Introduction
Dexmedetomidine, midazolam, and propofol, sedative
drugs commonly used in the intensive care unit, are widely used for
critical patients with tracheal intubation; most of these patients
suffer from sepsis or severe sepsis. Recently, the effects of these
sedative drugs on inflammation have been considered (1,2), and for
both clinical and in vitro studies have compared the effects
of these three drugs on inflammation models (3,4).
Propofol and midazolam has shown anti-inflammatory properties in a
variety of experimental models (1,5), while
dexmedetomidine, a highly selective agonist of α2-adrenergic
receptors, may have a biphasic effect on cells (6).
Dendritic cells (DCs) are one of the crucial immune
cells that bridge innate and adaptive immunity, in which DC process
antigens during innate immune responses to present them to naïve
T-cells, leading to an establishment of adaptive immunity (7). After they are stimulated, immature DCs
migrate to the draining lymph node where DCs present the processed
antigen peptides to lymphocytes, which then leads to the
establishment of adaptive immunity (8). Lipopolysaccharide (LPS), a bacterial
endotoxin contributing to sepsis, can stimulate immature DCs and
induce an inflammatory response; during this process, the
expression of inflammation cytokines, such as tumor necrosis factor
(TNF)-α, interleukin (IL)-1β, IL-6, and IL-10, increases (9,10).
Moreover, over-production of cyclooxygenase-2 (COX2) enzymes, which
are pro-inflammatory factors, can also be enhanced by LPS (11,12). The
nuclear factor-κB (NF-κB) and mitogen-activated protein kinase
(MAPK) pathways play a vital role in this inflammatory response
(10,13).
In vitro studies on the effect of sedatives
on inflammation have mostly concentrated on the effects on
macrophages (6,14,15) and
microglia (16), and few have
considered the effects on DCs (17,18).
Therefore, in this study, we investigated the effect of
dexmedetomidine, midazolam, and propofol, in doses based on the
typical blood levels achieved clinically, on the production of
inflammatory mediators in LPS-activated DCs, and then we explored
the underlying mechanism. We show that the three sedatives could
affect LPS-induced inflammation through different pathways.
Materials and methods
Cell culture
Mouse bone marrow-derived DCs (named DC2.4 cell)
were kindly donated by Cheng Hao (Department of Dermatology,
Medical College of Zhejiang University, Sir Run Run Shaw Hospital,
Hangzhou, China) (19). DC2.4 cells
were grown in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), in culture dishes (60×15 mm;
Corning Inc., Corning, NY, USA) at 37°C with 5% CO2 in a
humidified chamber. The culture medium was changed every day for
routine culture and each treatment was carried out when the cells
reached 80% confluence (6).
Experimental protocols
DC2.4 cells were stimulated with LPS (Escherichia
coli; serotype 055:B5; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany, 10 mg/ml) to induce the expression of inflammatory
molecules. A total of 26 groups were employed. Two groups of DC2.4
cells, treated with phosphate-buffered saline (PBS; denoted as the
control group) or LPS (1 µg/ml, denoted as the L group), which were
served as the negative or positive controls, respectively. To
elucidate the effect of dexmedetomidine (Heng Riu Inc., Jiangsu,
China), midazolam (Nhwa Inc., Jiangsu, China), and propofol
(Sigma-Aldrich; Merck KGaA) on DCs, 12 groups of DC2.4 cells were
divided into 12 groups and treated with different concentrations of
dexmedetomidine (0.5, 0.1, 0.01, or 0.001 µM), midazolam (50, 10,
1, or 0.1 µM), or propofol (100, 50, 15, or 1 µM) respectively,
which were named as group of D1, D2, D3, D4, M1, M2, M3, M4, P1,
P2, P3, P4, respectively. To compare the effect of dexmedetomidine,
midazolam, and propofol on LPS-stimulated DCs, DC2.4 cells were
divided into 12 groups again and treated with LPS (1 µg/ml) plus
different concentrations of dexmedetomidine (0.5, 0.1, 0.01, or
0.001 µM; denoted as L-D1, L-D2, L-D3, and L-D4, respectively), LPS
plus midazolam (50, 10, 1, or 0.1 µM; denoted as L-M1, L-M2, L-M3,
and L-M4, respectively), or LPS plus propofol (100, 50, 15, or 1
µM; denoted as L-P1, L-P2, L-P3, and L-P4, respectively). These
dosages included clinical dosages (0.001–1 µM dexmedetomidine,
0.1–10 µM midazolam, and 1–50 µM propofol) as well as a
supra-concentration of every drug (0.5 µM dexmedetomidine, 50 µM
midazolam, and 100 µM propofol) (18,20–22). The
concentration of LPS for all treatments was 1 µg/ml.
Cell viability assay
The cell counting kit-8 was used to determine cell
viability (CCK-8, Yiyuan Biotechnologies, Guangzhou, China), which
indicated the mitochondrial enzyme activity. To examine the effect
of the tested drugs on DC2.4 cell viability, the DC2.4
(3×104 cells/ml) were cultured with DMEM medium
containing 10% FBS in flat-bottom 96-well plates (200 µl/well) and
overnight at 37°C, in a humidified chamber with 5% CO2.
The cultured medium was replaced by the medium containing LPS
and/or anesthetic drugs, and then cells were cultured for 24 h.
After treatment, a new medium replaced the cultured medium, adding
20 µl/well of CCK-8 solution into each well, and followed by
incubation at 37°C for 2 h. The absorbance was measured at 490 nm,
with the correction wavelength set at 650 nm. The cell viablity
(%)=(ODdrug-ODblank
control)/(ODcontrol-ODblank control)
×100%. The the blank control was that had no cell while contained
medium and CCK-8. All experiments were performed in triplicate.
