Introduction
Breast cancer is the most lethal gynecological
malignancy and a leading cause of cancer deaths among women
worldwide. Prevention, early detection, and effective treatment are
the key intervention points to increase the survival rate (1,2). About
1.7 million new breast cancer patients were diagnosed in 2012,
representing approximately 12% of all cancer cases globally that
year (3). Currently, breast cancer
is classified into five major groups on the basis of molecular
profiling. Doxorubicin, gemcitabine, and taxanes are major clinical
chemotherapeutic agents (4,5) Side-effects and drug resistance are
common problems in patients taking long-term medication (6).
Triptolide (TPI) is an active compound with an
epoxy-diterpene structure, which can be extracted from the Chinese
herb Tripterygium wilfordii Hook F., and has been shown to
have potent anti-tumor activity (7).
TPI affects cell proliferation, growth and apoptosis through
upregulating apoptosis protein expression, inducing cell cycle
arrest, inhibiting tumor angiogenesis and inducing autophagy. TPI
has significant anti-tumor effects on various types of cancer cell
in vitro and in vivo, owing to its induction of cell
death, and has been found to be effective in lung cancer (8), prostate cancer (9), colon carcinoma (10,11), and
others. TPI also shows potent biological effects, such as reduction
of inflammatory and immunosuppression. With anti-inflammatory
property, TPI may protect dopaminergic neurons in vitro and
in vivo, which abolished the excessive production of
cytokines, such as tumor necrosis factor-α (TNF-α) and
interleukin-1β (IL-1β) (12). TPI
showed antiestrogenic activity (13), and induced S cell cycle arrest on
cell cycle distribution in MCF-7 cells (14). Characteristics of TPI-induced cell
death may include DNA damage, cytochrome C release, activation of
inflammatory pathways, promotion of autophagy and changes in
expression levels of apoptosis proteins (15). However, the anti-tumor effects and
mechanisms in breast cancer are unclear.
Mitogen-activated protein kinases (MAPK) are
involved in mediating cell survival (11). The role of extracellular
signal-regulated kinase (Erk) phosphorylation in cell proliferation
and death is controversial (16,17). It
has been reported that increased phosphorylation of Erk results in
more cells surviving. However, other studies have shown that the
activation of Erk may result in DNA damage and counteract
endoplasmic reticulum stress-induced apoptosis. Recently, there
have been many studies on the connection between TPI and MAPK
signaling pathways (16,18,19);
however, data on the effects of TPI-induced Erk activation and
autophagy in MCF-7 breast cancer cells remain limited.
Autophagy is a conserved catabolic mechanism to
maintain cell homeostasis and protein reuse, which occurs under
normal and stress conditions, including nutrient starvation,
metabolism variation, lack of energy and reactive oxygen
accumulation (20). Autophagy
degrades and recycles useless, variant and dysfunctional
macromolecules and organelles. This prevents cumulative damage and
cell aging, which are involved in neurodegenerative diseases.
Autophagy also has a developmental role, reducing cancer cell
instability and damage, and thereby preventing tumorigenesis.
Studies show that TPI can induce autophagy in prostate cells
(9); however, the relationship
between breast cancer cell death and autophagy is still
unclear.
In this study, we used TPI-treated MCF-7 human
breast cancer cells to investigate the effects on proliferation,
autophagy and apoptosis, and to explore the underlying molecular
mechanisms.
Materials and methods
Cell lines and culture conditions
Human breast cancer cell line MCF-7 was provided by
the School of Pharmacy, Jilin University (Jilin, China). MCF-7 was
incubated in RPMI 1640 medium, which contained 10% fetal bovine
serum, 100 U/l penicillin and 100 mg/ml streptomycin, at 37°C in 5%
CO2. Groups of cells were treated with TPI (0, 10, 50,
100, 200, 400 nmol/l) for 12, 24 and 48 h. TPI was purchased from
Preferred Biological Co. (16091402). All other reagents, including
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
(MTT), were purchased from Sigma Chemical, Co. (St. Louis, MO,
USA), unless otherwise indicated.
Chemicals and drugs
TPI was dissolved in DMSO to make a stock solution
(40 mmol/l) and diluted to various concentrations with cell culture
medium. Also used were: Caspase-3 antibody (p42574),
Bcl-2-associated X protein (Bax) antibody (Q07812), phospho-p44/42
MAPK (P-Erk1/2) antibody (p27361), phospho-p38 MAPK antibody
(Q16539), light chain-3B (LC3B) antibody (Q9GZQ8), SQSTM1/p62
antibody (Q13501), and Beclin-1 antibody (Q14457; all from Cell
Signaling Technology, Inc., Danvers, MA, USA); B-cell lymphoma 2
(Bcl-2) antibody (AF0060; Beyotime Institute of Biotechnology,
Haimen, China); glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
antibody (2118; Cell Signaling Technology, Inc.); goat anti-rabbit
IgG H&L (ab6720; Abcam, Cambridge, UK); Hoechasa Hoechst 33258
staining kit (C0003 Beyotime Institute of Biotechnology); and an
Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit
apoptosis detection kit (C1062; Beyotime Institute of
Biotechnology); 3-methyladenine and U0126 (T1879/T6223; Targeted
Molecules Corp., San Diego, CA, USA).
Cell viability and apoptosis
assay
Grouped according to the different culture periods,
4,000-6,000 MCF-7 cells in a logarithmic growth phase were seeded
to 96-well plates. After incubation for 12 h, cell culture medium
containing TPI was added to the cells, after which they were
further treated for 12, 24 and 48 h, respectively. The control
group was added to RMPI-1640 medium with 1% DMSO. MTT (100 µl of 5
mg/ml) was added to all wells, and cells were incubated for 4 h.
Formazan was dissolved by addition of 150 µl DMSO. Absorption at
490 nm was measured using a microplate reader. The data were
representative of at least three independent tests, and the cell
viability of study groups was expressed relative to the control
group (relative viability).
