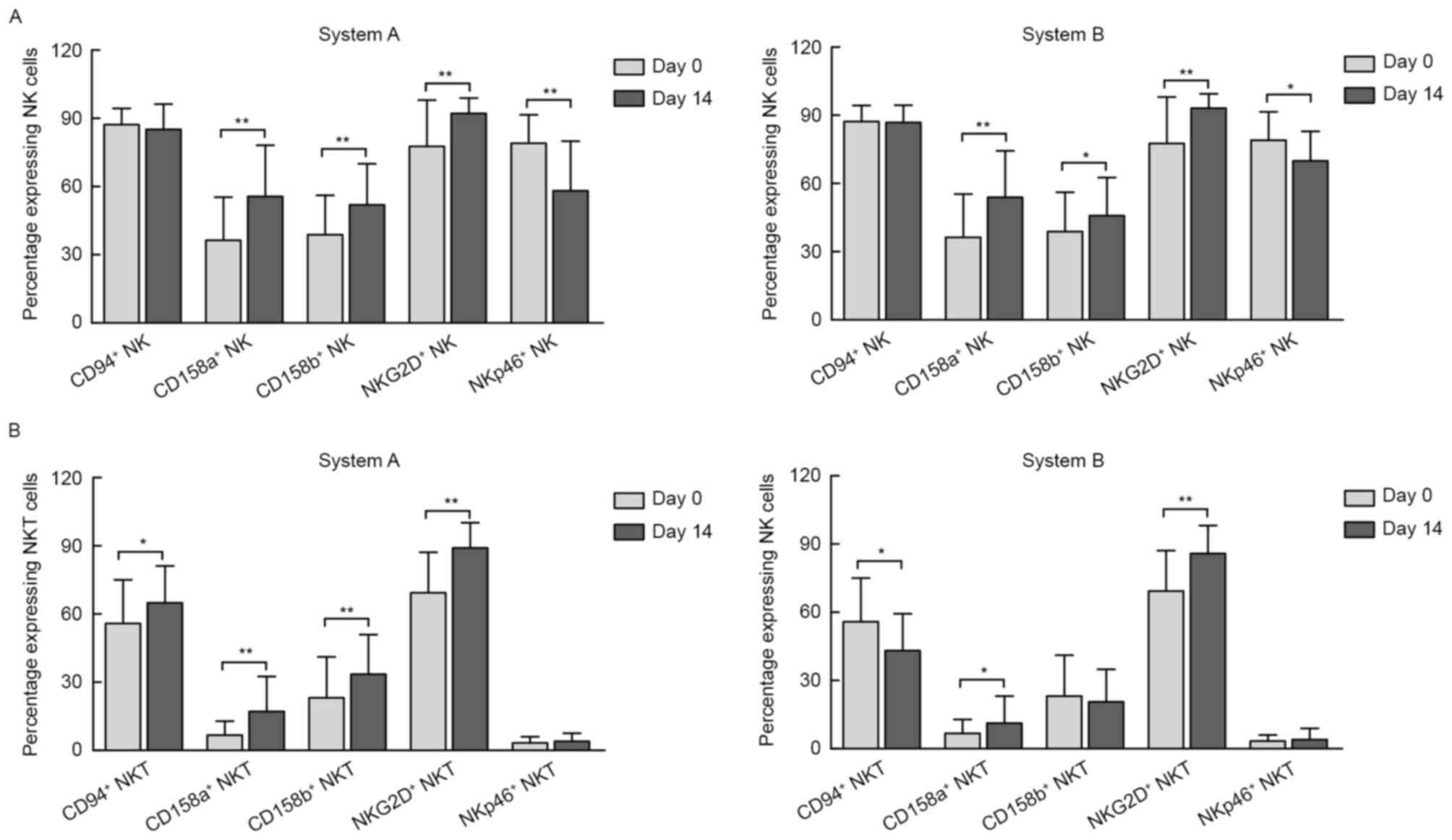
In the present study, three-color fluorescence flow
cytometry was used to analyze the phenotyping difference induced in
system A and B by verifying the expression of activating and
inhibitory NK cell surface-associated receptors on NK cells
(Fig. 2A) and NKT cells (Fig. 2B).
The results demonstrated that, compared with the
pre-culture conditions, the percentages of NK cells expressing
NKG2D, an NK-activating receptor, had significantly increased in
the two systems by day 14. By contrast, the percentage of NK cells
expressing NKp46, another NK-activating receptor, was significantly
decreased. However, the expression of the inhibitory receptors
CD158a and CD158b also increased post-induction in the two systems,
as presented in Fig. 2A. Of note,
compared with baseline, the expression of CD94/NKG2A, an inhibitory
receptor, was not changed on day 14 in either of the two
systems.
The expression of these NK-activating/inhibitory
receptors was also investigated in the subgroup of NKT cells. Of
note, although the percentages of NKG2D+ NKT cells
increased as they did in NK cells, there was no marked change in
the percentage of NKT cells expressing NKp46. In addition, the
percentages of NKT cells expressing CD94/NKG2A on day 14 were
opposite in the two systems [from 56±19 to 65±16% in system A and
from 56±19 to 43±16% in system B (P<0.01 for each; Fig. 2B)]. Furthermore, the expression of
CD158a increased in the two systems, while CD158b increased in
system A only (Fig. 2B).
