Introduction
Diabetic cardiomyopathy (DCM), a common complication
of diabetes, is a major cause of morbidity and mortality among
diabetic patients (1). Rubler et
al (2) was the first to
introduce the term ‘diabetic cardiomyopathy’. DCM is characterized
by a set of impairments, including oxidative stress, cardiomyocyte
hypertrophy, myocardial apoptosis and fibrosis, impaired diastolic
function and eventually cardiac failure (3). High glucose (HG)-mediated oxidative
stress has been considered as a major causal risk factor that
triggers hypertrophic and apoptotic responses, which ultimately
contributes to the pathogenesis of DCM (4,5).
Therefore, scavenging of reactive oxygen species (ROS) and reducing
cardiomyocyte hypertrophy and apoptosis may effectively prevent the
initiation or progression of DCM.
Relaxin (RLX), a pleiotropic hormone, was first
identified for its role in the reproduction and pregnancy (6), but is now best known for its
vasodilation effects, antiangiogenic and antifibrotic properties,
as well as cardioprotection in various experimental models
(7,8). Previous studies have indicated that
atrial cardiomyocytes produce RLX (9) and that the RLX family peptide
receptor-1 (RXFP1) is expressed by atrial ventricular cells
(10) and H9C2 cardiomyocytes
(11). In addition, the density of
RXFP1 in the heart was higher than that in uterine myometrium,
indicating that RLX has an important role in cardiovascular tissues
(12). It has been demonstrated that
HG promotes the expression of endogenous RLX and that RLX inhibited
cardiac fibrosis under HG conditions (13). Furthermore, RLX protected
cardiomyocytes from HG-mediated apoptosis by suppressing
apoptosis-associated pathways and endoplasmic reticulum stress
(14). RLX robustly inhibits
cardiomyocyte hypertrophy in vivo (15) and in vitro (16). These studies suggested the beneficial
effects of RLX on cardiovascular complications.
Notch1 signaling is mainly involved in the important
processes of cell fate determination, proliferation,
differentiation and regeneration (17). Studies have indicated that the
activation of Notch1 signaling protects against cardiac myocyte
injury, while inhibition of the Notch1 pathway exacerbates
myocardial hypertrophy, apoptosis (18) and fibrosis (19), and may eventually lead to dilated
myopathy (20). An increasing number
of studies have supported the association between RLX and Notch1 by
demonstrating that RLX prevents cardiac fibroblast to myofibroblast
and endothelial to mesenchymal transition involving the Notch1
pathway (21,22). Boccalini et al (11) also concluded that RLX-activated
Notch1 signaling protects cardiac myocyte cells from
hypoxia/reoxygenation injury. However, it has remained elusive
whether RLX ameliorates hyperglycemia-induced cardiomyocyte
hypertrophy and apoptosis via activation of the Notch1 pathway.
Hence, the present study was designed to explore the potential
protective mechanisms of RLX on H9c2 cells using an in vitro
model of hyperglycemia-induced myocardial injury. The present study
may provide possible molecular mechanisms underlying the effect of
RLX as a potent treatment for DCM.
Materials and methods
Cell culture and treatments
Embryonic rat cardiac H9c2 cells (Chinese Academy of
Sciences, Shanghai, China) were cultured in Dulbecco's modified
Eagle's medium (HyClone; GE Healthcare, Little Chalfont, UK)
supplemented with 100 U/ml penicillin and streptomycin, and 10%
fetal bovine serum (PAN Biotech, Aidenbach, Germany) at 37°C in a
humidified atmosphere containing 5% CO2. H9c2 cells have
numerous similarities with primary cardiomyocytes and are commonly
used as a model of hypertrophy or apoptosis. In addition, compared
with H9c2 cells, cardiomyocyte hypertrophy and apoptosis in primary
neonatal heart cells are difficult to discern due to varying shapes
and cell clumping. Therefore, the present study used the H9c2 cell
line. Cells between passages 3 and 5 were used for each experiment.
H9c2 cells were randomly divided into four groups for treatment:
Normal control group (NG, 5.5 mmol/l); high glucose group (HG, 33
mmol/l); high glucose + relaxin group (HG + RLX group); high
glucose + relaxin +
N-(N-(3,5-difluorophenacetyl)-L-alanyl)-S-phenylglycine t-butyl
ester (DAPT) group (HG + RLX + DAPT group). Cells in the
logarithmic growth phase were cultured in normal glucose medium for
24 h, and then stimulated with HG medium with or without RLX (100
nmol/ml; cat. no. 130-15; PeproTech, Inc., Rocky Hill, NJ, USA) for
48 h. The Notch1 inhibitor DAPT (10 µmol/l; cat. no. D5942;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was administered 4 h
prior to HG exposure and subsequent stimulation with RLX for 48 h.
The cells were then harvested and used in the following
experiments.
Cell counting Kit-8 (CCK-8) viability
assay
The viability of cells was measured using the CCK-8
assay (cat. no. CK04; Dojindo, Kumamoto, Japan) according to the
manufacturer's protocol. In brief, cells were seeded onto 96-well
plates overnight (1×103 cells per well) and subsequently
treated with different concentrations of RLX (0, 25, 50, 100 or 200
nmol/ml) and for different durations (24, 48 or 72 h).
Subsequently, cells were incubated with 10 µmol CCK-8 solution
under normal incubation conditions for 2 h. The absorbance at 450
nm, as an indicator of cell viability, was measured using a
microplate reader (Multiskan MK3; Thermo Fisher Scientific Inc.,
Waltham, MA, USA).
