Introduction
Lung cancer is a heterogeneous malignancy with
aggressive phenotypes (1,2). Non-small-cell lung cancer (NSCLC)
accounts for ~85% of lung cancer cases (2). Lung cancers harboring somatic mutations
in exons encoding the tyrosine kinase domain of the epidermal
growth factor receptor (EGFR) exhibit a significant tumor
regression when treated with the EGFR tyrosine kinase inhibitors
(TKIs) gefitinib or erlotinib in ~70% of cases (3–5).
However, acquired resistance inevitably develops in an overwhelming
majority of these patients.
Epithelial to mesenchymal transition (EMT) is
associated with the acquired resistance of NSCLC to gefitinib
(6,7). It is a process by which cells undergo a
morphological shift from the epithelial polarized to the
mesenchymal fibroblastoid phenotype. EMT has been recognized to
have pivotal roles in several diverse processes during embryonic
development, chronic inflammation and fibrosis, as well as tumor
progression (8–11). During EMT, epithelial cells lose
their defined cell-cell/cell-substratum contacts and their
structural/functional polarity, and become spindle-like.
Lung cancer stem cells (CSCs) or cancer-initiating
cells (CICs) have been identified and demonstrated to constitute a
primitive cell population capable of self-renewal and
differentiation that have the unique capacity to give rise to new
tumors upon serial transplantation (12–15).
They represent a small population of undifferentiated tumorigenic
cells responsible for tumor initiation, maintenance and spreading.
Resistance to conventional chemotherapeutic drugs is a common
characteristic of CICs (16). It has
been reported that lung CICs were associated with gefitinib
resistance (17).
TWIK-related acid-sensitive K(+) (TASK-1) is
expressed in a subset of NSCLCs, where it is functional, and
promoted the proliferation and inhibited apoptosis in a highly
TASK-1-expressing lung cancer cell line (18). The present study demonstrated that
TASK-1 induced gefitinib resistance in NSCLC A549 cells.
Overexpression of TASK-1 promoted the formation of CICs in A549
cells. CD133, octamer-binding transcription factor 4 (OCT-4) and
Nanong have been proposed as markers of CICs in lung cancer
(13,19,20). The
present study demonstrated that overexpression of TASK-1 promoted
CD133, OCT-4 and Nanong protein expression in A549 cells. Increased
formation of CSC-like populations may result in EMT of cancer cells
(21–24). The present study found that
overexpression of TASK-1 promotes EMT of A549 cells.
Materials and methods
Cell line and culture
The A549 NSCLC cell line was purchased from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China) within 3
months of performing the experiments. They were cultured in
Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum (both Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and antibiotics (100 mg/ml penicillin and 100 U/ml
streptomycin) in a 5% CO2 incubator at 37°C.
Cell transfection
The pGCMV/EGFP/TASK-1 plasmid and the pGCMV/EGFP/Neo
plasmid were constructed (Tiangen Biotech Co., Ltd., Beijing,
China). The two plasmids were transfected into A549 NSCLC cells
separately using Lipofectamine 2000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). In order to detect
the transfection efficiency of the plasmids, green fluorescent
signal was measured by fluorescence microscopy. Subsequent
experimentation was performed after 24 h.
Western blot analysis
Total proteins in cells were extracted using protein
lysis solution (Tiangen Biotech Co., Ltd.). Protein concentration
was measured using a bicinchoninic acid kit (Tiangen Biotech Co.,
Ltd.). Protein extracts (50 µg/lane) were resolved through 8%
SDS-PAGE, transferred onto polyvinylidene difluoride membranes
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and blocked with 5%
non-fat milk for 1 h at room temperature. Membranes were probed
with antibodies against human TASK-1 (ab135883), vimentin
(ab92547), E-cadherin (ab40772), CD133 (ab16518), OCT4 (ab109183),
Nanog (ab109250), matrix metalloproteinase (MMP)9 (ab76003), MMP2
(ab92536) or β-actin (ab8227) (all 1:500; Abcam, Cambridge, MA,
USA) at 4°C overnight. Membranes were then incubated with
anti-rabbit secondary antibodies (1:500; ab218695; Abcam) for 1 h
at room temperature. An enhanced chemiluminescence system (GE
Healthcare, Chicago, IL, USA) was used to detect the antibody
binding.