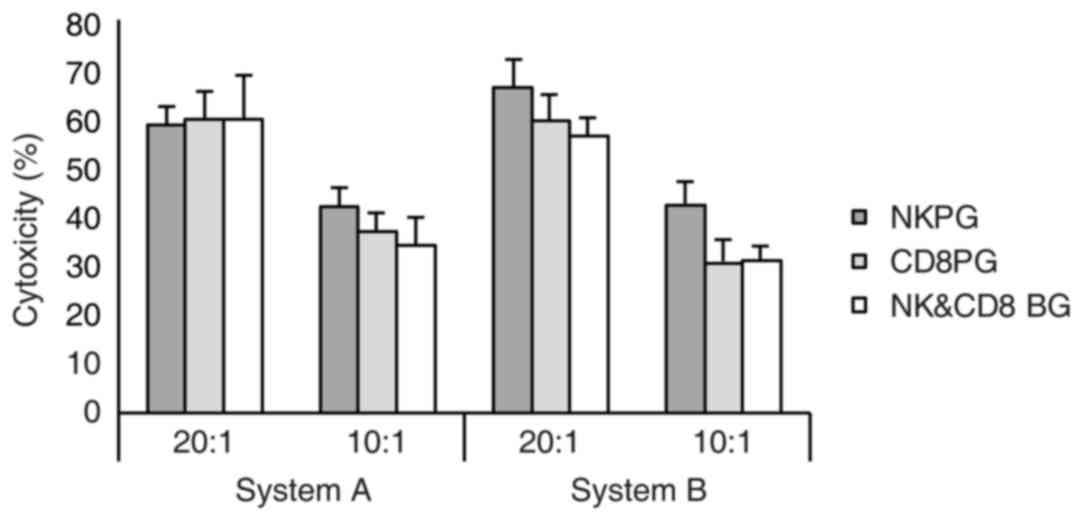
Cytotoxicity of expanded effector
cells against the K562 cell line
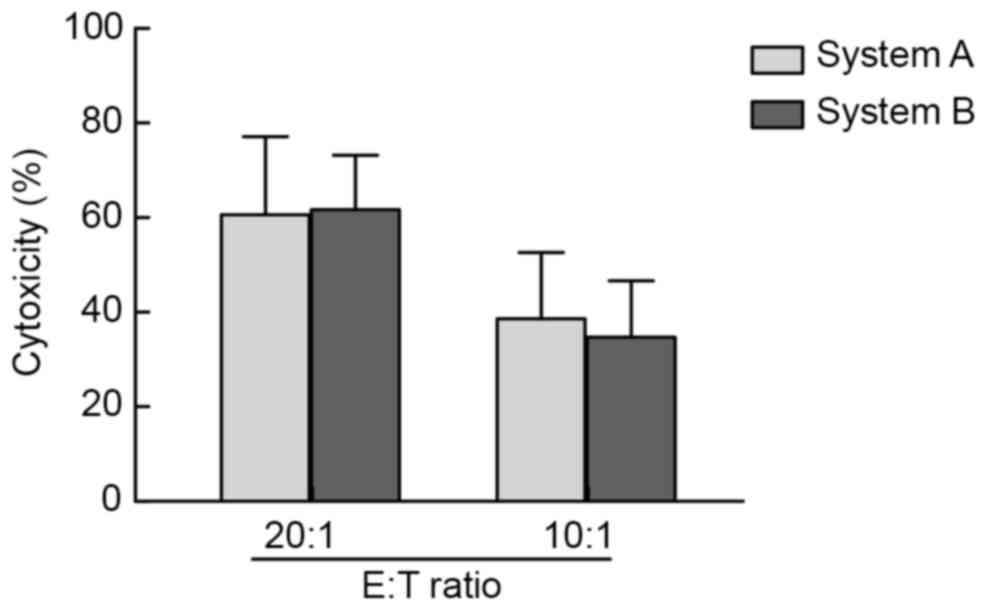
In the present study, K562 cells were used as target
cells and an LDH assay was applied to verify the in vitro
cytotoxicity on effector cells post-expansion in systems A and B.
When the ratio of effector cells vs. target cells was 20:1 and
10:1, strong cytotoxicity of effector cells against target cells
was detected in the two systems, however, there was no significant
difference between the two systems (Fig.
3).
Signaling pathways involved in cell
proliferation and differentiation
In a previous study, immobilized TTZ was revealed to
be effective in enhancing the growth of CD3-LAK cells and
increasing the numbers of NK cells and γδ+ T cells
(18). According to these published
results, the present study established two large-scale ex
vivo GMP-compliant culture systems based on immobilized TTZ and
revealed that the addition of OKT3 in system B increased the
expansion folds of total cells as well as CD8+ T cells
compared with those in system A. However, there was no significant
difference in the expansion folds of NK cells between the two
systems (Fig. 1).