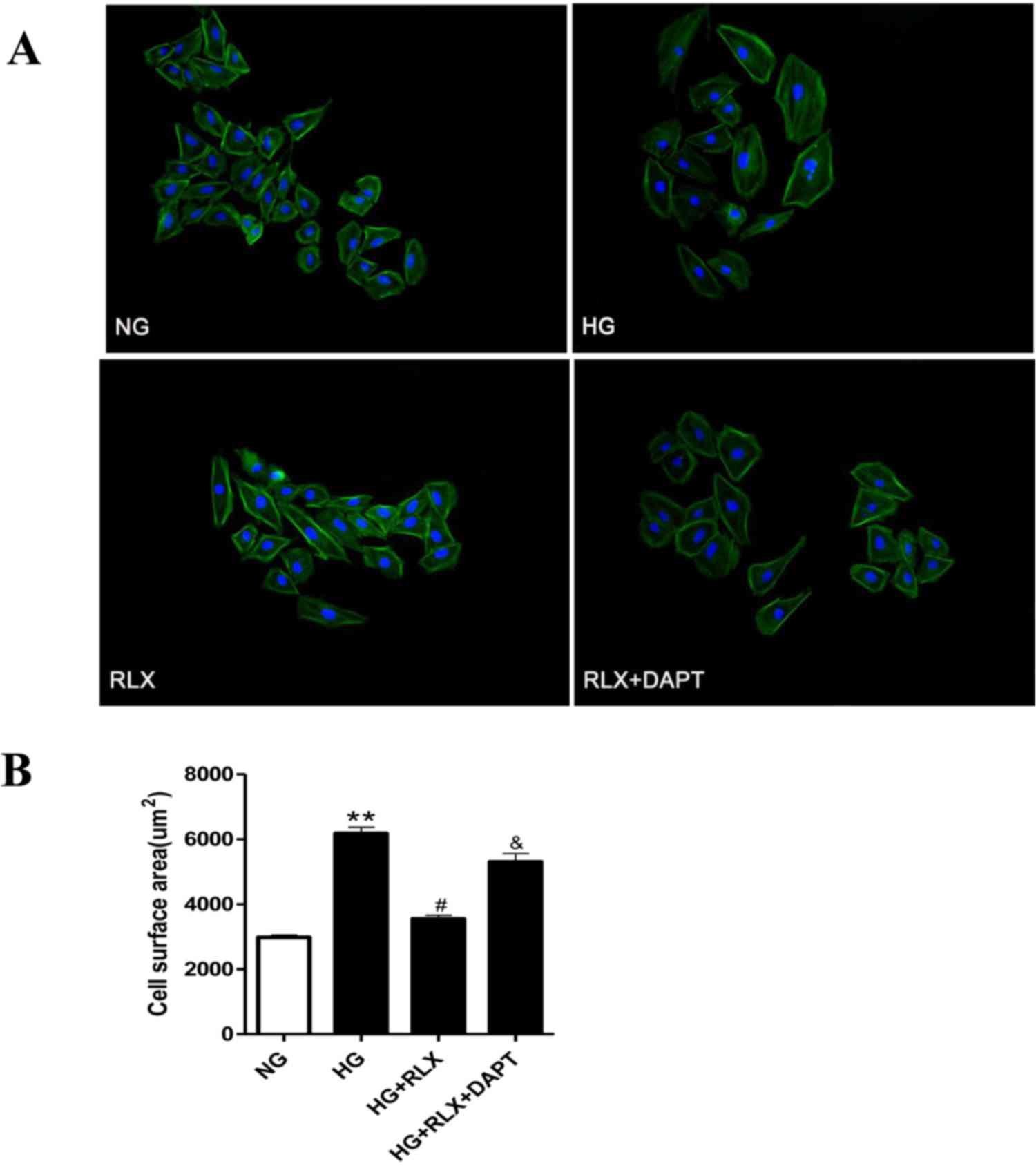
Cell surface area measurement
The surface area of cardiac myocytes, as a hallmark
of hypertrophy, was quantified using tetramethylrhodamine B
isothiocyanate-labeled phalloidin (cat. no. P1951; Sigma-Aldrich;
Merck KGaA). In brief, cells were grown on ordinary coverslips
(18×18 mm) in 24-well plates (1×104 cells per well) to
either 90% confluence or subconfluence, and then treated as
previously described. H9c2 cells were then fixed with 4%
paraformaldehyde and blocked with 10% goat serum (cat. no. AR1009;
Boster Biological Technology, Pleasanton, CA, USA) for 1 h at room
temperture. After washing with PBS three times, the cells were
stained using tetramethylrhodamine B isothiocyanate-labeled
phalloidin and incubated for 30 min at room temperature in the
dark, and then counterstained with DAPI (cat. no. C1006; Beyotime
Institute of Biotechnology, Shanghai, China) and imaged with a
fluorescence microscope (FLX 800; Olympus, Tokyo, Japan). Finally,
the cell size was analyzed using Image-Pro Plus image analysis
software (version 6.0; Media Cybernetics, Inc., Rockville, MD,
USA).
Cell apoptosis assay
Cell apoptosis was analyzed using a Annexin
V-fluorescein isothiocyanate (FITC) and propidium iodide (PI)
double staining apoptosis detection kit (cat. no. G003; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) and flow
cytometry. In brief, after incubation as previously described, the
H9c2 cells were collected, resuspended in binding buffer and then
stained with Annexin V-FITC and PI solution for 15 min at 37°C in
the dark. Finally, cell samples were quantified by flow cytometric
analysis (BD FACSVantage™ SE; BD Biosciences, Franklin
Lakes, NJ, USA). The index of apoptosis was expressed as the
percentage of total apoptotic cells, which included the percentage
of early apoptotic cells (Annexin V-positive and PI-negative) plus
the percentage of late apoptotic cells (Annexin V-positive and
PI-positive).
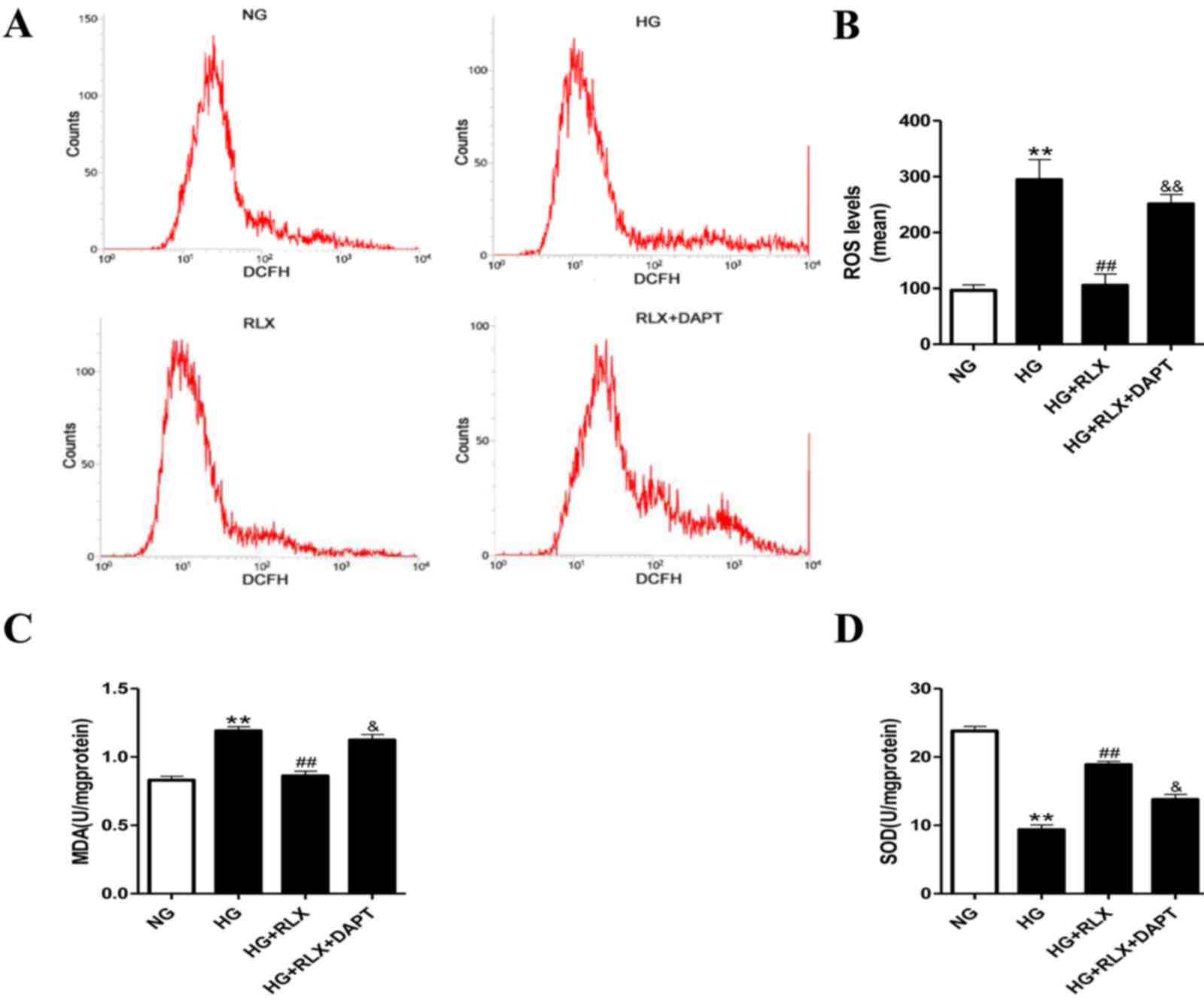
Intracellular ROS measurement
Intracellular ROS was assessed using the fluorescent
probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) (cat. no. S0033;
Beyotime Institute of Biotechnology). In brief, after incubation as
mentioned above, the H9c2 cells were stained with 10 µmol DCFH-DA
in PBS for 30 min at 37°C in the dark. The fluorescence intensity
was determined by flow cytometry and in addition, images were
captured with a fluorescence microscope (FLX 800; Olympus) with an
excitation and emission wavelength of 488 and 525 nm, respectively.