Sphere formation assay
Cells were seeded at 2.5×104 cells/well
on 0.5% agar pre-coated 6-well plates. After 1 week, half the
medium was exchanged every third day. After a total of 14 days,
single spheres were picked and counted (25). Sphere formation efficiency was
calculated by dividing the total number of spheres formed by the
total number of live cells seeded multiplied by hundred.
MTT assay
To monitor resistance to gefitinib, A549 cells were
treated with gefitinib (purity >99%; AstraZeneca, Cambridge, UK)
at different concentrations for 24 h. An MTT assay was performed as
described previously (2). Data were
analyzed with the software Origin 7.5 (OriginLab, Northampton, MA,
USA) to fit a sigmodial curve. The IC50 was the
gefitinib concentration that reduced the number of viable cells by
50%.
Immunofluorescence staining
Cells seeded on glass coverslips in 6-well plates
were fixed in 4% formaldehyde solution for 30 min at room
temperature and permeabilized with 0.5% Triton-X-100/PBS. Cells
were blocked with 5% BSA-PBS (BSA from Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 1 h at room temperature and incubated with
primary antibodies against E-cadherin (1:500; ab92547; Abcam) or
vimentin (1:500; ab40772; Abcam) at 4°C overnight. Cells were then
incubated with rhodamine- or fluorescein isothiocyanate-conjugated