Western blot analysis
MCF-7 cells (1.1×104) were seeded in a
6-well plate. After cell adherence and rapid growth, TPI was added
in different concentrations for 24 h, after which cold
phosphate-buffered saline (PBS) was used to stop each treatment.
Cells were lysed using RIPA buffer, which contains 1%
phenylmethanesulfonyl fluoride (PMSF). Lysed cells were placed on
ice, and protein was collected by centrifugation at 4°C and 12,000
× g for 15 min.
Protein concentrations were determined by Bio-Rad
protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Protein (30 µg) was separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 200 mA for
150 min and transferred on to a polyvinylidene difluoride (PVDF)
membrane. The membrane was blocked with 5% skimmed milk in
Tris-buffered saline with Tween-20-buffered solution (150 mM NaCl,
10 mM Tris-HCl, pH 7.5, 0.1% Tween-20) and probed with the primary
antibodies overnight at 4°C. The corresponding horseradish
peroxidase-conjugated secondary antibodies were combined with the
primary antibodies for 1 h. The relative protein band intensities
were detected using an enhanced chemiluminescence (ECL) reagent,
quantified by Image J (National Institute of Health, Bethesda, MD,
USA) and Quantity One software (Bio-Rad Laboratories, Inc.). The
data were representative of at least three independent tests, and a
representative immunoblot is shown.
Immunofluorescence staining assay
MCF-7 cells (1.1×104) were seeded in a
6-well plate. After cell adherence and rapid growth, cells were
treated with TPI at different concentrations for 24 h, after which
cold PBS was used to stop each treatment. Cells were added to a
Hoechst 33258 staining kit and incubated at room temperature for 15
min in the dark. Fluorescence microscopy was used to determine
nuclear condensation and chromatin fragmentation (Olympus IX81;
Olympus, Tokyo, Japan). Annexin V-FITC and propidium iodide (PI)
dye were used to confirm the results.
Dead and late-apoptosis cells, which had lost
membrane integrity, were stained with PI, which appears as red
fluorescence. Annexin V-FITC can enter the dead cells' cytoplasm
and appears as green fluorescence. The data were representative of
at least three independent tests, and a representative immunoblot
is shown.