The mean fluorescence intensity was calculated using Image-Pro Plus
software (version 6.0).
Malondialdehyde (MDA) and superoxide
dismutase (SOD) measurements
The activity of SOD (cat. no. A001-3) and the levels
of MDA (cat. no. A003-1) (both Nanjing Jiancheng Bioengineering
Institute) were detected with the relevant detection kits according
to the manufacturer's protocol. In brief, after incubation as
described above, the H9c2 cells were harvested, lysates were
prepared by ultrasonication at 24 KHz amplitude for 2 sec, all
steps were performed on ice and repeated five times, and further
centrifuged at 12,000 × g for 10 min at 4°C. The absorbance of the
colorimetric stains was determined at wavelengths of 532 and 450
nm, respectively. MDA and SOD levels were calculated using standard
curves and normalized to the normal control.
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using RNAiso Plus
reagent (cat. no. 9108; Takara Bio Inc., Dalian, China) and
complementary DNA was synthesized using a PrimeScript™
RT reagent kit (Perfect Real Time) (cat. no. RR037A; Takara Bio
Inc.). PCR amplification reactions were performed on an ABI-PRISM
7700 Sequence Detection system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with the SYBR® Premix Ex
Taq™ II (Tli RNase H Plus) (cat. no. RR820A; Takara Bio
Inc.) and the primers listed in Table
I. The PCR amplification conditions were as follows: 95°C for
30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec
for denaturation, annealing and elongation. The PCR products were
quantified using the 2−∆∆Cq comparative method (23) with GAPDH as an internal control.
 | Table I.Primers sequences used for
reverse-transcription quantitative polymerase chain reaction
analysis. |
Table I.
Primers sequences used for
reverse-transcription quantitative polymerase chain reaction
analysis.
| Gene | Primers 5′-3′ | Size (bp) |
|---|
| Notch1 | Forward:
CCTTGTCCCCGATTATCTACCA | 116 |
|
| Reverse:
CAGGTTCTGAGGCT GGATTTGT |
|
| hes1 | Forward:
GGCCAATTTGCTTTCCTCATC | 113 |
|
| Reverse:
GAAGGCGACACTGCGTTAGG |
|
| ANP | Forward:
CTGGGGAAGTCAACCCGTCT | 174 |
|
| Reverse:
TCTGGGCTCCAATCCTGTCA |
|
| BNP | Forward:
AGCCAGTCTCCAGAACAATCCA | 188 |
|
| Reverse:
TGTGCCATCTTGGAATTTCGA |
|
| GAPDH | Forward:
GACATGCCGCCTGGAGAAC | 92 |
|
| Reverse:
AGCCCAGGATGCCCTTTAGT |
|
Western blot analysis
Cells were lysed with radioimmunoprecipitation assay
buffer supplemented with a mixture of protease inhibitors (Beyotime
Institute of Biotechnology), and the lysates were clarified by
centrifugation at 12,000 × g for 15 min at 4°C. Protein
concentrations were quantified using a bicinchoninic acid assay kit
(Thermo Fisher Scientific, Inc.). Equal amounts of protein (10
µg/lane) were subjected to 12% SDS-PAGE and electrotransferred onto
polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA,
USA). Subsequently, the membranes were blocked with 5% skimmed milk
for 2 h at room temperature and incubated overnight at 4°C with
primary antibodies to Notch1 (cat. no. ab52301), which recognized
Notch-1 receptor and its activated form Notch-ICD, atrial
natriuretic polypeptide (ANP; cat. no. ab189921), hairy and
enhancer of split 1 (hes1; cat. no. ab108937) (all 1:1,000; Abcam,
Cambridge, MA, USA), brain natriuretic peptide (BNP; cat. no.
wl02126), MnSOD (cat. no. wl02506;), cytochrome C (cat. no.
wl01571), caspase-3 (cat. no. wl01992) (all 1:500; Wanlei
Biotechnology, Shenyang, China) and GAPDH (1:1,000; cat. no. 5174;
Cell Signaling Technology, Inc., Danvers, MA, USA). Finally, the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit immunoglobulin G (1:5,000; cat. no. BA1054; Boster
Biological Technology) for 2 h at room temperature. Proteins were
detected using enhanced chemiluminescent reagent (Beyotime
Institute of Biotechnology) and images were captured with a
ChemiDoc XRS detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The values were normalized to GAPDH and
densitometric analysis of the bands was performed using Fusion-Capt
analysis software (Vilber Lourmat Deutschland GmbH, Eberhardzell,
Germany).
Statistical analysis
Statistical analysis was performed using SPSS 19.0
software (IBM Corp., Armonk, NY, USA). Values are expressed as the
mean ± standard deviation. Comparisons between groups were made
using one-way analysis of variance followed by the Tukey's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RLX increases HG-induced cell
viability and decreases apoptosis
The cell viability in the normal control group was
defined as 100%, while that in the other groups was presented as
the percentage of the control group. The CCK8 assay revealed that
HG caused a marked reduction in the viability of the H9c2 cells
when compared with that in the control group (P<0.01; Fig. 1A). However, the viability of H9c2
cells was statistically increased by RLX in the 50 nmol/ml group
(P<0.05). With a further increase of the RLX concentration, the
viability of cells was considerably increased by RLX in the 100 and
200 nmol/ml groups (P<0.01). In addition, with the prolongation
of the incubation time, the viability of H9c2 cells was gradually
increased by RLX and significantly increased after 48 h (P<0.01)
(data not shown). These results indicated that RLX increased the
cell viability in a dose- and time-dependent manner. Accordingly,
100 nmol/ml RLX was applied to H9c2 cardiomyocytes for a duration
of 48 h in the following experiments.