secondary antibodies (1:500; ab150077; Abcam) for 1 h at room
temperature. The coverslips were counterstained with DAPI and
imaged under a confocal microscope (TCS SP5; Lecia Microsystems,
Wetzlar, Germany).
Wound healing assay
Cells (5×105) were seeded onto each 35-mm
glass bottom dish (MatTek Co., Ashland, MA, USA) and cultured at
37°C with 5% CO2 for 24 h. The confluent monolayer of
cells was wounded with yellow 200 µl pipette tips (Tiangen Biotech
Co., Ltd.). After washing with warm PBS, the cells were incubated
in fresh serum-free culture medium. Images of the wounded areas
were captured at different time-points with an inverted microscope
(Eclipse TE-2000U; Nikon, Tokyo, Japan) equipped with a video
camera (DS-U1; Nikon). Results were examined at five randomly
selected fields in each field, at ×20 magnification. The wound
areas were calculated by ImageJ 1.43b software (NIH, Bethesda, MD,
USA).
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) was
used for the analysis of experimental data. Student's t-test
(two-tailed) was used for comparison between two groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
Overexpression of TASK-1 promotes
gefitinib resistance
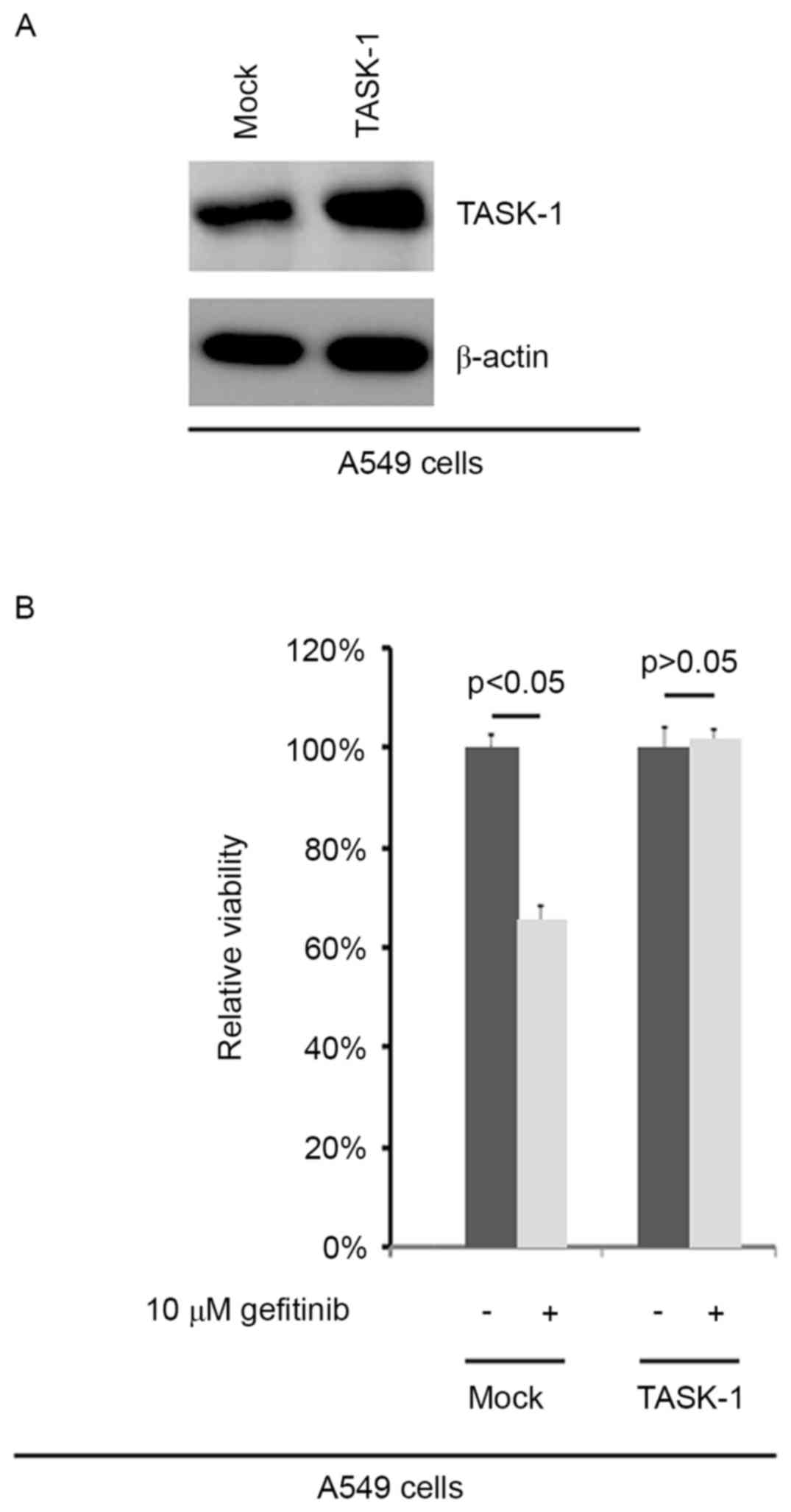
In order to confirm the efficiency of
plasmid-mediated TASK-1-expression, A549 cells transfected with
TASK-1-expressing plasmid and empty vector were subjected to