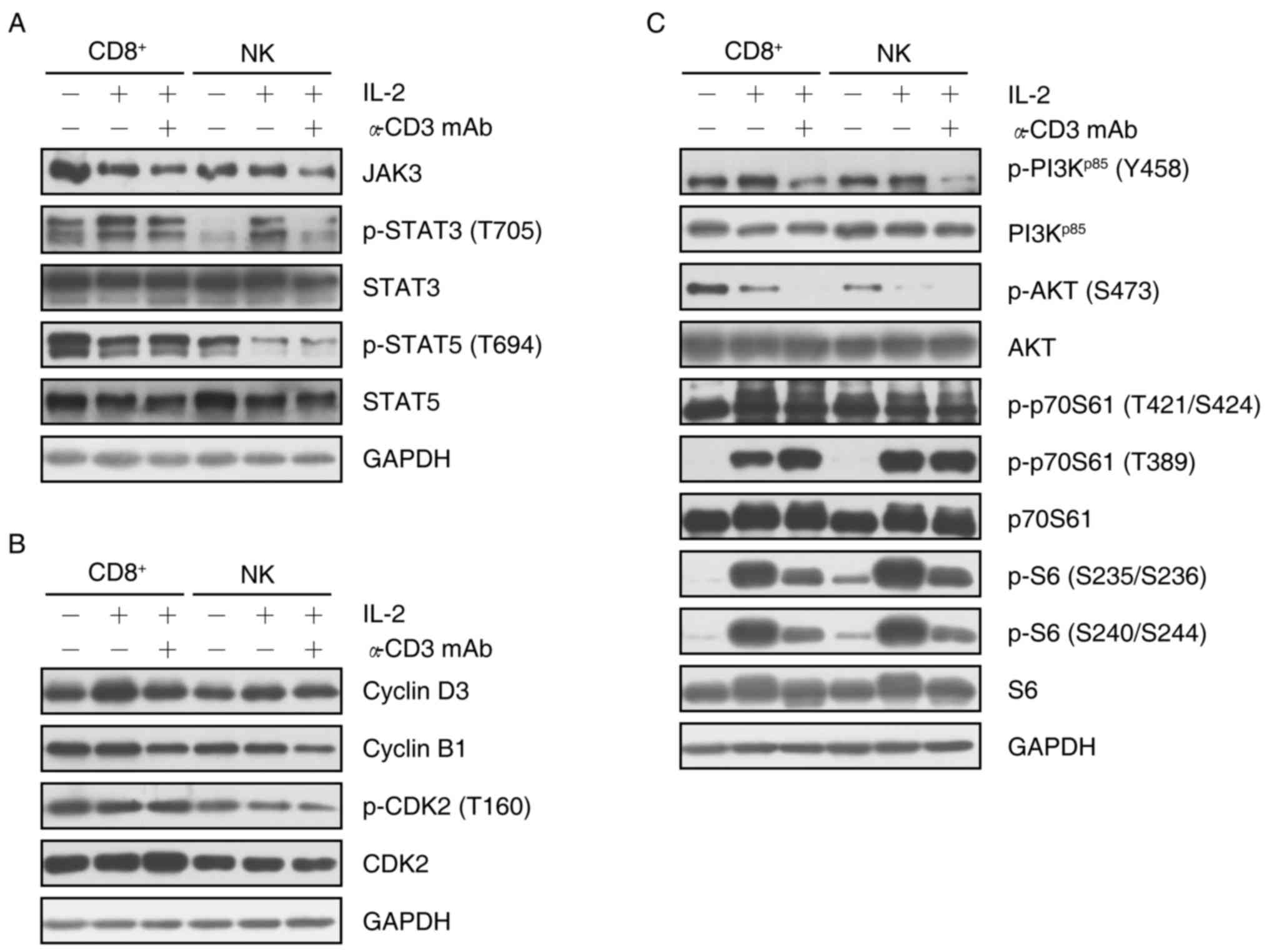
To explore different mechanisms involved in the
proliferation of specific cell subsets, the present study analyzed
the relevant protein expression and phosphorylation in the Janus
kinase (JAK)/signal transducer and activator of transcription
(STAT) and phosphoinositide-3 kinase (PI3K)/AKT/p70S6K1 signaling
pathways in isolated CD8+ T cells or NK cells
re-stimulated by IL-2 or a combination of IL-2 and OKT3. The
results demonstrated that IL-2 re-stimulation decreased the
expression of JAK3 in CD8+ T cells, and subsequent
incubation with OKT3 enhanced this decrease. Furthermore, the
combination of IL-2 and OKT3 decreased the expression of JAK3 in NK
cells. Of note, although the expression of STAT3 phosphorylated at
T705 was enhanced by IL-2, additional treatment with OKT3 reversed
this increase in CD8+ T cells and NK cells. However, the
expression of STAT3 did not show any change with IL-2 or, IL-2 and
OKT3 in CD8+ T cells and NK cells. Notably, although
IL-2 reduced STAT5 expression, and phosphorylation at T694 in
CD8+ T cells and NK cells, no marked change was observed
following addition of anti-CD3 mAb. These results indicated that
IL-2 stimulated transcription by downregulating the expression of
JAK3 in CD8+ T cells, and the phosphorylation of STAT5
at T694 in CD8+ T cells and NK cells, while upregulating
of the phosphorylation of STAT3 at T705. Furthermore, OKT3 was
involved in regulating transcription primarily through inhibiting
the JAK3/STAT3 (T705) pathway (Fig.