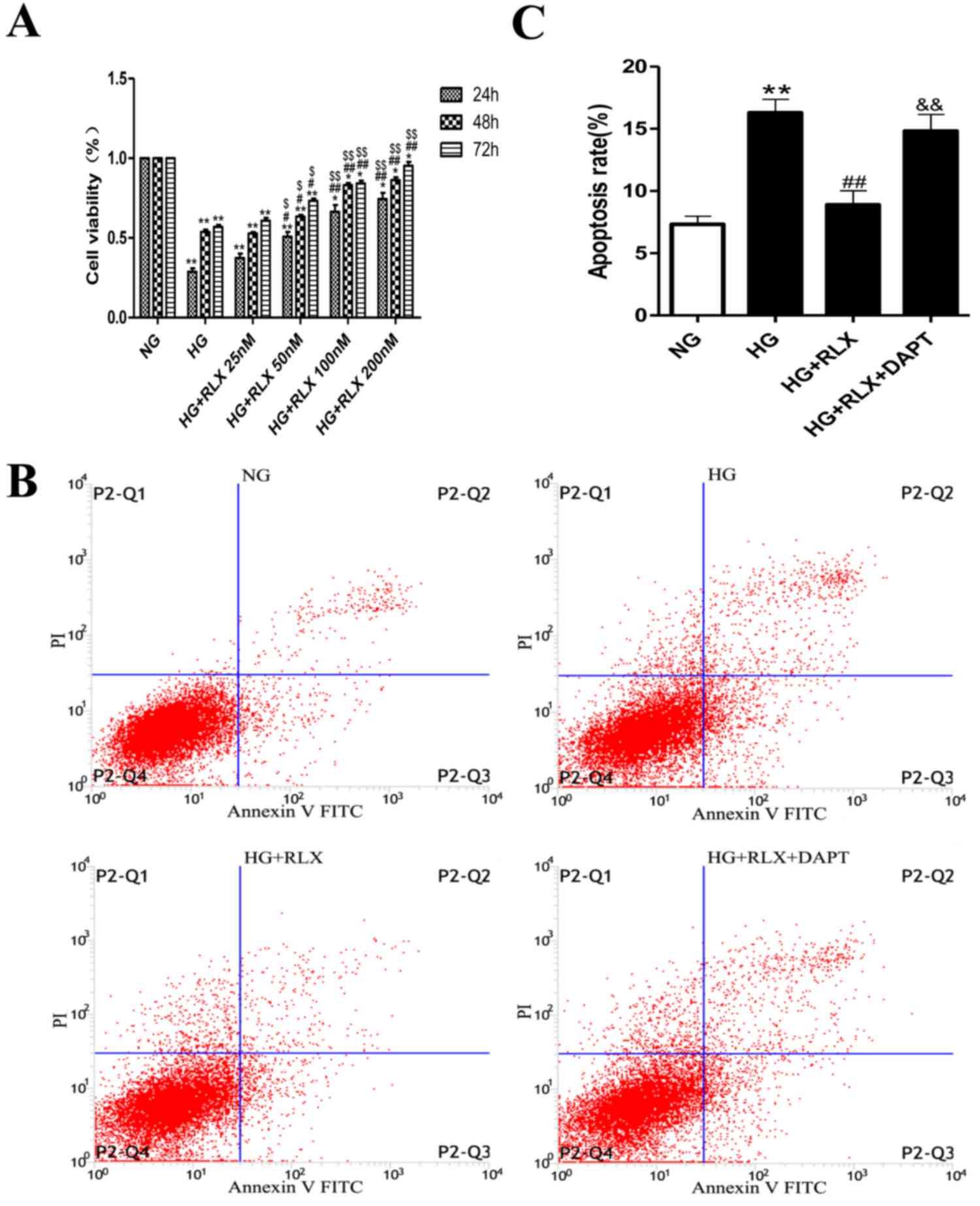 | Figure 1.RLX increases cell viability in a
dose- and time-dependent manner and reduces apoptosis of H9c2 cells
exposed to HG. The cell viability in the normal control group was
defined as 100%, while that in the other groups was presented as
the percentage of the control group. (A) Cell viability was
measured by a Cell Counting Kit-8 assay. (B) Cell apoptosis was
measured using Annexin-V/PI staining and flow cytometry. P2-Q1
represents the percentages of dead cells, P2-Q2 represents the
percentages of cells in early apoptosis, P2-Q3 represents the
percentages of cells in late apoptosis and P2-Q4 represents the
percentages of viable cells. (C) Quantitative analysis of flow
cytometry results. Values are expressed as the mean ± standard
deviation (n=6 in each group). *P<0.05 and **P<0.01 vs. NG
group; #P<0.05 and ##P<0.01 vs. HG
group; $P<0.05 and $$P<0.01 vs. HG +
RLX (25 nM) group; &&P<0.01 vs. HG + RLX
group. FITC, fluorescein isothiocyanate; P2, the number of cells;
PI, propidium iodide; Q, quadrant; NG, normal glucose; HG, high
glucose; RLX, relaxin. |
To test the effect of RLX on cardiac apoptosis in
H9c2 cells exposed to HG, Annexin V-FITC/PI dual staining and flow
cytometry were used. As presented in Fig. 1B and C. the early and late apoptotic
rates were significantly increased in the HG group compared with
those in the control group (P<0.01). When compared with the HG
group, addition of RLX resulted in a noticeable decrease in
apoptotic cells (P<0.01), which was consistent with the results
of previous studies (14,16). Furthermore, H9c2 cells were incubated
with the Notch inhibitor DAPT for 4 h prior to treatment with HG.
The number of apoptotic cells was increased when compared with that
in the HG + RLX group (P<0.01).
RLX attenuates HG-induced
cardiomyocyte hypertrophy
To assess cellular hypertrophy, the cell surface
area was determined by fluorescence microscopy. As displayed in
Fig. 2A and B, H9c2 cells treated
with HG for 48 h demonstrated a marked increase in cell size
compared with that in the control group (P<0.01), which was
reduced by simultaneous treatment with RLX (P<0.05). However,
pretreatment with DAPT markedly enhanced the cell surface area
compared with that in the HG+RLX group (P<0.05).
RLX prevents HG-induced oxidative
stress
To determine whether the HG-mediated increase in
oxidative stress is critical for the effects of RLX on myocardial
injury, the intracellular levels of oxidative stress were
determined by DCFH-DA staining. It was demonstrated that compared
with the control group, incubation under HG significantly enhanced
ROS (P<0.01; Fig. 3A and B) and
MDA (P<0.01; Fig. 3C) production
and reduced SOD activity (P<0.01; Fig. 3D). Treatment with RLX significantly
suppressed ROS and MDA generation with a concomitant increase in
SOD activity (all P<0.01) compared with the HG group. Of note,
compared with the HG+RLX group, pretreatment with DAPT almost
abolished the protective effects of RLX against oxidative stress
(P<0.05).