western blot analysis. The results showed that TASK-1 protein was
significantly upregulated in A549 cells transfected with
TASK-1-expressing plasmid (Fig. 1A).
To further identify whether TASK-1 affected the efficacy of
gefitinib in A549 cells, A549 cells transfected with
TASK-1-expressing plasmids or empty vector were subjected to an MTT
assay (Fig. 1B). The results
demonstrated that TASK-1 transformed native, gefitinib-sensitive
A549 cells into gefitinib-resistant A549 cells, suggesting that its
overexpression induced gefitinib resistance (Fig. 1B).
TASK-1 gives A549 cells CIC-like
traits
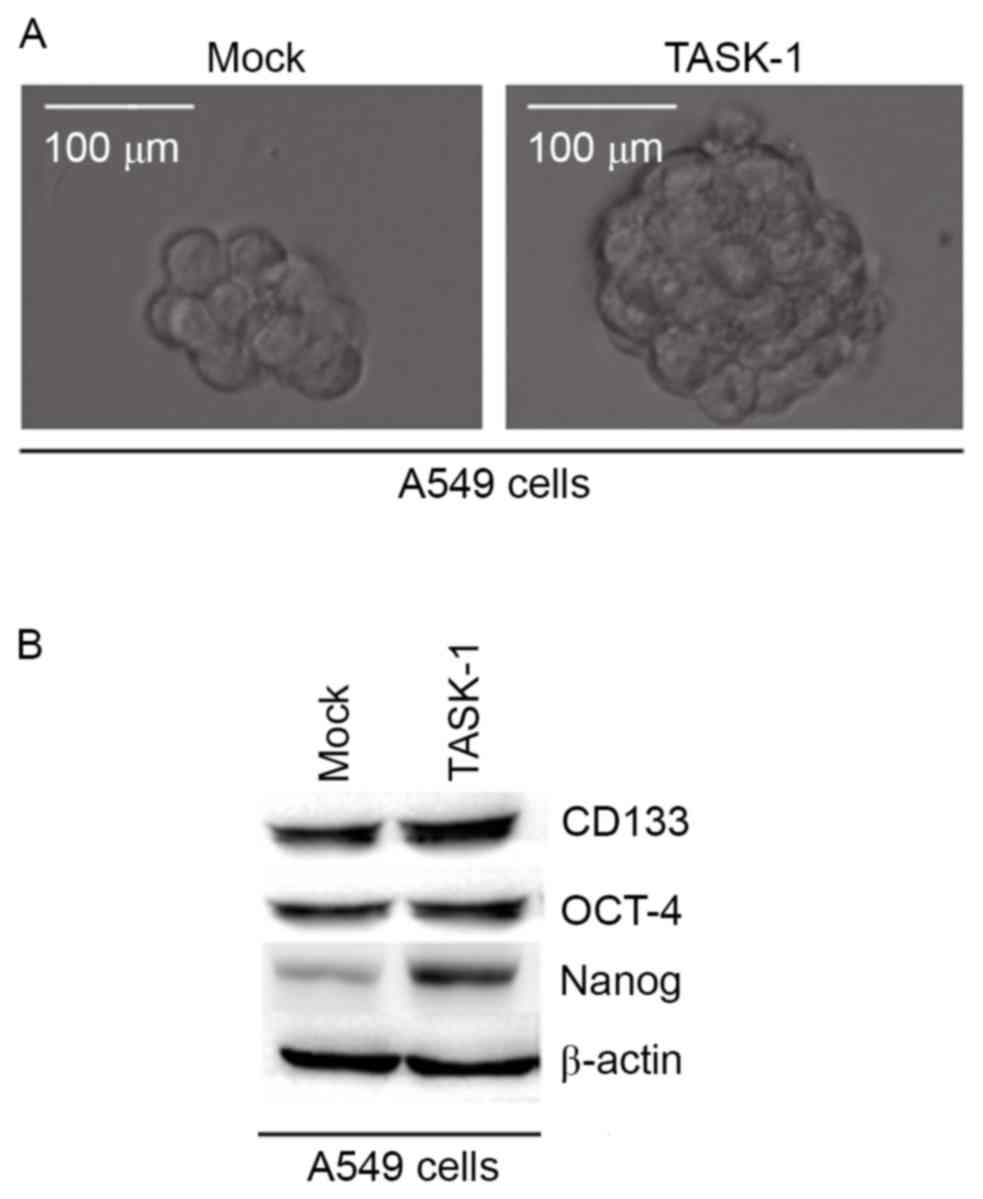
In order to identify whether TASK-1 affects CIC-like
traits in A549 cells, a sphere formation assay was performed to
assess the capacity of A549 cells for self renewal, which is
associated with CICs and CSCs. The results demonstrated that after
14 days of culture TASK-1-overexpressing cells formed bigger
spheres than control cells, indicating markedly increased CIC-like
traits provided by the TASK-1-expressing plasmids (Fig. 2A). To identify whether TASK-1
regulates CD133, OCT-4 and Nanog protein expression, western blot
analysis of A549 cells transfected with TASK-1-expressing plasmids
and empty vector was performed. The results revealed that CD133,
OCT-4 and Nanog protein were upregulated in A549 cells by TASK-1
(Fig. 2B).