Flow cytometry analysis
After treatment at different concentrations, cells
were collected and suspended in PBS. Cells (5×105) were
taken into tubes and suspended in binding buffer, then stained
using an Annexin V-FITC/PI apoptosis detection kit (Beyotime
Institute of Biotechnology) and evaluated using a flow cytometer
(FACS; BD Bioscience, Sparks, Maryland, USA).
Statistical analysis
Data were expressed as the mean ± standard deviation
and analyzed via one-way analysis of variance among treatment
groups with Tukey's post hoc test for multiple comparisons. All
data in this study were processed in this way at least three times
independently. For all statistical analyses, differences were
considered significant at P<0.05. Analyses were performed using
GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA).
Results
TPI has a significant toxic effect on
breast cancer MCF-7 cells
The effect of TPI on the viability of MCF-7 cells
was investigated as a function of exposure time and drug
concentration. TPI reduced the viability of MCF-7 breast cancer
cells in a dose- and time-dependent manner compared with the
vehicle control group (Fig. 1A).
When cells were treated with 50 nmol/l TPI for 12 h, the cell
viability was 70%, a significant decrease compared with the vehicle
group. At the maximum concentration tested (400 nmol/l), cell
viability decreased to 60, 50, and 25% when treated for 12, 24, and
48 h, respectively. Treatment with 400 nmol/l TPI for 48 h resulted
in marked inhibitory effects on the cell viability compared with
treatment at the same TPI concentration for 12 h, indicating that
the anti-cancer activity of TPI occurred in a time-dependent
manner.
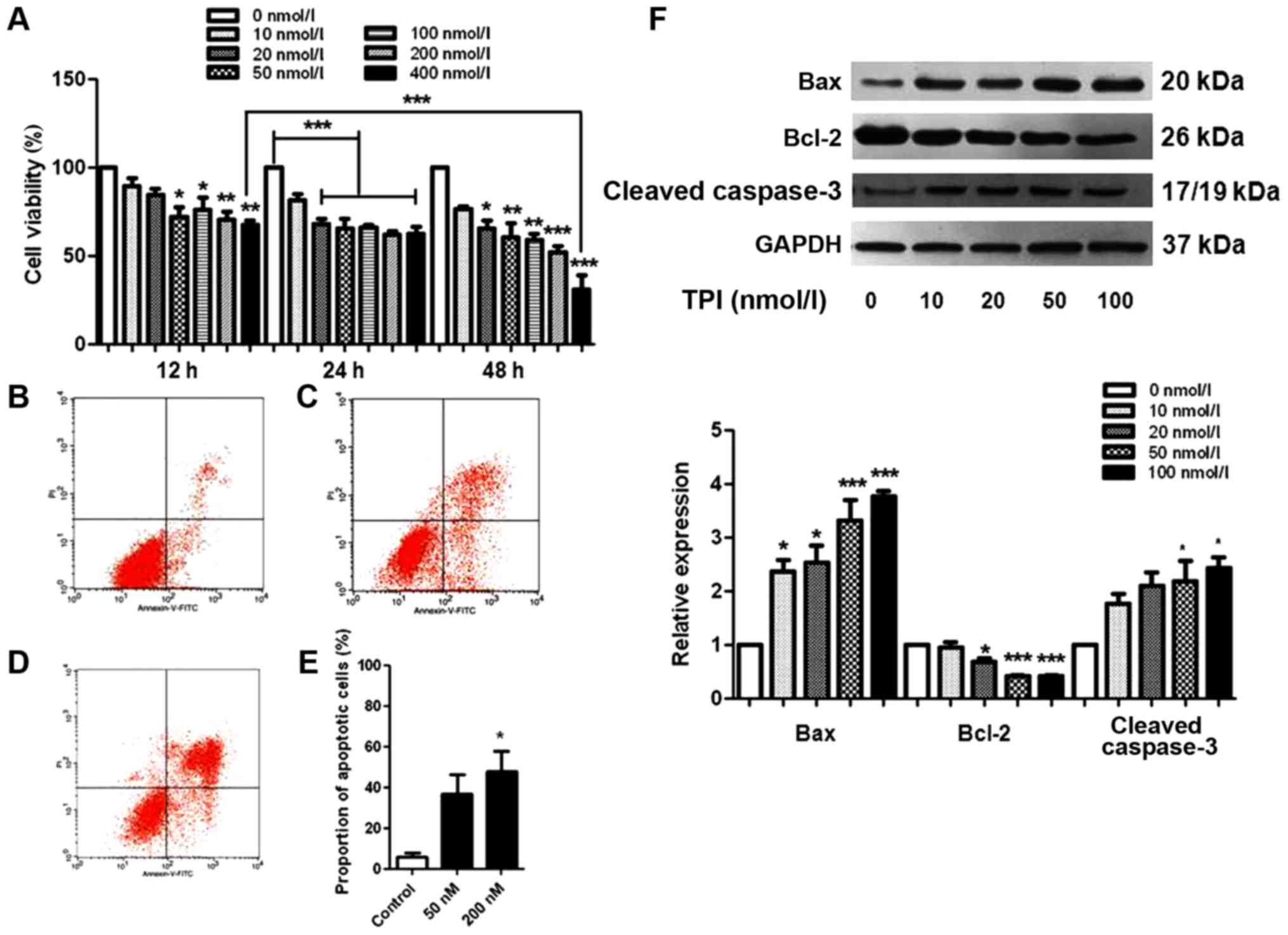 | Figure 1.TPI induced apoptosis in human breast
cancer MCF-7 cells. (A) Cytotoxicity was studied by MTT assay and
the average was presented as a percent of viability. MCF-7 cells
were treated with different concentrations of TPI (0, 10, 20, 50,
100, 200 and 400 nmol/l) for 12, 24 and 48 h. *P<0.05,
**P<0.01 and ***P<0.001 vs. 0 nmol/l, and as indicated.
Representative fluorescent images labeled with Annexin V-FITC/PI
following treat with (B) 0 nmol/l, (C) 100 nmol/l and (D) 200
nmol/l TPI for 24 h. Flow cytometric assay was performed and the