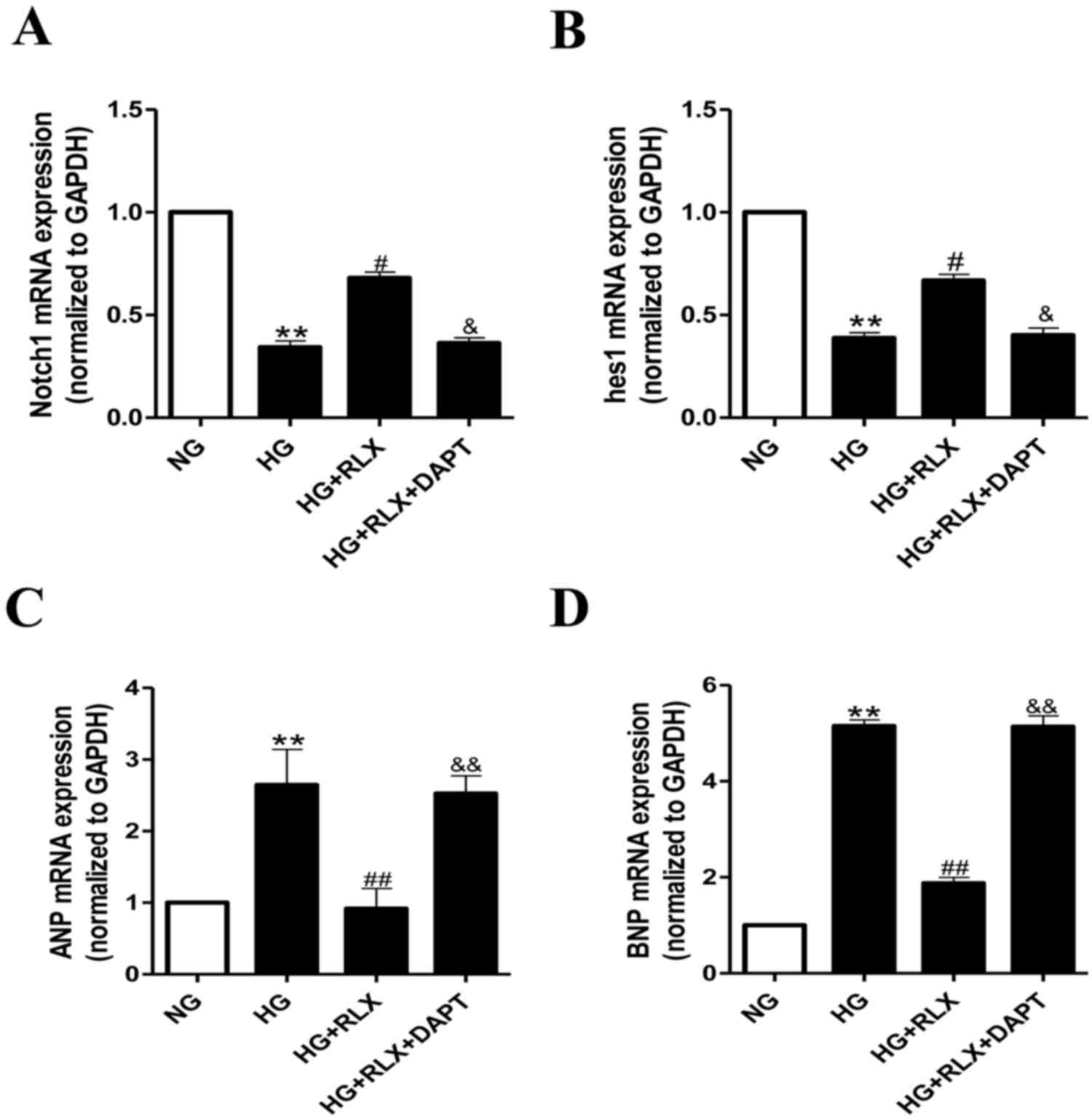
RLX attenuates HG-induced
cardiomyocyte hypertrophy and apoptosis via Notch1 signaling
To confirm whether Notch1 signaling is critical for
the effects of RLX on HG-induced cardiac hypertrophy and apoptosis,
alterations in the expression of Notch1 and its downstream protein,
hes1, were examined by RT-qPCR and western blot analysis. As
presented in Fig. 4A and B, the
expression of Notch1 and hes1 mRNA was significantly decreased in
H9c2 cells exposed to HG compared with that in the control group
(all P<0.01). Addition of RLX significantly enhanced the gene
expression of Notch1 and hes1 when compared with that in the HG
group (all P<0.05). Furthermore, pretreatment with DAPT
significantly reduced the expression of Notch1 and hes1 mRNA
compared with that in the HG+RLX group (all P<0.05).
As presented in Fig. 4C
and D, the mRNA expression levels of ANP and BNP were increased
in the HG group compared with those in the control group (all
P<0.01). Addition of RLX led to a downregulation of ANP and BNP
mRNA expression as compared with that in the HG group (all
P<0.01). Pretreatment with DAPT led to an upregulation of the
mRNA expression of ANP and BNP as compared with that in the HG+RLX
group (all P<0.01).
These results were further confirmed by western blot
analysis (Fig. 5A-D). Overall, it
was indicated that RLX attenuated HG-induced cardiomyocyte
hypertrophy and apoptosis via mechanisms involving the Notch1
signaling pathway.
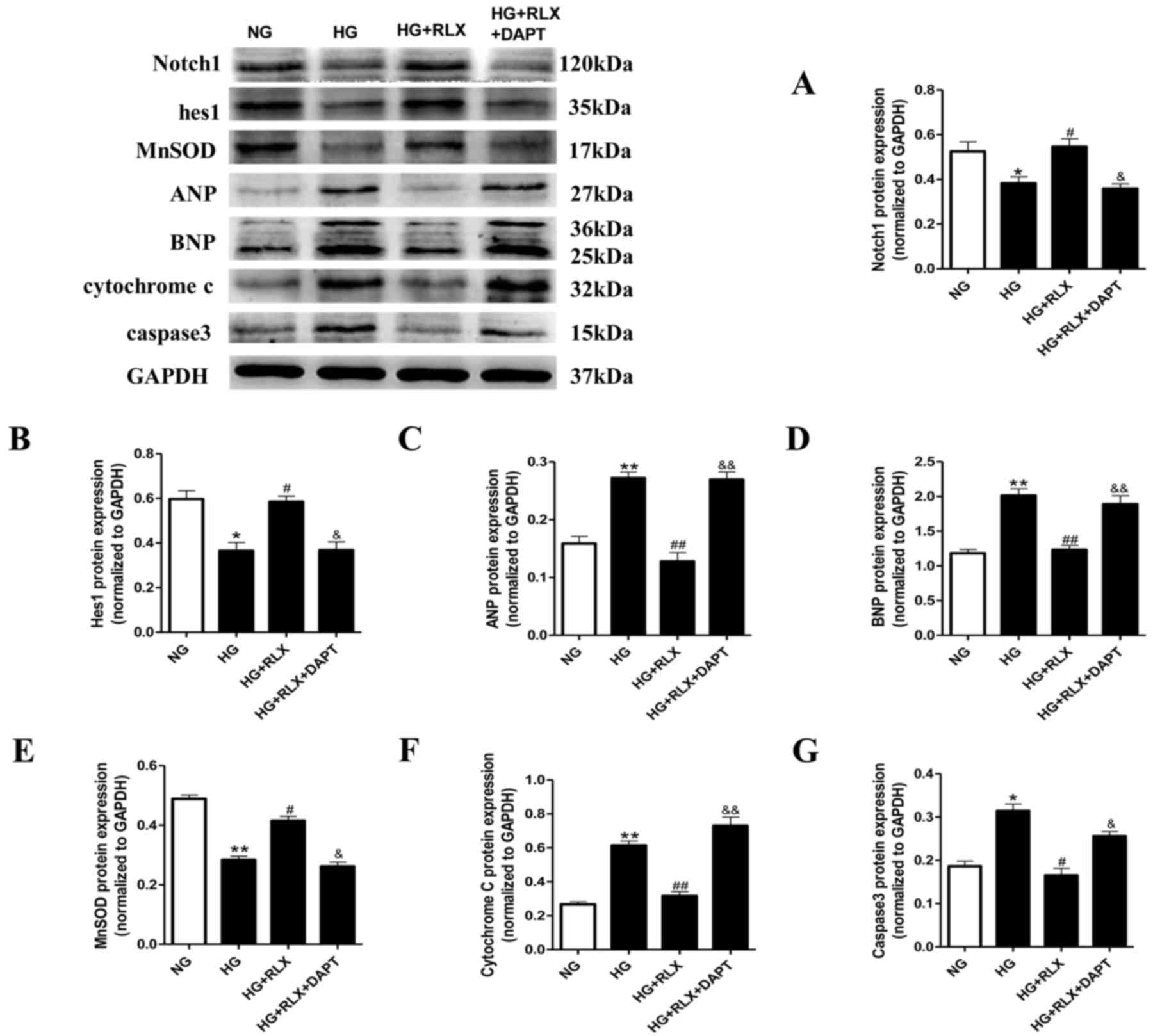 | Figure 5.RLX decreases ANP, BNP, caspase-3 and
cytochrome C protein levels, and increases MnSOD activity, as well
as activator of Notch1 and hes1 protein expression in H9c2 cells
exposed to HG. Protein expression was measured by western blot
analysis. (A-G) Quantitative analysis of (A) Notch1, (B) hes1, (C)
ANP and (D) BNP, (E) MnSOD, (F) cytochrome C and (G) caspase 3. For
the quantitative evaluation of BNP, the two bands obtained were for
two different glycosylated BNP precursors and both were taken into
account. Values are expressed as the mean ± standard deviation (n=6
in each group). *P<0.05 and **P<0.01 vs. NG group;
#P<0.05 and ##P<0.01 vs. HG group;
&P<0.05 and &&P<0.01 vs. HG
+ RLX group. NG, normal glucose; HG, high glucose; RLX, relaxin;
ANP, atrial natriuretic polypeptide; BNP, brain natriuretic
polypeptide; hes1, hairy and enhancer of split 1; MnSOD, manganese
superoxide dismutase. |
Oxidative damage was further assessed through
quantitating the protein expression of MnSOD and cytochrome C by
western blot analysis. As presented in Fig. 5E and F, the expression of MnSOD
(P<0.01) in cardiomyocytes exposed to HG was significantly
decreased compared with that in the control group, accompanied with
a marked increase in cytochrome C (P<0.01). Application of RLX
significantly upregulated the expression of MnSOD (P<0.05) and
inhibited cytochrome C (P<0.01) compared with the HG group.
Pretreatment with DAPT significantly abrogated the protective
effects of RLX.
Furthermore, the protein levels of caspase-3, which
is representative of the extrinsic and intrinsic pathways of
apoptosis, were detected by western blot analysis (Fig. 5G). The expression of caspase-3 was
significantly increased in the HG group compared with that in the
control group (P<0.05). However, when compared with that in the
HG group, administration of RLX led to a significant downregulation
of the expression of caspase-3 (P<0.05). Pretreatment with DAPT
largely abrogated the protective effects of RLX (P<0.05). These
results demonstrated that Notch1 signaling may be involved in the
protective role of RLX against cardiac cell apoptosis.
Discussion
The present study found that incubation under HG
decreased cell viability, enhanced oxidative stress and increased
cardiomyocyte hypertrophy and caused apoptosis in H9c2 cells.
However, RLX treatment increased the cell viability in a dose- and
time-dependent manner, reduced intracellular oxidative stress,
which was concomitant with an inhibition of cardiomyocyte
hypertrophy, manifested as increased cell surface area, reduced ANP