TASK-1 induces EMT in A549 cells
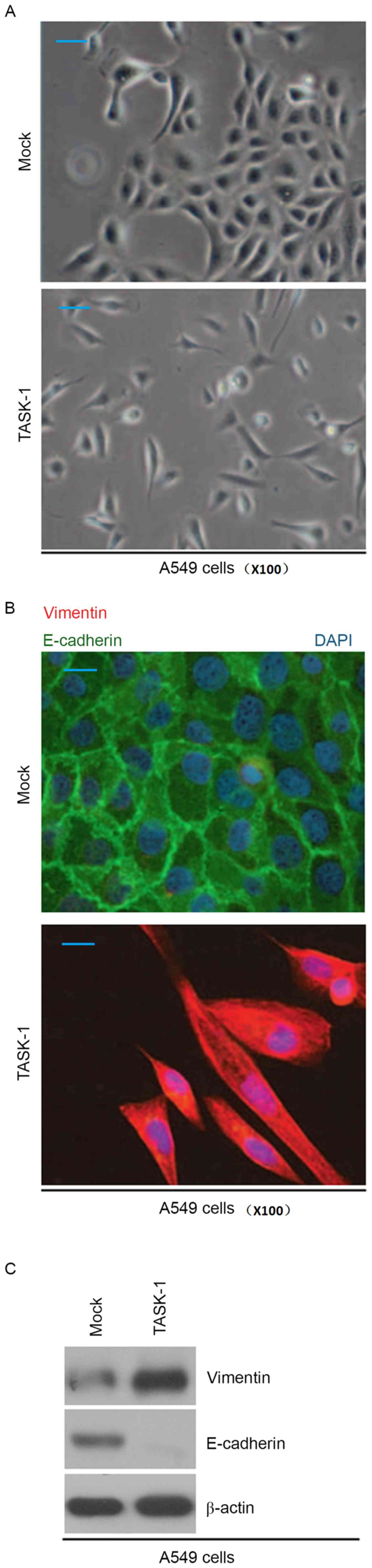
Cell morphological observation revealed that
overexpression of TASK-1 induced EMT phenotypes in A549 cells
(transition from a cobblestone-like to a spindle-like morphology;
Fig. 3A). In order to detect whether
TASK-1 affects E-cadherin (epithelial marker) and vimentin
(mesenchymal marker) protein, immunofluorescence analysis of A549
cells transfected with TASK-1 and empty vector was performed. It
was found that overexpression of TASK-1 promoted vimentin
expression and inhibited E-cadherin expression in A549 cells
(Fig. 3B). Western blot analysis was
also performed to detect E-cadherin and vimentin protein in A549
cells transfected with TASK-1 and empty vector. The results
revealed that vimentin protein is upregulated, while E-cadherin is
downregulated in A549 cells transfected with TASK-1 overexpression
vector (Fig. 3C).
Overexpression of TASK-1 promotes
migration of A549 cells
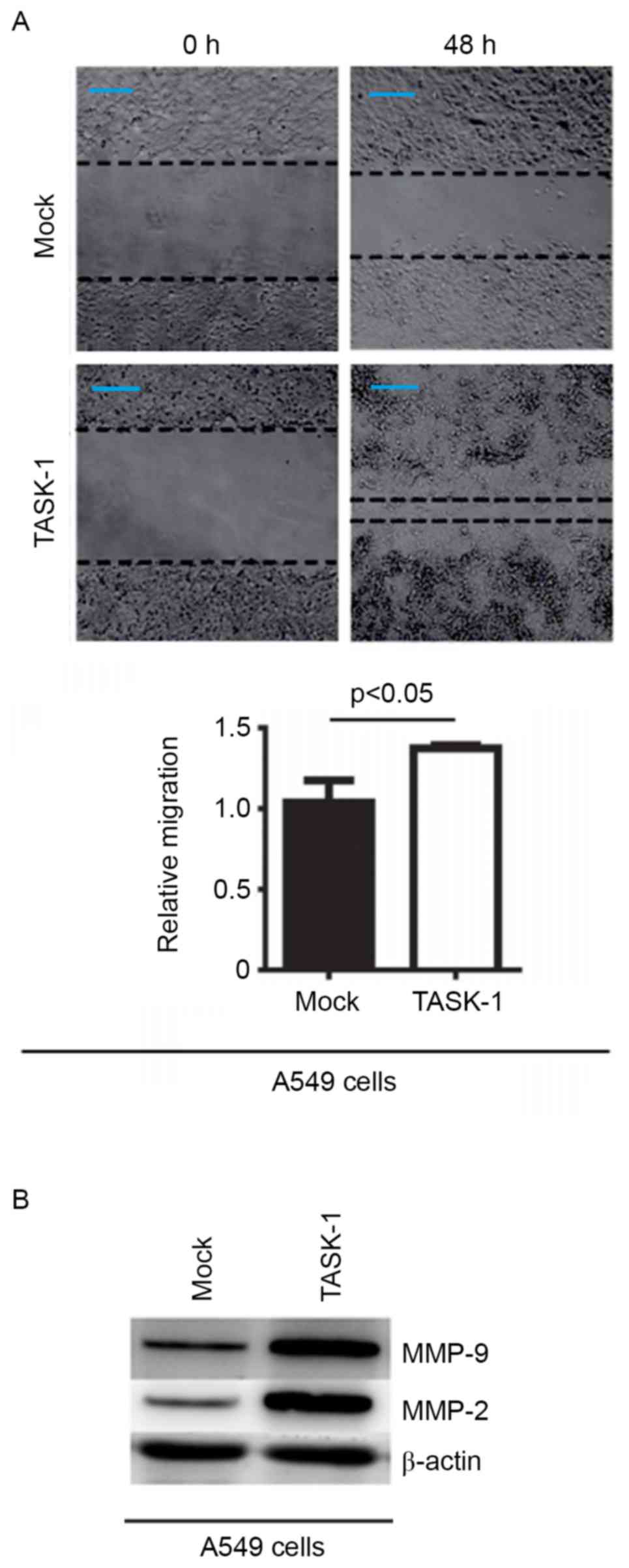
In an attempt to identify the role of TASK-1 in
regulating the migration of A549 cells, a would healing assay was
performed to detect the migration of A549 cells transfected with
TASK-1-expressing plasmid and empty vector. Overexpression of
TASK-1 was found to promote the migration in the cells (Fig. 4A). In order to detect whether TASK-1
affects MMP-2 and MMP-9 protein expression, western blot analysis
of A549 cells transfected with TASK-1 and empty vector was
performed. It was revealed that expression of TASK-1 promotes MMP-2
and MMP-9 expression in A549 cells (Fig.
4B).
Discussion
NSCLC patients with early-stage disease are treated
by surgery, and 30–60% develop recurrent tumors, which results in
mortality (26,27). Chemotherapeutic agents, including
gemcitabine, platinum compounds and taxanes, improve survival to a
limited extent, but overall survival rates remain low due to
recurrence of more aggressive, drug-resistant tumors (28,29).
NSCLC in non-smokers predominantly has mutations in EGFR (30); such patients respond well to EGFR
inhibitors such as gefitinib, but eventually develop resistance and
succumb to the disease (31). The
recurrence may be local or metastatic, and commonly occurs after a
period of clinical dormancy. Resistance to EGFR inhibitors occurs
through various mechanisms, including the appearance of a T790M