statistical results were presented in (E). (F) Expression of
apoptosis-associated proteins following treatment with 0, 10, 20,
50 and 100 nmol/l TPI. Data are representative of three independent
experiments. *P<0.05 and ***P<0.001 vs. 0 nmol/l (control).
TPI, triptolide; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; FITC,
fluorescein isothiocyanate; PI, propidium iodide; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X protein. |
Consistent with the MTT assay results, the
proportion of apoptotic cells gradually increased when TPI
concentration was increased, which was confirmed by the statistical
data from flow cytometry analysis (Fig.
1B-E). The proportions of Annexin V-FITC-positive and
PI-positive cells significantly increased after TPI treatment,
compared with the untreated cells. Approximately 40% of MCF-7 cells
were apoptotic at 100 nmol/l, a significant increase compared with
the vehicle group, in which the early apoptotic cell proportion was
17%.
When cells undergo apoptosis, caspase-3 is activated
and cleaved caspase-3 expression is increased, stimulating
programmed cell death (21). To
evaluate apoptotic cell death in MCF-7 cells comprehensively, we
carried out a western blot with apoptosis-related proteins Bax,
Bcl-2 and caspase-3 as apoptosis markers. Fig. 1F shows the changes in levels of Bax,
Bcl-2 and caspase-3 with different TPI concentrations. Bax and
cleaved caspase-3 were significantly increased when cells were
incubated with 50 nmol/l TPI for 24 h. A low concentration of TPI
(10 nmol/l) markedly increased the expression of Bax; however, the
trend for Bcl-2 was unchanged. These results indicate a
dose-dependent mechanism for the effects of TPI on cell
survival.
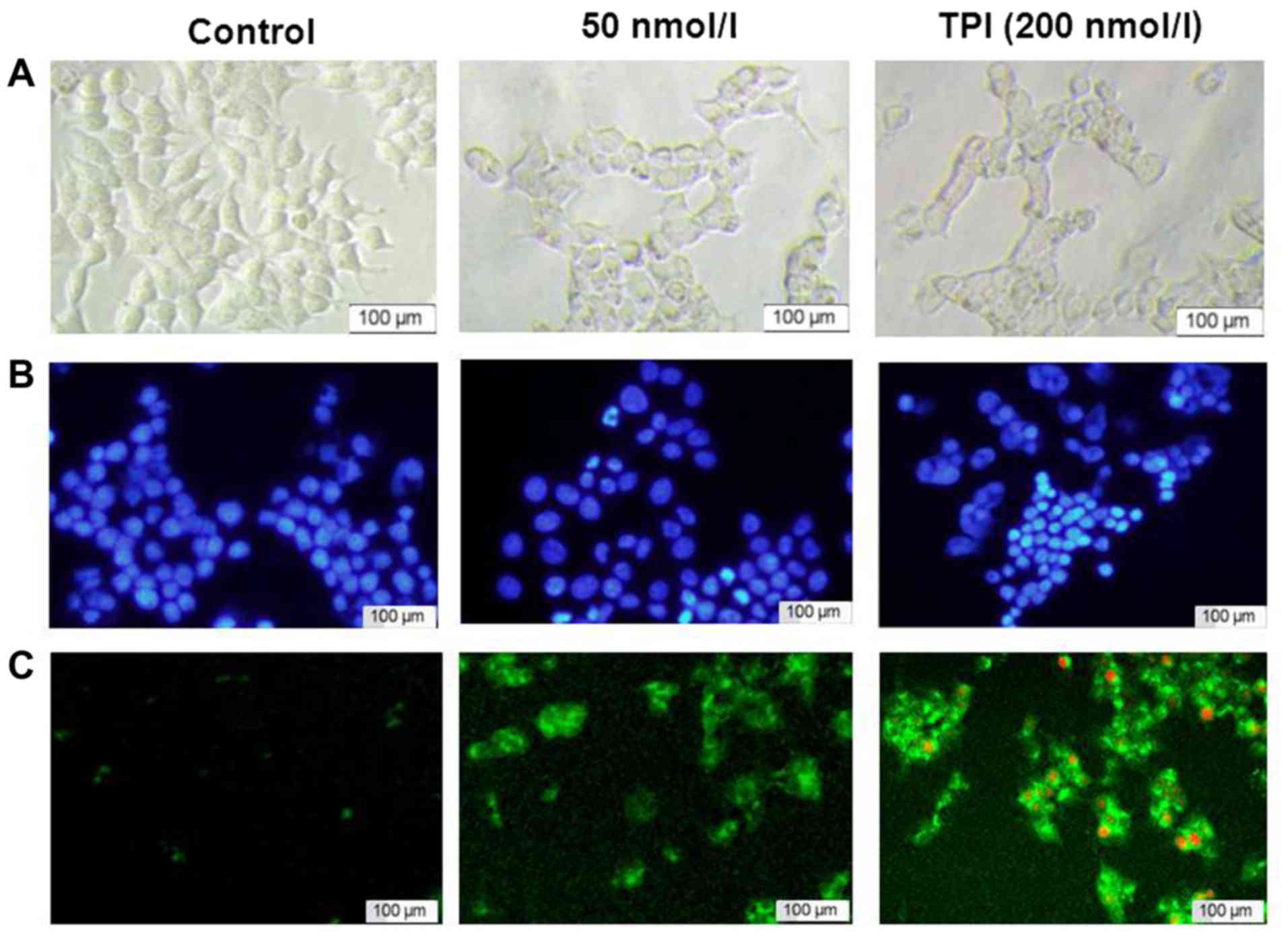
TPI induces apoptosis of breast cancer
cells
Having determined that TPI has toxic effects on
MCF-7 breast cancer cells, we further investigated the apoptotic
effects by immunofluorescence assay. Cells were treated with TPI
(0, 100 and 200 nmol/l) for 24 h. Fluorescence microscopy was
performed following staining with Hoechst 33258 and Annexin
V-FITC/PI. The proportion of cells undergoing positive or
morphological changes such as membrane shrinking was significantly
increased after TPI treatment, compared with the untreated cells.