and BNP content, and decreased apoptosis manifested as increased
apoptotic cell death and increased caspase-3 protein expression.
Furthermore, RLX treatment activated the expression of Notch1 and
hes1 under HG conditions, while the Notch1 inhibitor DAPT largely
abolished RLX-induced increases in Notch1 and hes1 and prevented
RLX-mediated cardiomyocyte protection, suggesting that RLX has a
role in cardioprotection at least in part through the Notch1/hes1
signaling pathway.
RLX, an insulin-like peptide hormone, does not mimic
the metabolic effects of insulin, but increased RLX levels were
demonstrated to improve insulin sensitivity in diabetic patients
(24). Previous studies have
indicated that RLX levels were significantly lower in patients with
diabetes, whereas a compensatory increase in RLX levels helped
pregnant women with diabetes mellitus overcome increased insulin
resistance (25,26). In fact, clinical and experimental
studies have suggested that RLX protects the heart from HG-induced
myocardial damage, possibly through its vasodilative,
antiangiogenic (7), antifibrotic
(13) and antiapoptotic actions
(14). Although the effects of RLX
on HG-induced cardiomyocyte apoptosis were previously investigated,
the molecular mechanism has remained to be determined. In addition,
the effect of RLX on the interaction between hyperglycemia and
hypertrophy, as well as the underlying intracellular signaling
pathways have remained elusive.
Increased oxidative stress is closely associated
with cardiac hypertrophy and apoptosis, which has been reported in
numerous studies (5,27). Evidence from animal and cell culture
experiments indicated that ROS elimination inhibited
hyperglycemia-induced hypertrophy and apoptosis (4). Several studies have demonstrated that
RLX is a potent antioxidant effector that protects against the
development of certain diseases by reducing ROS production and
maintaining nitric oxide availability (28,29). In
addition, studies have revealed that RLX prevents HG- (14) or ischemia/reperfusion-induced
(11) myocardial apoptosis in
neonatal rat ventricular myocytes by reducing oxidative damage and
inhibiting apoptosis-associated pathways. The present findings are
in accordance with a previous study indicating that RLX treatment
inhibited HG-induced cardiomyocyte apoptosis, manifested as an
increased apoptotic cell index, and upregulated the protein
synthesis of caspase-3, as a key mediator of apoptosis (30).
Previous studies indicated that endogenous RLX is
upregulated in the hypertrophic heart and that RLX treatment
reversed hypertrophy in spontaneously hypertensive rats; RLX
treatment was also demonstrated to counteract phenylephrine-induced
cardiomyocyte hypertrophy by inhibiting activated extracellular
signal-regulated kinase (ERK)-1/2 (15,16).
However, Moore et al (16)
reported that RLX alone failed to suppress phenylephrine-induced
cardiomyocyte hypertrophy, whereas RLX indirectly inhibited cardiac
fibroblast-conditioned medium-induced hypertrophy and directly
inhibited H2O2-induced apoptosis through
activation of the Akt and ERK pathways. In addition, Xu et
al (31) demonstrated that
endogenous RLX had no significant effect on chronic pressure
overload-induced cardiac hypertrophy and fibrosis. Therefore, the
effect of RLX on cardiac hypertrophy is currently controversial.
The present study reported that administration of RLX inhibited
HG-induced increases in cell surface area and the expression of ANP
and BNP as markers of hypertrophy at the mRNA and protein level. To
our knowledge, our study has revealed, for the first time, RLX
inhibited HG-induced cardiomyocyte hypertrophy.
In order to verify whether the prevention of
HG-induced hypertrophy and apoptosis by RLX was through inhibition
of oxidative stress, the ROS-sensitive DCFH-DA dye was used to
measure intracellular ROS levels. MDA as a byproduct of lipid
peroxidation and SOD as an antioxidant enzyme were assessed as
typical biomarkers of oxidative insults. In addition, MnSOD (a
mitochondrial antioxidant enzyme) and cytochrome C (32) are important members of the intrinsic
mitochondrial pathway. The mitochondria are a major source of ROS.