4A).
The proliferation of CD8+ T cells and NK
cells appeared to be associated with cyclin D3 and cyclin B1, since
the expression of cyclin D3 was markedly upregulated by IL-2
re-stimulation, while the expression of cyclin B1 was downregulated
following addition of OKT3. Although cyclin D3 is a target of the
cyclin E/cyclin-dependent kinase (CDK)2 complex via p27Kip1
(25), the proliferation was
possibly not dependent on the cyclin E/CDK2 complex, since the
expression of p-CDK2 (T160) did not exhibit any change with IL-2 in
CD8+ T cells and NK cells. However, the combination of
IL-2 and OKT3 decreased the phosphorylation of CDK2 at T160 in NK
cells (Fig. 4B).
The regulation of protein translation was also
investigated via the PI3K/AKT/p70S6K1/S6 axis. IL-2 increased the
phosphorylation of PI3Kp85 at Y458 and decreased the
phosphorylation of AKT at S473. Similarly, the phosphorylation of
p70S6K1 at T389 and S6 at S235/S236 and S240/S244 was upregulated
by IL-2 re-stimulation. Of note, additional incubation with OKT3
reversed the upregulation of the phosphorylation on PI3K p85 and
S6, but reduced the phosphorylation of AKT at S473. Notably, the
addition of OKT3 enhanced the phosphorylation of p70S6K1 at T389 in
CD8+ T cells but not NK cells (Fig. 4C).
These results indicated that during the process of
proliferation and differentiation of PBMCs, apart from regulating
the cell cycle, OKT3 was typically involved in the regulation of
transcription and protein translation.
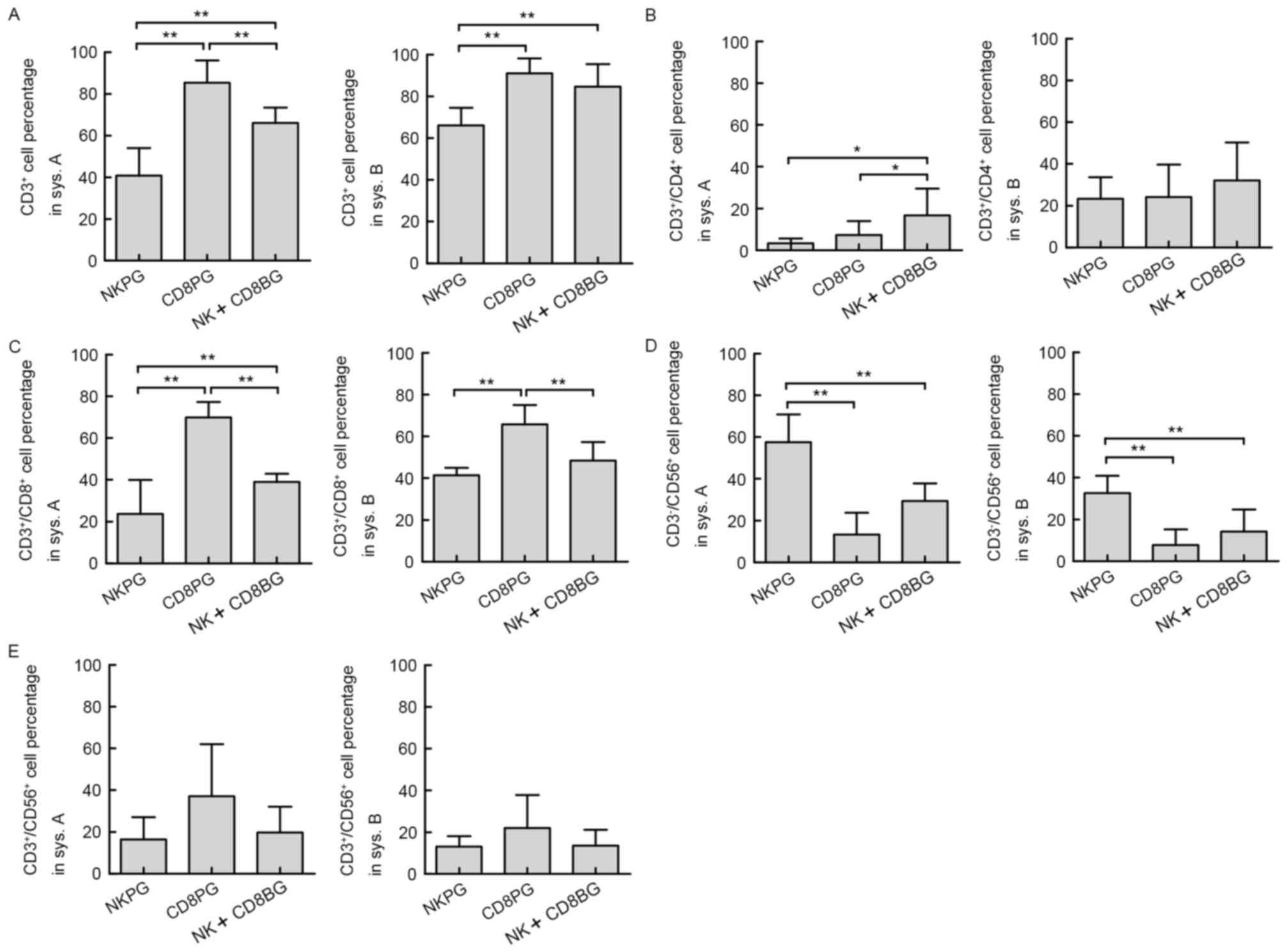
Subgroup analysis
Of notable interest was the finding that the main
effector cell subsets varied amongst cell cultures from different
individuals even in the same culture system. Accordingly, the 22
healthy donors were divided into three groups based on the results
of the cell phenotype analysis of the final product in system A: NK
cell-predominant growth group (NKPG, proportion of NK cells ≥45%,
n=6); CD8+ cell-predominant growth group (CD8PG,
proportion of CD3+/CD8+ T cells ≥50%, n=8);
and NK and CD8+ T cell-balanced growth group (NK+CD8BG,
proportion of NK cells <45% and the proportion of
CD3+/CD8+ T cells <50%, n=8). The subsets
of effector cells (CD3+,
CD3+/CD4+, CD3+/CD8+,
CD3−/CD56+ and
CD3+/CD56+) in different subgroups in system
A and system B were then compared. The results demonstrated that
the percentage of total effector T cells (CD3+) in the
CD8PG group was significantly higher than that in the NKPG group in
system A as well as in system B (Fig.