Fig. 2A shows the morphological
changes in visible light. Nuclear condensation and chromatin
fragmentation were increased after treatment with 200 nmol/l TPI
for 24 h (Fig. 2B). During the
apoptosis process, phosphatidylserine is translocated to the outer
layer of the plasma membrane, where it is recognized and bound by
Annexin V; the complex appears as green fluorescence after
activation of apoptosis. PI can pass through disordered areas of
the dead cell's membrane, where it intercalates with the DNA double
helix. Thus, necrotic cells will appear red and green at the same
time, as shown in Fig. 2C. The above
results indicate that TPI inhibits the proliferation of MCF-7 cells
by inducing apoptosis.
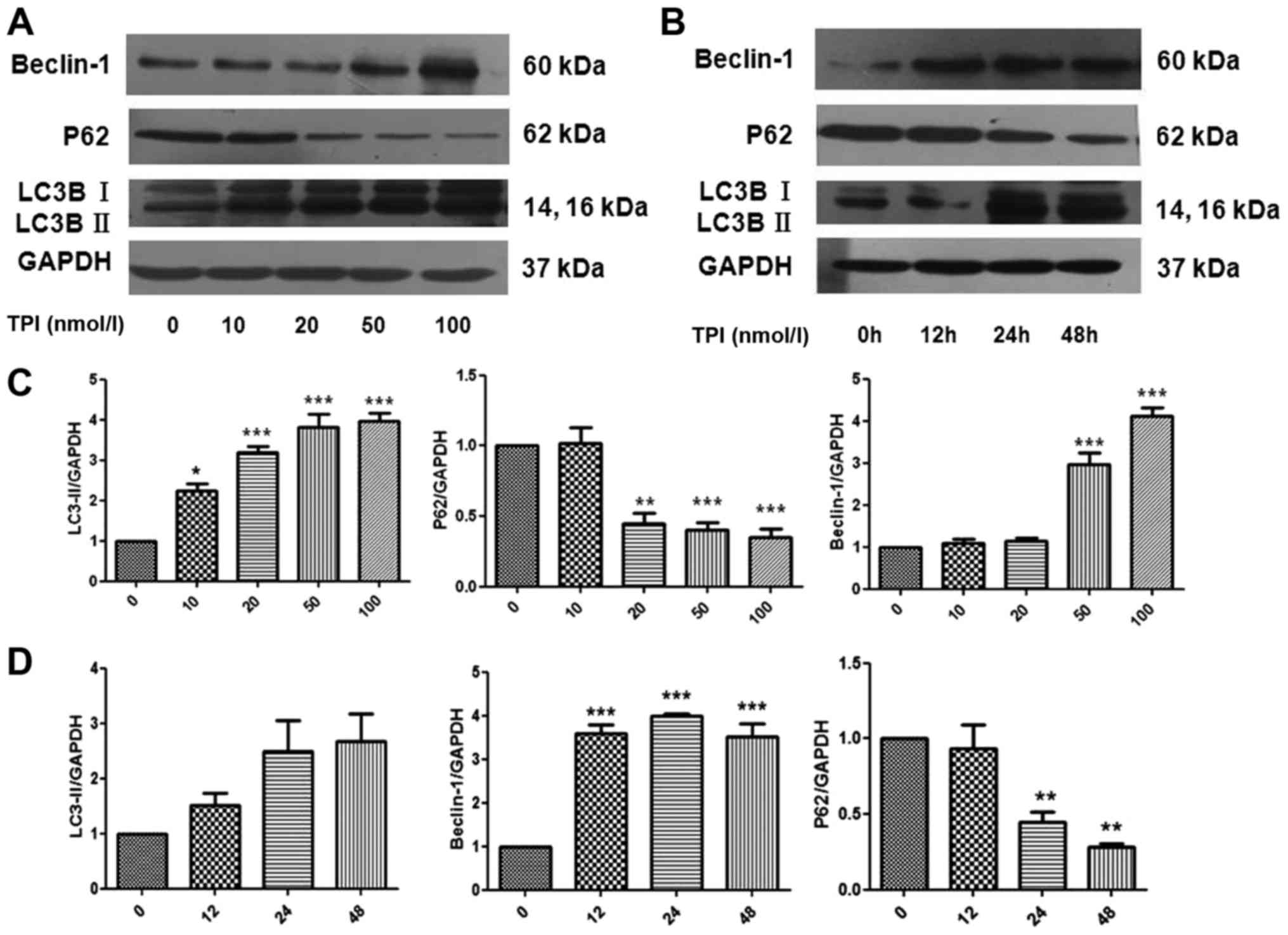
TPI likely induces autophagy through
LC3B protein activation
Autophagy is a conserved process aimed at
maintaining cell homeostasis under normal and stress conditions
(22). Above experiments showed that
TPI induced MCF-7 cell apoptosis by Bax upregulation, as well as
cleaved caspase-3 activation, which indicates that TPI induces
cytotoxicity through the mitochondrial pathway. Moreover,
TPI-induced cell variations, such as membrane damage, organelle
aging and mutant protein accumulation, are also associated with
triggering autophagy.
Here, induction of autophagy was evaluated by
western blotting to detect p62 protein downregulation and increased
Beclin-1 and LC3B protein levels. LC3B-II can interact with p62
allowing autophagosomal engulfment of mitochondria, and subsequent
degradation via fusion with the lysosome (23). Treatment with TPI at different
concentrations (0, 10, 20, 50 and 100 nmol/l) for 24 h led to
increased LC3B-II and Beclin-1 accumulation (Fig. 3), indicating the conversion of LC3B-I
to LC3B-II. The p62 protein level decreased. The results showed
that TPI induced autophagy, and indicated that this took place in a
time- and dose-dependent manner. MCF-7 breast cancer cell growth
may be inhibited by TPI through simultaneous induction of autophagy
and programmed cell death.
TPI affects cell autophagy and
apoptosis through the p38/Erk pathway
As well as the effects on LC3B, p62 and Beclin-1,
our western blot results showed that TPI activated Erk1/2,