The present study indicated that RLX treatment reduced the
intracellular ROS and MDA levels, enhanced SOD activity, and
increased the protein synthesis of MnSOD, as well as decreased the
protein expression of cytochrome C, which was concomitant with the
inhibition of cardiomyocyte hypertrophy and apoptosis in H9c2 cells
under HG treatment. Therefore, the present results collectively
demonstrated that RLX attenuated HG-induced cardiomyocyte
hypertrophy and apoptosis via oxidative stress.
Previous studies have indicated that the activation
of Notch1 signaling improves diabetic wound healing (33) and diabetes-induced microvasculopathy
(34) in a streptozotocin-induced
mouse model of diabetes mellitus. Another study also demonstrated
that activation of the Notch1 and phosphoinositide-3 kinase/Akt
signaling pathways is critical for the prevention of DCM by
inhibiting HG-induced cardiomyocyte apoptosis (35). Basu et al (36) indicated that hyperglycemia inhibited
the expression of Notch1 and its downstream effectors in chick
embryos, as well as H9c2 and endocardial-derived cells, and
increased the risk of congenital heart defects in maternal mice
with diabetes mellitus. In addition, animal studies have revealed
that Notch1 overexpression protects against myocardial
ischemia-reperfusion and ischemic preconditioning, as well as
ischemic postconditioning injury by reducing cardiomyocyte
apoptosis, which involves reduction of oxidative/nitrative stress
(37,38). The involvement of Notch1 in cardiac
hypertrophy has been reported previously. For instance, the
expression of Notch1 and hes1 is downregulated in the one-kidney
one clip mouse model (39) and in
the adult heart under pressure overload-induced cardiac hypertrophy
(19). Conversely, Notch inhibition
accelerates the development of cardiac hypertrophy and fibrosis.
Therefore, the aforementioned studies collectively indicated that
Notch1 signaling has a pivotal role in cardiac hypertrophy and
apoptosis associated with various cardiac diseases.
Boccalini et al (11) and Zhou et al (22) reported that the protective effects of
RLX may depend on the mechanism of Notch1 activation. Their studies
emphasized the antiapoptotic and antifibrotic actions of RLX using
a model of hypoxia/reoxygenation injury or a model of cardiac
fibrosis. However, the focus of the present study was to
investigate the effect of RLX on HG-induced cardiomyocyte
hypertrophy and apoptosis using a model of hyperglycemia-induced
injury in vitro, and to assess the possible molecular
mechanism, which was different from the studies by Boccalini et
al (11) and Zhou et al
(22). In addition, Boccalini et
al (11) demonstrated that RLX
inhibits hypoxia/reoxygenation-induced apoptosis in H9c2 rat
cardiomyoblasts. While RLX was previously reported to regulate
HG-induced cardiac apoptosis, it had remained elusive whether the
underlying mechanism involves the Notch1 signaling pathway. Of
note, the present study was the first, to the best of our
knowledge, to reveal that RLX inhibits HG-induced cardiomyocyte
apoptosis through activation of Notch1 signaling.
To investigate the Notch1 pathway and whether it has
a pivotal role in the effects of RLX on HG-induced cardiac
hypertrophy and apoptosis, the highly active γ-secretase inhibitor
DAPT was used to block the Notch1 pathway. The results indicated
that HG decreased the expression of Notch1 and hes1, and blocking
the Notch1 pathway with DAPT increased cardiac hypertrophy and
apoptosis. It was revealed that DAPT largely, but not fully offset
the cytoprotective effects of RLX, suggesting that multiple
signaling pathways may be activated by RLX in cardiac myocytes.
Therefore, these results collectively indicated that RLX-mediated
inhibition of HG-induced cardiac hypertrophy and apoptosis is at
least partly due to activation of the Notch1 signaling pathway.
However, the present study had several limitations.
First, all experiments were performed using the H9c2 cell line, and
further exploration should be done using primary neonatal
cardiomyocytes or in vivo experiments. Furthermore, the
expression of ANP and BNP was examined in the present study, while
these proteins do not adequately represent hypertrophy in
vivo. Therefore, more specific markers for the hypertrophic
heart should be considered in the future. Additional studies are
required to clarify whether the anti-hypertrophic and
anti-apoptotic action of RLX is mediated via multiple signaling
pathways, and to investigate how the Notch1 pathway is interlinked
and cooperates to mediate the effects of RLX. Further studies are
required to address these questions.
In conclusion, the results of the present study
demonstrated that RLX protects H9c2 cells from HG-induced
hypertrophy and apoptosis at least partly through the activation of
Notch1 signaling, and may be associated with the inhibition of
oxidative stress. The present findings provided novel insight into
the molecular mechanisms of RLX-mediated cardioprotection as well
as further evidence for selecting RLX as a novel therapeutic drug
for the treatment of DCM.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81300140).
Glossary
Abbreviations
Abbreviations:
|
RLX
|
relaxin
|
|
HG
|
high glucose
|
|
ANP
|
atrial natriuretic polypeptide
|
|
BNP
|
brain natriuretic peptide
|
|
ROS
|
reactive oxygen species
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
References
|
1
|
Chavali V, Tyagi SC and Mishra PK:
Predictors and prevention of diabetic cardiomyopathy. Diabetes
Metab Syndr Obes. 6:151–160. 2013.PubMed/NCBI
|
|
2
|
Rubler S, Dlugash J, Yuceoglu YZ, Kumral
T, Branwood AW and Grishman A: New type of cardiomyopathy
associated with diabetic glomerulosclerosis. Am J Cardiol.
30:595–602. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hayat SA, Patel B, Khattar RS and Malik
RA: Diabetic cardiomyopathy: Mechanisms, diagnosis, and treatment.
Clin Sci (Lond). 107:539–557. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou X and Lu X: The role of oxidative
stress in high glucose-induced apoptosis in neonatal rat
cardiomyocytes. Exp Biol Med (Maywood). 238:898–902. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huynh K, Bernardo BC, McMullen JR and
Ritchie RH: Diabetic cardiomyopathy: Mechanisms and new treatment
strategies targeting antioxidant signaling pathways. Pharmacol
Ther. 142:375–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sherwood OD: Relaxin's physiological roles
and other diverse actions. Endocr Rev. 25:205–234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bitto A, Irrera N, Minutoli L, Calò M, Lo
Cascio P, Caccia P, Pizzino G, Pallio G, Micali A, Vaccaro M, et
al: Relaxin improves multiple markers of wound healing and
ameliorates the disturbed healing pattern of genetically diabetic
mice. Clin Sci. 125:575–585. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Samuel CS, Unemori EN, Mookerjee I,
Bathgate RA, Layfield SL, Mak J, Tregear GW and Du XJ: Relaxin
modulates cardiac fibroblast proliferation, differentiation, and
collagen production and reverses cardiac fibrosis in vivo.
Endocrinology. 145:4125–4133. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taylor MJ and Clark CL: Evidence for a
novel source of relaxin: Atrial cardiocytes. J Endocrinol.
143:R5–R8. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Formigli L, Francini F, Nistri S, Margheri
M, Luciani G, Naro F, Silvertown JD, Orlandini SZ, Meacci E and
Bani D: Skeletal myoblasts overexpressing relaxin improve
differentiation and communication of primary murine cardiomyocyte
cell cultures. J Mol Cell Cardiol. 47:335–345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boccalini G, Sassoli C, Formigli L, Bani D
and Nistri S: Relaxin protects cardiac muscle cells from
hypoxia/reoxygenation injury: Involvement of the Notch-1 pathway.