gatekeeper mutation, expression of the c-Met gene or activation of
alternate signaling pathways (32,33).
Development of strategies to combat resistance to EGFR inhibitors
in NSCLC will provide an immense benefit to a large number of
patients (34).
A recent study reported that knockdown of TASK-1 by
small interfering RNA significantly enhanced apoptosis and reduced
proliferation in A549 cells (18).
For the first time, to the best of our knowledge, the present study
demonstrated that overexpression of TASK-1 induced gefitinib
resistance in A549 cells, implying that TASK-1 may represent a
novel target to reverse gefitinib resistance.
Resistance to radiation therapies and conventional
chemotherapeutic drugs is a common characteristic of CSCs (35–37). It
has been reported that aldehyde dehydrogenase 1 family member A1
(ALDH1A1)-positive CSCs promote gefitinib resistance in lung cancer
(17). The present study found that
overexpression of TASK-1 promoted A549 cells to adopt CIC-like
properties. In addition, overexpression promoted CD133, OCT-4 and
Nanog protein expression in A549 cells, indicating that TASK-1
induces the development of CIC-like traits. However, it remains
elusive whether TASK-1 promotes the formation of ALDH1A1-positive
lung CICs.
Increased formation of a CIC population may result
in EMT of cancer cells and EMT has been shown to contribute to the
formation of CIC-like characteristics (38). It has been suggested that the
reversal of the EMT phenotype potentially enhances the sensitivity
of lung cancer cells to gefitinib (39). Consistent with these results, the
present study demonstrated that overexpression of TASK-1 induced
EMT of A549 cells. The degradation of ECM and the destruction of
basement membrane are prerequisites for tumor infiltration and
metastasis (40). Tumor cells must
have the ability to degrade ECM and basement membranes, and this
degradation process depends on proteolytic enzymes, mainly
serine-degrading enzymes, cysteine proteases and MMPs. MMPs are
considered to be the most important. MMP2 and MMP9 are important
members of the MMP family, encoding 72 KDa gelatinase A and 92 KDa
gelatinase B, respectively, which have a partial co-acting
substrate that degrades the major constituents of the basement
membrane IV type collagen. This is conducive to the migration of
tumor cells (41,42).