mammalian target of rapamycin (mTOR) and p38 MAPK signaling in
MCF-7 cells, which are also related to autophagy induction
(Fig. 4) (24). Erk1/2 has a critical role in cell
proliferation, differentiation, skeleton construction and
apoptosis. The protein levels were quantified as p-p38/p38 and
p-Erk/Erk expression ratios, which were increased with increasing
drug concentration, with the most marked changes found in the 100
nmol/l groups. However, we found that the phosphorylation of mTOR
was decreased, which indicated that the activation of mTOR
suppressed by TPI.
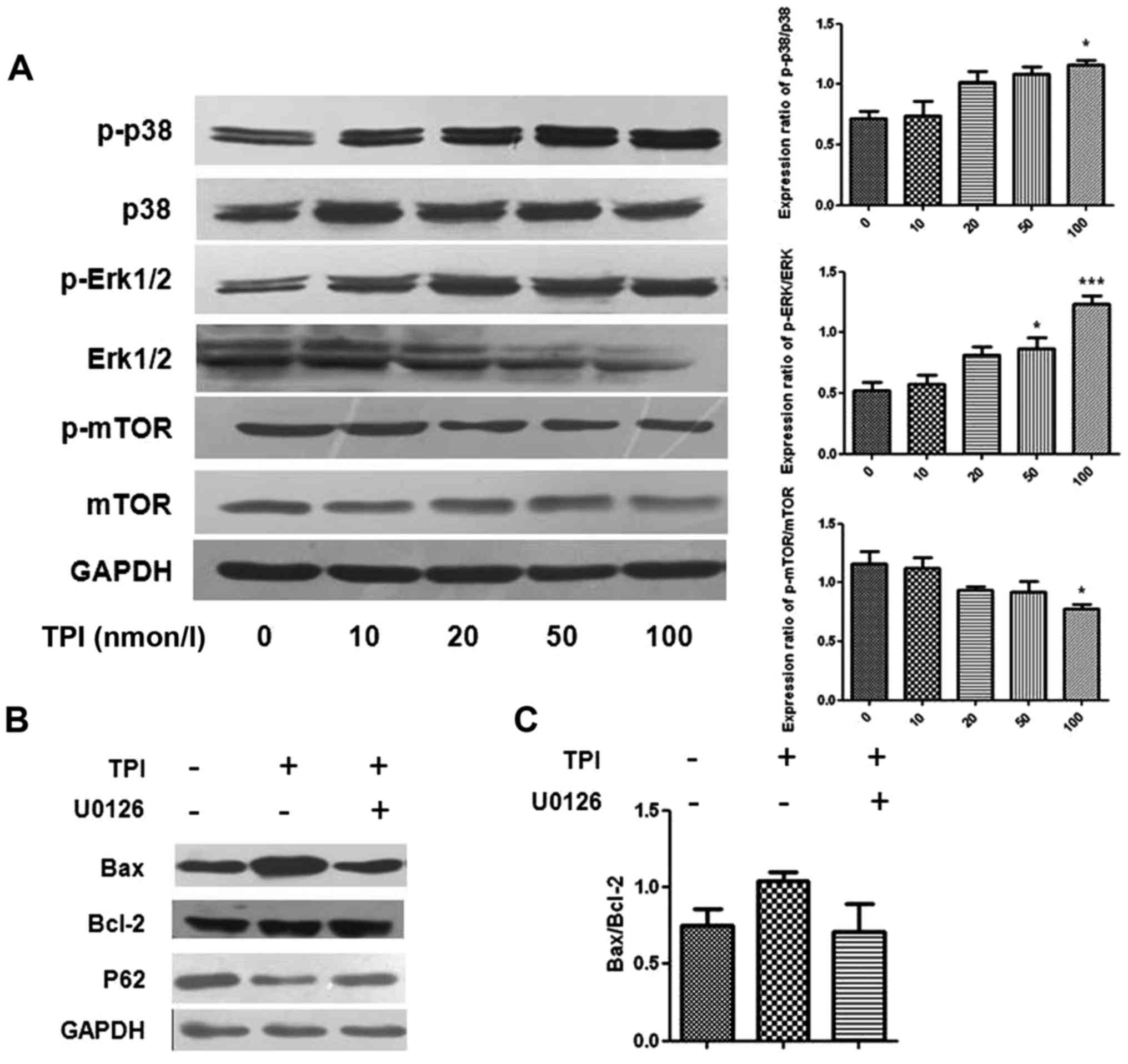 | Figure 4.Detection of autophagy induction and
p-Erk1/2, p38 and mTOR protein in different concentrations of TPI
by western blot analysis. (A) Representative immunoblot and the
quantified protein expression levels of p-P-38, p-Erk 1/2 and
p-mTOR following treatment with increasing concentrations of TPI,
presented as p-p38/p38, p-Erk/Erk and p-mTOR/mTOR expression
ratios. (B) The expression of Bax, Bcl-2 and p62 were affected by
the Erk inhibitor U0126 (20 µmol/l) and TPI (50 nmol/l). (C)
Quantitative analyses for Bax protein expression. Data are
representative of three independent experiments. *P<0.05 and
***P<0.001 vs. 0 nmol/l TPI. TPI, triptolide; p-,
phosphorylated; Erk, extracellular signal-regulated kinase; mTOR,
mammalian target of rapamycin; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein. |
After co-treatment with the Erk1/2 inhibitor U0126
and TPI, p62 expression levels were higher than with TPI alone and
upregulation of Bax was inhibited, indicating that activation of
Erk1/2 influenced autophagy induction, as pharmacological
inhibition of Erk1/2 activation attenuated TPI-induced autophagy
(Fig. 4). The results indicated that
Bax and p62 protein expression were affected by Erk1/2 protein.
Discussion
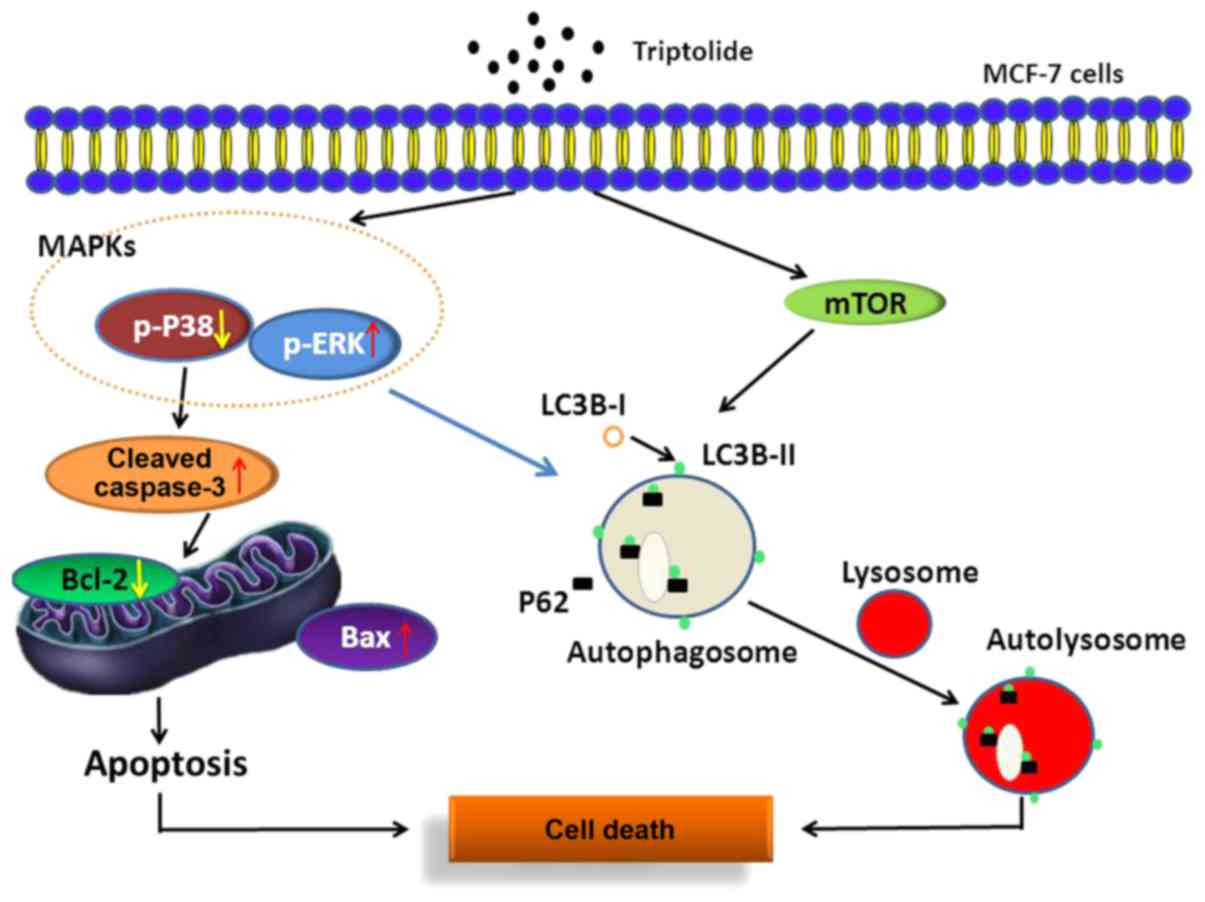
In this study, we propose that TPI induces MCF-7
cell apoptosis through the mitochondrial pathway, the mTOR/Erk/p38
MAPK signaling pathway and autophagy. We have shown that Erk
activation is associated with autophagy induction and apoptosis;
U0126 reversed the mediating effect of TPI on protein expression,
indicating that the regulation of Erk affected autophagy induction
by TPI in MCF-7 cells (Fig. 5).