5A). Although in system B, no significant difference was
present between the CD8PG and CD8+NKBG groups, the percentage of
CD3+ cells in the CD8+NKBG group was still significantly
greater than that in NKPG group in system A. The percentage of
CD3+/CD4+ cells did not exhibit any
difference between different subgroups in system B, although in
system A, it was higher in the CD8+NKBG group compared with that in
the NKPG and CD8PG groups (Fig. 5B).
Differences in the percentages of CD3+/CD8+
and CD3−/CD56+ cells were consistent between
the two systems, since in the CD8PG group,
CD3+/CD8+ cells were significantly enhanced
compared with those in the other two groups (Fig. 5C), while
CD3−/CD56+ cells predominated in the NKPG
group (Fig. 5D). Furthermore, no
difference was observed in the percentage of NKT
(CD3+/CD56+) cells between different
subgroups in either system (Fig.
5E). These results assured that the major subset of effector
cells expanded from different subgroups was consistent between the
two culture systems.
Additional comparison of the expression of
activating (NKG2D+ and NKp46+) and inhibitory
(CD94+, CD158a+ and CD158b+) NK
cell surface-associated receptors on the NK and NKT cells derived
from different individuals indicated that no significant difference
was present among the three groups following the expansion in
systems A and B (data not shown). Similarly, no substantial
difference in cytotoxicity of the expanded effector cells was
identified among the three subgroups in either system (Fig. 6).
Discussion
Large quantities of activated effector cells are
required for successful adoptive immunotherapy. The present study
aimed to optimize the ex vivo expansion systems for
clinical-grade effector cells. Two large-scale ex vivo
effector cell culture systems were established, in which PBMCs
derived from healthy donors were stimulated with HER-2 antibody or
co-stimulated with HER-2 antibody and anti-CD3 antibody. It was
revealed that HER-2 antibody stimulus promoted the preferential
proliferation of NK cells. It is known that NK cells recognize
tumor cells through Fcγ receptors combining with Fcγ of TTZ on
tumor cell surfaces, and then kill the tumor cells via ADCC effects
(26–30). A TTZ coating on the surface of
culture flasks may recruit NK cells from PBMCs via Fcγ to activate
NK cells and promote NK cell proliferation. This effect may be
similar to the role of feeder cells. OKT3 has been commonly used in
PBMCs to induce the proliferation of NK cells, suggesting that OKT3
does not only induce T-cell proliferation but also promotes T-cell
generating cytokines that support NK cell proliferation (31). However, in the present study, OKT3
co-stimulation evidently enhanced the expansion of the total cells,
but primarily induced PBMCs to differentiate into CD3+ T
cells, as the proportion of CD3+CD8+ and
CD3+CD4+ T cells was higher in the HER-2 and
anti-CD3 antibody co-stimulation system.
The signaling pathways involved in the proliferation
and differentiation of PBMCs induced by OKT3 co-stimulation were
investigated. A previous study demonstrated that after 168 h of
induced activation of PMBCs via anti-CD3 monoclonal antibody, they
responded to re-stimulation by anti-CD3 monoclonal antibody. IL-2
and IL-2Rβγ are known combine to activate JAK, thereby activating
the three downstream signaling pathways STAT, PI3K-AKT and MAPK
(32). The present study indicated
that IL-2 stimulation promoted the proliferation of CD8+
T cells and NK cells via the PI3K/p70S6K1 signaling pathway, while
the stimulation effect of anti-CD3 monoclonal antibody on cell
proliferation was relatively weak. Therefore, anti-CD3 monoclonal
antibody may primarily serve as an inducer of cell differentiation.
Stimulation by addition of OKT3 may enhance total cell
proliferation through stimulating the secretion of
growth-associated cytokines.