In conclusion, the results of the present study
provided strong molecular evidence demonstrating that TASK-1
promotes gefitinib resistance, EMT and CIC-like properties of
NSCLCs. Thus, downregulation of TASK-1 appears to be a novel
strategy for the reversal of the EMT, decreasing CIC-like traits
and increasing gefitinib sensitivity of NSCLCs.
References
|
1
|
Ding L, Getz G, Wheeler DA, Mardis ER,
McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan
MB, et al: Somatic mutations affect key pathways in lung
adenocarcinoma. Nature. 455:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanders HR and Albitar M: Somatic
mutations of signaling genes in non-small-cell lung cancer. Cancer
Genet Cytogenet. 203:7–15. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Costa DB, Kobayashi S, Tenen DG and
Huberman MS: Pooled analysis of the prospective trials of gefitinib
monotherapy for EGFR-mutant non-small cell lung cancers. Lung
Cancer. 58:95–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller VA, Riely GJ, Zakowski MF, Li AR,
Patel JD, Heelan RT, Kris MG, Sandler AB, Carbone DP, Tsao A, et
al: Molecular characteristics of bronchioloalveolar carcinoma and
adenocarcinoma, bronchioloalveolar carcinoma subtype, predict
response to erlotinib. J Clin Oncol. 26:1472–1478. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jackman DM, Miller VA, Cioffredi LA, Yeap
BY, Jänne PA, Riely GJ, Ruiz MG, Giaccone G, Sequist LV and Johnson
BE: Impact of epidermal growth factor receptor and KRAS mutations
on clinical outcomes in previously untreated non-small cell lung
cancer patients: Results of an online tumor registry of clinical
trials. Clin Cancer Res. 15:5267–5273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li D, Zhang L, Zhou J and Chen H:
Cigarette smoke extract exposure induces EGFR-TKI resistance in
EGFR-mutated NSCLC via mediating Src activation and EMT. Lung
Cancer. 93:35–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grünert S, Jechlinger M and Beug H:
Diverse cellular and molecular mechanisms contribute to epithelial
plasticity and metastasis. Nat Rev Mol Cell Biol. 4:657–665. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pine SR, Marshall B and Varticovski L:
Lung cancer stem cells. Dis Markers. 24:257–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eramo A, Lotti F, Sette G, Pilozzi E,
Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C and De
Maria R: Identification and expansion of the tumorigenic lung
cancer stem cell population. Cell Death Differ. 15:504–514. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim CF, Jackson EL, Woolfenden AE,
Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT and Jacks T:
Identification of bronchioalveolar stem cells in normal lung and
lung cancer. Cell. 121:823–835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang F, Qiu Q, Khanna A, Todd NW, Deepak
J, Xing L, Wang H, Liu Z, Su Y, Stass SA and Katz RL: Aldehyde
dehydrogenase 1 is a tumor stem cell-associated marker in lung
cancer. Mol Cancer Res. 7:330–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang CP, Tsai MF, Chang TH, Tang WC, Chen
SY, Lai HH, Lin TY, Yang JC, Yang PC, Shih JY and Lin SB:
ALDH-positive lung cancer stem cells confer resistance to epidermal
growth factor receptor tyrosine kinase inhibitors. Cancer Lett.
328:144–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leithner K, Hirschmugl B, Li Y, Tang B,
Papp R, Nagaraj C, Stacher E, Stiegler P, Lindenmann J, Olschewski
A, et al: TASK-1 regulates apoptosis and proliferation in a subset
of non-small cell lung cancers. PLoS One. 11:e01574532016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen YC, Hsu HS, Chen YW, Tsai TH, How CK,
Wang CY, Hung SC, Chang YL, Tsai ML, Lee YY, et al: Oct-4
expression maintained cancer stem-like properties in lung
cancer-derived CD133-positive cells. PLoS One. 3:e26372008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong
CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS and Wu CW:
Coexpression of Oct4 and nanog enhances malignancy in lung
adenocarcinoma by inducing cancer stem cell-like properties and
epithelial-mesenchymal transdifferentiation. Cancer Res.