The MTT assay results showed that TPI significantly
reduced the MCF-7 cell survival rate when its concentration was
greater than 10 nmol/l. TPI activated the caspase cascade through
regulating Bax, caspase-3 and Bcl-2, and promoted programmed cell
death via the mitochondrial pathway. The change in cell viability
was confirmed via flow cytometry assay, and morphology variation
was observed by fluorescent staining with Hoechst 33258 and Annexin
V-FITC/PI, consistent with previous reports (25). Much research has focused on the
ability of TPI to inhibit proliferation and migration of tumors,
and its potential applications in drug resistance and
radiation/chemotherapy sensitization (26). The anti-cancer mechanisms of TPI have
been shown to be related to multiple signals and pathways,
including the estrogen receptor signaling pathway (25) and phospholipase D1 expression
(27). TPI induced autophagy by
inhibiting the mTORC1 complex and activating both ULK complex and
Beclin-1 in PC3 cells, then finally activated the CaMkkβ-AMPK
pathway (24). Our data demonstrated
for the first time that TPI is able to induce apoptosis via the
mTOR/p38 MAPK/Erk signaling pathway in MCF-7 cells, indicating that
it may affect inflammation, oxidative stress and caspase-associated
proteins both upstream and downstream.
Cell autophagy is a regulatory mechanism to maintain
a stable internal environment through degrading mutated proteins
and damaged organelles. LC3B expression levels are positively
correlated with the number of intracellular autophagy vesicles,
which reflects the developmental degree of autophagy. Autophagy
maturity is regulated by Beclin-1. After treatment with TPI, the
changes in LC3B, p62 and Beclin-1 levels revealed that TPI induces
MCF-7 cell autophagy induction. mTOR can inhibit autophagy, and
acts as a critical negative regulator (28,29). We
found that TPI decreased mTOR phosphorylation, and thus has an
antithetical role in the inhibition of autophagy initiation
(23).
Phosphorylated Erk1/2 can suppress cell growth, and
also promotes the expression of Beclin-1 (30). Studies have found that
phosphorylation of Erk induces autophagy in human cervical cancer
Hela cells (31) and liver cancer
cells (22). We found that 40 nmol/l
TPI could increase p-p38 MAPK and p-Erk expression after treatment
for 24 h. Erk activation is closely related to autophagy induction
(16,30,32).
When combined with Erk inhibitor U0126, as shown in Fig. 4B and C, downregulation of P62 and
upregulation of Bax were inhibited, which showed that the
inhibition of Erk1/2 reversed autophagy changes induced by TPI.
In contrast to other reports, these data suggest
that TPI-induced Erk activation initiates apoptosis and autophagy
rather than promoting survival. TPI may inhibit tumor cell survival
in multiple ways.
In conclusion, we have shown that TPI induces
mitochondrial apoptosis and autophagy, which together lead to cell
death. TPI activates the mTOR/Erk/p38 MAPK signaling pathway and
regulates Bax and caspase-3, thereby inducing caspase cascade
reactions and autophagy in MCF-7 cells. We have shown for the first
time that there is a close connection between Erk activation and
TPI induced autophagy in MCF-7 breast cancer cells, which may be a
novel mechanism. The data suggest that TPI might be an effective
therapeutic option for breast cancer via multiple pathways.
Acknowledgements
The present study was supported by National Natural
Science Foundation of China (grant no. 81503168), International
Standard for Clinical Trial Technology Platform Construction of
Liver Diseases (grant no. 2014ZX09303303) and Graduate Innovation
Fund of Jilin University (grant no. 2017049).
References
|
1
|
Cedolini C, Bertozzi S, Londero AP,
Bernardi S, Seriau L, Concina S, Cattin F and Risaliti A: Type of
breast cancer diagnosis, screening, and survival. Clin Breast
Cancer. 14:235–240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de la Mare JA, Contu L, Hunter MC, Moyo B,
Sterrenberg JN, Dhanani KC, Mutsvunguma LZ and Edkins AL: Breast
cancer: Current developments in molecular approaches to diagnosis
and treatment. Recent Pat Anticancer Drug Discov. 9:153–175. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robijns J, Censabella S, Bulens P, Maes A
and Mebis J: The use of low-level light therapy in supportive care
for patients with breast cancer: Review of the literature. Lasers
Med Sci. 32:229–242. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ikeda H, Taira N, Nogami T, Shien K, Okada
M, Shien T, Doihara H and Miyoshi S: Combination treatment with
fulvestrant and various cytotoxic agents (doxorubicin, paclitaxel,
docetaxel, vinorelbine and 5-fluorouracil) has a synergistic effect
in estrogen receptor-positive breast cancer. Cancer Sci.