The cytotoxicity of NK cells and NKT cells is
determined by the balance of signals from activating and inhibitory
receptors on the effector cell surface. NKG2D was first reported to
be an activated receptor of NK cells. It is not only expressed on
the NK cell surface, but also on NKT-cells and γδ+ T
cell subsets (33–36). In vitro studies have
demonstrated that binding of NKG2D and its ligands provides signals
for NK cell activation and co-stimulatory signals for T-cells
(35,37,38).
Through the binding of MHC class I chain-related molecule (MIC)A,
MICB or human cytomegalovirus glycoprotein UL16 binding proteins to
NKG2D, the receptor recognizes tumor cells and secretes perforin
and granzymes to kill tumor cells (39–44). The
results of the present study indicated that although the effector
cell phenotypes of PBMCs derived from different individuals varied
in systems A and B, the expression of a variety of activating and
inhibitory receptors of harvested NK and NKT cells were not
different based on which system was used. Despite the different
phenotypic characteristics, effector cells generated in the two
culture systems had similarly strong cytotoxicity against tumor
cells, which may be interpreted in the light of the results of
in vivo and in vitro studies reporting that NK cells
continuously promoted the response of CD8+ T cells
(45,46). A clinical study also confirmed that a
high proportion of CD3+/CD8+ T cell subsets
in CIK cell transfusion was associated with improved the overall
survival rate in patients with hepatocellular carcinoma, lung
cancer and colorectal cancer (47).
The present study revealed that PBMCs derived from
different individuals exhibited three distinct proliferation and
differentiation patterns under the same culture conditions. This
phenomenon was reported in a previous study demonstrating that
PBMCs derived from healthy donors had a high proportion of NK cells
and a relatively low proportion of CD3+ T cell following
the in vitro expansion; however, no other subgroup analysis
was performed (31). It was reported
that the number of harvested NK cells in the culture was associated
with the number of NK cells in the peripheral blood sample
originally drawn (48). By contrast,
the proportion of NK cell subsets in the NKPG group prior to in
vitro culture was not significantly different from that in the
other two groups of the present study. Thus, the significant NK
cell proliferation in the NKPG group was unlikely to have been
caused by the high proportion of NK cell subsets in PBMCs prior to
the in vitro culture, but may have been associated with the
genetic background of the population, such as the expression of NK
cell surface Fcγ receptors.
In conclusion, the two in vitro GMP-compliant
culture systems used in the present study effectively induced the
activation, proliferation and differentiation of immune effector
cells. The method of TTZ immobilization was easy and safe to
operate without the requirement of feeder cells to induce NK cell
expansion from unselected PBMCs. Addition of OKT3 promoted the
total cell proliferation and primarily induced the PBMC to
differentiate into CD3+ T cells. Thus, the present study
provided GMP-compliant methods for the generation of large-scale
clinical-grade effector cells for adoptive immunotherapy. However,
the composition of produced effector cells varied largely among the
donors, which must be taken into consideration when a high purity
of specific cell subset is required for treatment. The mechanisms
underlying variations among donors require additional
investigation.
Glossary
Abbreviations
Abbreviations:
|
GMP
|
good manufacturing practice
|
|
PBMC
|
peripheral blood mononuclear cells
|
|
IL
|
interleukin
|
|
NK
|
natural killer
|
|
CIK
|
cytokine-induced killer
|
|
CAR-T
|
chimeric antigen receptor-engineered T
cells
|
|
MHC
|
major histocompatibility complex
|
|
HLA-I
|
human leukocyte antigen class I
|
|
ADCC
|
antibody-dependent cell-mediated
cytotoxicity
|
|
TTZ
|
trastuzumab
|
|
LDH
|
lactate dehydrogenase
|
|
NKPG
|
NK cell-predominant phenotyping
group
|
|
CD8PG
|
CD8+ cell-predominant
phenotyping group
|
|
NK + CD8BG
|
NK and CD8+ T cell-balanced
phenotyping group
|
References
|
1
|
Ruella M and Kalos M: Adoptive
immunotherapy for cancer. Immunol Rev. 257:14–38. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shimasaki N, Coustan-Smith E, Kamiya T and
Campana D: Expanded and armed natural killer cells for cancer
treatment. Cytotherapy. 18:1422–1434. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Introna M: CIK as therapeutic agents
against tumors. J Autoimmun. Jul 2–2017.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bollino D and Webb TJ: Chimeric antigen
receptor-engineered natural killer and natural killer T cells for
cancer immunotherapy. Transl Res. 187:32–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ping Y, Liu C and Zhang Y: T-cell
receptor-engineered T cells for cancer treatment: Current status
and future directions. Protein Cell. Jan 20–2017.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang
YS, Wang W and Wei YQ: Hurdles of CAR-T cell-based cancer
immunotherapy directed against solid tumors. Sci China Life Sci.
59:340–348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi FD, Ljunggren HG, La Cava A and Van
Kaer L: Organ-specific features of natural killer cells. Nat Rev
Immunol. 11:658–671. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Konjević G, Mirjacić Martinović K, Vuletić
A, Jurisić V and Spuzić I: Distribution of several activating and
inhibitory receptors on CD3-CD16+ NK cells and their correlation
with NK cell function in healthy individuals. J Membr Biol.