70:10433–10444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kurrey NK, Jalgaonkar SP, Joglekar AV,
Ghanate AD, Chaskar PD, Doiphode RY and Bapat SA: Snail and slug
mediate radioresistance and chemoresistance by antagonizing
p53-mediated apoptosis and acquiring a stem-like phenotype in
ovarian Cancer cells. Stem Cells. 27:2059–2068. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morel AP, Lièvre M, Thomas C, Hinkal G,
Ansieau S and Puisieux A: Generation of breast cancer stem cells
through epithelial-mesenchymal transition. PLoS One. 3:e28882008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Santisteban M, Reiman JM, Asiedu MK,
Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC,
Manjili MH, et al: Immune-induced epithelial to mesenchymal
transition in vivo generates breast cancer stem cells. Cancer Res.
69:2887–2895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tiran V, Stanzer S, Heitzer E, Meilinger
M, Rossmann C, Lax S, Tsybrovskyy O, Dandachi N and Balic M:
Genetic profiling of putative breast cancer stem cells from
malignant pleural effusions. PLoS One. 12:e01752232017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Demicheli R, Fornili M, Ambrogi F, Higgins
K, Boyd JA, Biganzoli E and Kelsey CR: Recurrence dynamics for
non-small-cell lung cancer: Effect of surgery on the development of
metastases. J Thorac Oncol. 7:723–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Senthi S, Lagerwaard FJ, Haasbeek CJ,
Slotman BJ and Senan S: Patterns of disease recurrence after
stereotactic ablative radiotherapy for early stage non-small-cell
lung cancer: A retrospective analysis. Lancet Oncol. 13:802–809.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sève P and Dumontet C: Chemoresistance in
non-small cell lung cancer. Curr Med Chem Anticancer Agents.
5:73–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lara PN Jr, Lau DH and Gandara DR:
Non-small-cell lung cancer progression after first-line
chemotherapy. Curr Treat Options Oncol. 3:53–58. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun S, Schiller JH and Gazdar AF: Lung
cancer in never smokers-a different disease. Nat Rev Cancer.
7:778–790. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brugger W and Thomas M: EGFR-TKI resistant
non-small cell lung cancer (NSCLC): New developments and
implications for future treatment. Lung Cancer. 77:2–8. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nurwidya F, Takahashi F, Murakami A,
Kobayashi I, Kato M, Shukuya T, Tajima K, Shimada N and Takahashi
K: Acquired resistance of non-small cell lung cancer to epidermal
growth factor receptor tyrosine kinase inhibitors. Respir Investig.
52:82–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wangari-Talbot J and Hopper-Borge E: Drug
resistance mechanisms in non-small cell lung carcinoma. J Can Res
Updates. 2:265–282. 2013.PubMed/NCBI
|
|
34
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou BB, Zhang H, Damelin M, Geles KG,
Grindley JC and Dirks PB: Tumour-initiating cells: Challenges and
opportunities for anticancer drug discovery. Nat Rev Drug Discov.
8:806–823. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Frank NY, Schatton T and Frank MH: The
therapeutic promise of the cancer stem cell concept. J Clin Invest.
120:41–50. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Wakeman TP, Lathia JD, Hjelmeland
AB, Wang XF, White RR, Rich JN and Sullenger BA: Notch promotes
radioresistance of glioma stem cells. Stem Cells. 28:17–28.
2010.PubMed/NCBI
|
|
38
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xie M, Zhang L, He CS, Xu F, Liu JL, Hu
ZH, Zhao LP and Tian Y: Activation of notch-1 enhances
epithelial-mesenchymal transition in gefitinib-acquired resistant
lung cancer cells. J Cell Biochem. 113:1501–1513. 2012.PubMed/NCBI
|
|
40
|
Hirvonen R, Talvensaari-Mattila A, Pääkkö
P and Turpeenniemi-Hujanen T: Matrix metalloproteinase-2 (MMP-2) in
T(1–2)N0 breast carcinoma. Breast Cancer Res Treat. 77:85–91. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kalhori V and Törnquist K: MMP2 and MMP9
participate in S1P-induced invasion of follicular ML-1 thyroid
cancer cells. Mol Cell Endocrinol. 404:113–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu B, Cui J, Sun J, Li J, Han X, Guo J,
Yi M, Amizuka N, Xu X and Li M: Immunolocalization of MMP9 and MMP2
in osteolytic metastasis originating from MDA-MB-231 human breast
cancer cells. Mol Med Rep. 14:1099–1106. 2016. View Article : Google Scholar : PubMed/NCBI
|