102:2038–2042. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zheng RR, Hu W, Sui CG, Ma N and Jiang YH:
Effects of doxorubicin and gemcitabine on the induction of
apoptosis in breast cancer cells. Oncol Rep. 32:2719–2725. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tacar O, Sriamornsak P and Dass CR:
Doxorubicin: An update on anticancer molecular action, toxicity and
novel drug delivery systems. J Pharm Pharmacol. 65:157–170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li XJ, Jiang ZZ and Zhang LY: Triptolide:
Progress on research in pharmacodynamics and toxicology. J
Ethnopharmacol. 155:67–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reno TA, Kim JY and Raz DJ: Triptolide
inhibits lung cancer cell migration, invasion, and metastasis. Ann
Thorac Surg. 100:1817–1825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Z, Sangwan V, Banerjee S, Chugh R,
Dudeja V, Vickers SM and Saluja AK: Triptolide sensitizes
pancreatic cancer cells to TRAIL-induced activation of the death
receptor pathway. Cancer Lett. 348:156–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Xiao E, Yuan L and Li G: Triptolide
synergistically enhances antitumor activity of oxaliplatin in colon
carcinoma in vitro and in vivo. DNA Cell Biol. 33:418–425. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grossi V, Peserico A, Tezil T and Simone
C: p38α MAPK pathway: A key factor in colorectal cancer therapy and
chemoresistance. World J Gastroenterol. 20:9744–9758. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou HF, Liu XY, Niu DB, Li FQ, He QH and
Wang XM: Triptolide protects dopaminergic neurons from
inflammation-mediated damage induced by lipopolysaccharide
intranigral injection. Neurobiol Dis. 18:441–449. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang Y, Wang J, Cheng J and Wang L:
Antiestrogenic activity of triptolide in human breast cancer cells
MCF-7 and immature female mouse. Drug Dev Res. 78:164–169. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu J, Jiang Z, Xiao J, Zhang Y, Lin S,
Duan W, Yao J, Liu C, Huang X, Wang T, et al: Effects of triptolide
from Tripterygium wilfordii on ERalpha and p53 expression in two
human breast cancer cell lines. Phytomedicine. 16:1006–1013. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng X, Shi W, Zhao C, Zhang D, Liang P,
Wang G and Lu L: Triptolide sensitizes human breast cancer cells to
tumor necrosis factor-α-induced apoptosis by inhibiting activation
of the nuclear factor-κB pathway. Mol Med Rep. 13:3257–3264. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan BJ and Chiu GN: Role of oxidative
stress, endoplasmic reticulum stress and ERK activation in
triptolide-induced apoptosis. Int J Oncol. 42:1605–1612. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lewinska A, Adamczyk-Grochala J,
Kwasniewicz E, Deregowska A and Wnuk M: Diosmin-induced senescence,
apoptosis and autophagy in breast cancer cells of different p53
status and ERK activity. Toxicol Lett. 265:117–130. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu M, Chen J, Huang Y, Ke J, Li L, Huang
D and Wu W: Triptolide alleviates isoprenaline-induced cardiac
remodeling in rats via TGF-β1/Smad3 and p38 MAPK signaling pathway.
Pharmazie. 70:244–250. 2015.PubMed/NCBI
|
|
19
|
Meng G, Wang W, Chai K, Yang S, Li F and
Jiang K: Combination treatment with triptolide and
hydroxycamptothecin synergistically enhances apoptosis in A549 lung
adenocarcinoma cells through PP2A-regulated ERK, p38 MAPKs and Akt
signaling pathways. Int J Oncol. 46:1007–1017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang Q, Liu X, Cao C, Lei J, Han D, Chen
G, Yu J, Chen L, Lv D and Li Z: Apelin-13 induces autophagy in
hepatoma HepG2 cells through ERK1/2 signaling pathway-dependent
upregulation of Beclin1. Oncol Lett. 11:1051–1056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koleini N and Kardami E: Autophagy and
mitophagy in the context of doxorubicin-induced cardiotoxicity.
Oncotarget. 8:46663–46680. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao F, Huang W, Zhang Z, Mao L, Han Y,
Yan J and Lei M: Triptolide induces protective autophagy through
activation of the CaMKKβ-AMPK signaling pathway in prostate cancer
cells. Oncotarget. 7:5366–5382. 2016.PubMed/NCBI
|
|
25
|
Li H, Pan GF, Jiang ZZ, Yang J, Sun LX and
Zhang LY: Triptolide inhibits human breast cancer MCF-7 cell growth
via downregulation of the ERα-mediated signaling pathway. Acta
Pharmacol Sin. 36:606–613. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang N, Dong XP, Zhang SL, You QY, Jiang
XT and Zhao XG: Triptolide reverses the Taxol resistance of lung
adenocarcinoma by inhibiting the NF-κB signaling pathway and the
expression of NF-κB-regulated drug-resistant genes. Mol Med Rep.
13:153–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang DW, Lee JY, Oh DH, Park SY, Woo TM,
Kim MK, Park MH, Jang YH and Min do S: Triptolide-induced
suppression of phospholipase D expression inhibits proliferation of
MDA-MB-231 breast cancer cells. Exp Mol Med. 41:678–685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meijer AJ, Lorin S, Blommaart EF and
Codogno P: Regulation of autophagy by amino acids and
MTOR-dependent signal transduction. Amino Acids. 47:2037–2063.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen P, Chen M, He M, Chen L, Song Y, Xiao
P, Wan X, Dai F, Pan T and Wang Q: Inhibition of ERα/ERK/P62
cascades induces ‘autophagic switch’ in the estrogen
receptor-positive breast cancer cells exposed to gemcitabine.
Oncotarget. 7:48501–48516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yue X, Zhao P, Wu K, Huang J, Zhang W, Wu
Y, Liang X and He X: GRIM-19 inhibition induced autophagy through
activation of ERK and HIF-1α not STAT3 in Hela cells. Tumour Biol.
37:9789–9796. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Innajak S, Mahabusrakum W and
Watanapokasin R: Goniothalamin induces apoptosis associated with
autophagy activation through MAPK signaling in SK-BR-3 cells. Oncol
Rep. 35:2851–2858. 2016. View Article : Google Scholar : PubMed/NCBI
|