230:113–123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caligiuri MA: Human natural killer cells.
Blood. 112:461–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang J, Wu C and Lu B: Cytokine-induced
killer cells promote antitumor immunity. J Transl Med. 11:832013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmidt-Wolf IG, Lefterova P, Mehta BA,
Fernandez LP, Huhn D, Blume KG, Weissman IL and Negrin RS:
Phenotypic characterization and identification of effector cells
involved in tumor cell recognition of cytokine-induced killer
cells. Exp Hematol. 21:1673–1679. 1993.PubMed/NCBI
|
|
12
|
Lu PH and Negrin RS: A novel population of
expanded human CD3+CD56+ cells derived from T cells with potent in
vivo antitumor activity in mice with severe combined
immunodeficiency. J Immunol. 153:1687–1696. 1994.PubMed/NCBI
|
|
13
|
Maniar A, Zhang X, Lin W, Gastman BR,
Pauza CD, Strome SE and Chapoval AI: Human gammadelta T lymphocytes
induce robust NK cell-mediated antitumor cytotoxicity through CD137
engagement. Blood. 116:1726–1733. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pittari G, Filippini P, Gentilcore G,
Grivel JC and Rutella S: Revving up natural killer cells and
cytokine-induced killer cells against hematological Malignancies.
Front Immunol. 6:2302015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Berg M, Lundqvist A, McCoy P Jr..Samsel L,
Fan Y, Tawab A and Childs R: Clinical-grade ex vivo-expanded human
natural killer cells up-regulate activating receptors and death
receptor ligands and have enhanced cytolytic activity against tumor
cells. Cytotherapy. 11:341–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fujisaki H, Kakuda H, Shimasaki N, Imai C,
Ma J, Lockey T, Eldridge P, Leung WH and Campana D: Expansion of
highly cytotoxic human natural killer cells for cancer cell
therapy. Cancer Res. 69:4010–4017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siegler U, Meyer-Monard S, Jörger S, Stern
M, Tichelli A, Gratwohl A, Wodnar-Filipowicz A and Kalberer CP:
Good manufacturing practice-compliant cell sorting and large-scale
expansion of single KIR-positive alloreactive human natural killer
cells for multiple infusions to leukemia patients. Cytotherapy.
12:750–763. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gong W, Xiao W, Hu M, Weng X, Qian L, Pan
X and Ji M: Ex vivo expansion of natural killer cells with high
cytotoxicity by K562 cells modified to co-express major
histocompatibility complex class I chain-related protein A, 4–1BB
ligand and interleukin-15. Tissue Antigens. 76:467–475. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Childs RW and Berg M: Bringing natural
killer cells to the clinic: Ex vivo manipulation. Hematology Am Soc
Hematol Educ Program. 2013:234–246. 2013.PubMed/NCBI
|
|
20
|
Cobleigh MA, Vogel CL, Tripathy D, Robert
NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman
G and Slamon DJ: Multinational study of the efficacy and safety of
humanized anti-HER2 monoclonal antibody in women who have
HER2-overexpressing metastatic breast cancer that has progressed
after chemotherapy for metastatic disease. J Clin Oncol.
17:2639–2648. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Albanell J, Bellmunt J, Molina R, García
M, Caragol I, Bermejo B, Ribas A, Carulla J, Gallego OS, Español T
and Solé Calvo LA: Node-negative breast cancers with
p53(−)/HER2-neu(−) status may identify women with very good
prognosis. Anticancer Res. 16:1027–1032. 1996.PubMed/NCBI
|
|
22
|
Steplewski Z, Lubeck MD and Koprowski H:
Human macrophages armed with murine immunoglobulin G2a antibodies
to tumors destroy human cancer cells. Science. 221:865–867. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shields RL, Namenuk AK, Hong K, Meng YG,
Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al: High
resolution mapping of the binding site on human IgG1 for Fc gamma
RI, Fc gamma RII, Fc gamma RIII and FcRn and design of IgG1
variants with improved binding to the Fc gamma R. J Biol Chem.
276:6591–6604. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakagawa S, Matsuoka Y, Ichihara H,
Yoshida H, Yoshida K and Ueoka R: New cancer immunotherapy using
autologous lymphocytes activated with trastuzumab. Biol Pharm Bull.
35:1213–1215. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barnouin K, Fredersdorf S, Eddaoudi A,
Mittnacht S, Pan LX, Du MQ and Lu X: Antiproliferative function of
p27kip1 is frequently inhibited in highly malignant Burkitt's
lymphoma cells. Oncogene. 18:6388–6397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Whenham N, D'Hondt V and Piccart MJ:
HER2-positive breast cancer: From trastuzumab to innovatory
anti-HER2 strategies. Clin Breast Cancer. 8:38–49. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baselga J, Gianni L, Geyer C, Perez EA,
Riva A and Jackisch C: Future options with trastuzumab for primary
systemic and adjuvant therapy. Semin Oncol. 31 5 Suppl 10:S51–S57.