FASEB J. 29:239–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan YY, Wade JD, Tregear GW and Summers
RJ: Quantitative autoradiographic studies of relaxin binding in rat
atria, uterus and cerebral cortex: Characterization and effects of
oestrogen treatment. Br J Pharmacol. 127:91–98. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang P, Li HW, Wang YP, Chen H and Zhang
P: Effects of recombinant human relaxin upon proliferation of
cardiac fibroblast and synthesis of collagen under high glucose
condition. J Endocrinol Invest. 32:242–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X, Ma X, Zhao M, Zhang B, Chi J, Liu
W, Chen W, Fu Y, Liu Y and Yin X: H2 and H3 relaxin inhibit high
glucose-induced apoptosis in neonatal rat ventricular myocytes.
Biochimie. 108:59–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parikh A, Patel D, McTiernan CF, Xiang W,
Haney J, Yang L, Lin B, Kaplan AD, Bett GC, Rasmusson RL, et al:
Relaxin suppresses atrial fibrillation by reversing fibrosis and
myocyte hypertrophy and increasing conduction velocity and sodium
current in spontaneously hypertensive rat hearts. Circ Res.
113:313–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moore XL, Tan SL, Lo CY, Fang L, Su YD,
Gao XM, Woodcock EA, Summers RJ, Tregear GW, Bathgate RA and Du XJ:
Relaxin antagonizes hypertrophy and apoptosis in neonatal rat
cardiomyocytes. Endocrinology. 148:1582–1589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
High FA and Epstein JA: The multifaceted
role of Notch in cardiac development and disease. Nat Rev Genet.
9:49–61. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dabral S, Tian X, Kojonazarov B, Savai R,
Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, et
al: Notch1 signalling regulates endothelial proliferation and
apoptosis in pulmonary arterial hypertension. Eur Respir J.
48:1137–1149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nemir M, Metrich M, Plaisance I, Lepore M,
Cruchet S, Berthonneche C, Sarre A, Radtke F and Pedrazzini T: The
Notch pathway controls fibrotic and regenerative repair in the
adult heart. Eur Heart J. 35:2174–2185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Urbanek K, Cabral-da-Silva MC, Ide-Iwata
N, Maestroni S, Delucchi F, Zheng H, Ferreira-Martins J, Ogórek B,
D'Amario D, Bauer M, et al: Inhibition of notch1-dependent
cardiomyogenesis leads to a dilated myopathy in the neonatal heart.
Circ Res. 107:429–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sassoli C, Chellini F, Pini A, Tani A,
Nistri S, Nosi D, Zecchi-Orlandini S, Bani D and Formigli L:
Relaxin prevents cardiac fibroblast-myofibroblast transition via
notch-1-mediated inhibition of TGF-β/Smad3 signaling. PLoS One.
8:e638962013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou X, Chen X, Cai JJ, Chen LZ, Gong YS,
Wang LX, Gao Z, Zhang HQ, Huang WJ and Zhou H: Relaxin inhibits
cardiac fibrosis and endothelial-mesenchymal transition via the
Notch pathway. Drug Des Devel Ther. 9:4599–4611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bani D, Pini A and Yue SK: Relaxin,
insulin and diabetes: An intriguing connection. Curr Diabetes Rev.
8:329–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Zhu M, Zhao M, Chen W, Fu Y, Liu
Y, Liu W, Zhang B, Yin X and Bai B: The plasma levels of relaxin-2
and relaxin-3 in patients with diabetes. Clin Biochem.
46:1713–1716. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Whittaker PG, Edwards JR, Randolph C,
Büllesbach EE, Schwabe C and Steinetz BG: Abnormal relaxin
secretion during pregnancy in women with type 1 diabetes. Exp Biol
Med (Maywood). 228:33–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gupta MK, Neelakantan TV, Sanghamitra M,
Tyagi RK, Dinda A, Maulik S, Mukhopadhyay CK and Goswami SK: An
assessment of the role of reactive oxygen species and redox
signaling in norepinephrine-induced apoptosis and hypertrophy of
H9c2 cardiac myoblasts. Antioxid Redox Signal. 8:1081–1093. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ng HH, Jelinic M, Parry LJ and Leo CH:
Increased superoxide production and altered nitric oxide-mediated
relaxation in the aorta of young but not old male relaxin-deficient
mice. Am J Physiol Heart Circ Physiol. 309:H285–H296. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Willcox JM and Summerlee AJ: Relaxin
protects astrocytes from hypoxia in vitro. PLoS One. 9:e908642014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lakhani SA, Masud A, Kuida K, Porter GA
Jr, Booth CJ, Mehal WZ, Inayat I and Flavell RA: Caspases 3 and 7:
Key mediators of mitochondrial events of apoptosis. Science.
311:847–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Q, Lekgabe ED, Gao XM, Ming Z, Tregear
GW, Dart AM, Bathgate RA, Samuel CS and Du XJ: Endogenous relaxin
does not affect chronic pressure overload-induced cardiac
hypertrophy and fibrosis. Endocrinology. 149:476–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cai L, Li W, Wang G, Guo L, Jiang Y and
Kang YJ: Hyperglycemia-induced apoptosis in mouse myocardium:
mitochondrial cytochrome C-mediated caspase-3 activation pathway.
Diabetes. 51:1938–1948. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang RH, Qi SH, Shu B, Ruan SB, Lin ZP,
Lin Y, Shen R, Zhang FG, Chen XD and Xie JL: Epidermal stem cells
(ESCs) accelerate diabetic wound healing via the Notch signalling
pathway. Biosci Rep. 36:pii: e003642016. View Article : Google Scholar
|
|
34
|
Yoon CH, Choi YE, Cha YR, Koh SJ, Choi JI,
Kim TW, Woo SJ, Park YB, Chae IH and Kim HS: Diabetes-induced
jagged1 overexpression in endothelial cells causes retinal
capillary regression in a murine model of diabetes mellitus:
Insights into diabetic retinopathy. Circulation. 134:233–247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Li B, Zheng Z, Kang T, Zeng M,
Liu Y and Xia B: Protective effects of Notch1 signaling activation
against high glucose-induced myocardial cell injury: Analysis of
its mechanisms of action. Int J Mol Med. 36:897–903. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Basu M, Bosse K and Garg V: Notch1
Haploinsufficiency Increases Risk of Congenital Heart Defects in
the Setting of Maternal Diabetes by an Epigenetic Mechanism.
Circulation. 130:192852014.
|
|
37
|
Pei H, Yu Q, Xue Q, Guo Y, Sun L, Hong Z,
Han H, Gao E, Qu Y and Tao L: Notch1 cardioprotection in myocardial
ischemia/reperfusion involves reduction of oxidative/nitrative
stress. Basic Res Cardiol. 108:3732013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou XL, Wan L, Xu QR, Zhao Y and Liu JC:
Notch signaling activation contributes to cardioprotection provided
by ischemic preconditioning and postconditioning. J Transl Med.
11:2512013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nemir M, Jordan N, Croquelois A,
Domenighetti A and Pedrazzini T: Notch signaling: A potential
regulator of cardiac response to hypertrophic stimuli. J Mol Cell
Cardiol. 42 Suppl:S134–S135. 2007. View Article : Google Scholar
|