2004. View Article : Google Scholar
|
|
28
|
Hudis CA: Trastuzumab-mechanism of action
and use in clinical practice. N Engl J Med. 357:39–51. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nimmerjahn F and Ravetch JV: Antibodies,
Fc receptors and cancer. Curr Opin Immunol. 19:239–245. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Beano A, Signorino E, Evangelista A, Brusa
D, Mistrangelo M, Polimeni MA, Spadi R, Donadio M, Ciuffreda L and
Matera L: Correlation between NK function and response to
trastuzumab in metastatic breast cancer patients. J Transl Med.
6:252008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huijskens MJ, Walczak M, Sarkar S, Atrafi
F, Senden-Gijsbers BL, Tilanus MG, Bos GM, Wieten L and Germeraad
WT: Ascorbic acid promotes proliferation of natural killer cell
populations in culture systems applicable for natural killer cell
therapy. Cytotherapy. 17:613–620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Malek TR: The biology of interleukin-2.
Annu Rev Immunol. 26:453–479. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Groh V, Rhinehart R, Secrist H, Bauer S,
Grabstein KH and Spies T: Broad tumor-associated expression and
recognition by tumor-derived gamma delta T cells of MICA and MICB.
Proc Natl Acad Sci USA. 96:pp. 6879–6884. 1999, View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vivier E, Tomasello E and Paul P:
Lymphocyte activation via NKG2D: Towards a new paradigm in immune
recognition? Curr Opin Immunol. 14:306–311. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Groh V, Rhinehart R, Randolph-Habecker J,
Topp MS, Riddell SR and Spies T: Costimulation of CD8alphabeta T
cells by NKG2D via engagement by MIC induced on virus-infected
cells. Nat Immunol. 2:255–260. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang H, Yang D, Xu W, Wang Y, Ruan Z, Zhao
T, Han J and Wu Y: Tumor-derived soluble MICs impair CD3(+)CD56(+)
NKT-like cell cytotoxicity in cancer patients. Immunol Lett.
120:65–71. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bauer S, Groh V, Wu J, Steinle A, Phillips
JH, Lanier LL and Spies T: Activation of NK cells and T cells by
NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Diefenbach A, Jamieson AM, Liu SD, Shastri
N and Raulet DH: Ligands for the murine NKG2D receptor: Expression
by tumor cells and activation of NK cells and macrophages. Nat
Immunol. 1:119–126. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Meyer A, Carapito R, Ott L, Radosavljevic
M, Georgel P, Adams EJ, Parham P, Bontrop RE, Blancher A and Bahram
S: High diversity of MIC genes in non-human primates.
Immunogenetics. 66:581–587. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pende D, Rivera P, Marcenaro S, Chang CC,
Biassoni R, Conte R, Kubin M, Cosman D, Ferrone S, Moretta L and
Moretta A: Major histocompatibility complex class I-related chain A
and UL16-binding protein expression on tumor cell lines of
different histotypes: Analysis of tumor susceptibility to
NKG2D-dependent natural killer cell cytotoxicity. Cancer Res.
62:6178–6186. 2002.PubMed/NCBI
|
|
41
|
Friese MA, Platten M, Lutz SZ, Naumann U,
Aulwurm S, Bischof F, Bühring HJ, Dichgans J, Rammensee HG, Steinle
A and Weller M: MICA/NKG2D-mediated immunogene therapy of
experimental gliomas. Cancer Res. 63:8996–9006. 2003.PubMed/NCBI
|
|
42
|
Salih HR, Antropius H, Gieseke F, Lutz SZ,
Kanz L, Rammensee HG and Steinle A: Functional expression and
release of ligands for the activating immunoreceptor NKG2D in
leukemia. Blood. 102:1389–1396. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Watson NF, Spendlove I, Madjd Z, McGilvray
R, Green AR, Ellis IO, Scholefield JH and Durrant LG: Expression of
the stress-related MHC class I chain-related protein MICA is an
indicator of good prognosis in colorectal cancer patients. Int J
Cancer. 118:1445–1452. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vetter CS, Groh V, Thor Straten P, Spies
T, Brocker EB and Becker JC: Expression of stress-induced MHC class
I related chain molecules on human melanoma. J Invest Dermatol.
118:600–605. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Assarsson E, Kambayashi T, Schatzle JD,
Cramer SO, von Bonin A, Jensen PE, Ljunggren HG and Chambers BJ: NK
cells stimulate proliferation of T and NK cells through 2B4/CD48
interactions. J Immunol. 173:174–180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Martin-Fontecha A, Thomsen LL, Brett S,
Gerard C, Lipp M, Lanzavecchia A and Sallusto F: Induced
recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1
priming. Nat Immunol. 5:1260–1265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pan K, Wang QJ, Liu Q, Zheng HX, Li YQ,
Weng DS, Li JJ, Huang LX, He J, Chen SP, et al: The phenotype of ex
vivo generated cytokine-induced killer cells is associated with
overall survival in patients with cancer. Tumour Biol. 35:701–707.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Meyer-Monard S, Passweg J, Siegler U,
Kalberer C, Koehl U, Rovó A, Halter J, Stern M, Heim D, Gratwohl
Alois JR and Tichelli A: Clinical-grade purification of natural
killer cells in haploidentical hematopoietic stem cell
transplantation. Transfusion. 49:362–371. 2009. View Article : Google Scholar : PubMed/NCBI
|