Data are representative of the mean and standard deviation of three
replicates within an experiment.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
In order to observe the changes in inflammatory
molecules at mRNA level, we used RT-qPCR. We extracted total RNA
from treated DC2.4 cells using an Ultrapure RNA kit (CWBIO,
Beijing, China) according to the manufacturer's protocol. In brief,
we synthesized first-strand cDNA with a HiFiScript 1st Strand cDNA
Synthesis kit (CWBIO). Then, we performed RT-qPCR with SYBR-Green
(SYBR Premix ExTaq GC; Takara Biotechnology Co., Ltd., Dalian,
China) on a LightCycler480 Real-Time PCR System. The primer
sequences were as follows: TNF-α-F: 5′-AGGGTCTGGGCCATAGAACT-3′,
TNF-α-R: 5′-CCACCACGCTCTTCTGTCTAC-3′; IL-1β-F:
5′-TGGCAACTGTTCCTGAACTCAA-3′, IL-1β-R: 5′-AGCAGCCCTTCATCTTTTGG3′;
IL-6-F: 5′-AGTTGCCTTCTTGGGACTGA3′, IL-6-R:
5′-TCCACGATTTCCCAGAGAAC3′; IL-10-F: 5′-GCCAAGCCTTATCGGAAATG3′,
IL-10-R: 5′-CACCCAGGGAATTCAAATGC3′; 36B4-F:
5′-GCCCTGCACTCTCGCTTTCT-3′, 36B4-R: 5′-CAACTGGGCACCGAGGCAACAGTTG-3′
(23). The RT-PCR reaction involved
initial denaturation (95°C, 30 sec), followed by 42 cycles each
consisting of denaturation (95°C, 5 sec), and extension (60°C, 30
sec), and then a final extension step at 95°C for 30 sec.
Cytokine assays
To test the inflammatory cytokine levels, DC2.4
cells were cultured in 96-well plates (200 µl/well) at
3×104 cells/ml overnight. With LPS treatment, the
detection time are set at 0, 0.5, 1, 2, 4, 6, 8, 12, 18 and 24 h.
The highest expression levels of IL-6, IL-10, TNF-α and IL-1β were
observed at 4, 6, 2 and 2 h, respectively. The pre-experiment
showed that, the difference between the TNF-α/IL-1β expression
level at 2 h and at the peak time (respective, 4 and 6 h) is small,
while their expression level at 2 h LPS treatment are obviously
higher than the blank group (data not shown). In order to unify
experimental environment and convenient experiment, 2 h was
selected for subsequent experiments. So the cell was all cultured
with drugs for 2 h. After adding LPS (1 µg/ml) and/or anesthetic
drugs in the culture media for 2 h, the culture media were renewed
and cells were cultured further for 18 h. And then the media were
collected and stored at −80°C until required for analysis.
Enzyme-linked immunosorbent assay (ELISA) kits for mouse TNF-α,
IL-6, and IL-10 (R&D Systems, Inc., Minneapolis, MN, USA) were
used according to the manufacturer's instructions. IL-1β, as an
endocellularly secreted inflammatory cytokine, was barely secreted
to the outside of cells after LPS stimulation. When the cells were
ruptured by repeated freezing and thawing (24), and IL-1β could be detected using an
IL-1β Colorimetric ELISA kit (R&D Systems, Inc.). According to
the ELISA reagents assay kit protocols, the minimum detection dose
ranges for TNF-α, IL-6, IL-10 and IL-1β were 0.36–7.21, 1.3–1.8,
0.46–4.80 and 0.652–5.22 pg/ml, and the sensitivity of the ELISA
for them were 7.21, 1.8, 5.22 and 4.8 pg/ml, respectively. The
concentrations of TNF-α, IL-6, IL-10, and IL-1β were respectively
assayed using a microplate reader set to 450 nm, with the
correction wavelength set at 540 nm. All experiments were performed
in triplicate. Data are representative of the mean and standard
deviation of three replicates within an experiment.
Western blot analysis
Western blotting was used to assess the protein
expression of COX2, IκB-α, ERK-MAPK, p38-MAPK, and JNK-MAPK in
DC2.4 cells. Based on a pre-experiment, 15 min was chosen as the
treatment time for all proteins, except for COX2, whose treatment
time was 10 h. In short, after drug treatments, proteins were
extracted from the cultured cells using RIPA buffer (Boster, Wuhan,
China) containing PMSF (BOSTER) and phosphatase inhibitors
(Boster). Protein concentrations were quantified using a BCA
protein assay kit (Boster). Equal amounts of total protein were
separated electrophoretically by SDS-PAGE and the corresponding
blots probed with the relevant primary antibodies (Cell Signaling
Technology, Inc., Danvers, MA, USA), followed by incubation with
appropriate secondary antibodies. The target proteins were then
visualized using a chemiluminescence method (ECL kit; Amersham
Biosciences, Foster City, CA, USA) and imaged with a digital
imaging system (Image Quant LAS-4000; Fujifilm, Tokyo, Japan).
Multi-Gauge Software was used for determining band densities
(Fujifilm).
Statistical analysis
Results are representative of at least 3 independent
experiments. The data are presented as mean ± SEM and were analyzed
using the GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA,
USA) and SPSS software (SPSS, Inc., Chicago, IL, USA). The
experiments were analyzed using One-way analysis of variance
(ANOVA) with Dunnett's test for the comparaison of more groups of
data and Unpaired Student t-test for the comparaison of two groups
of data. In all cases, P<0.05 was considered to indicate a
statistically significant difference.
Results
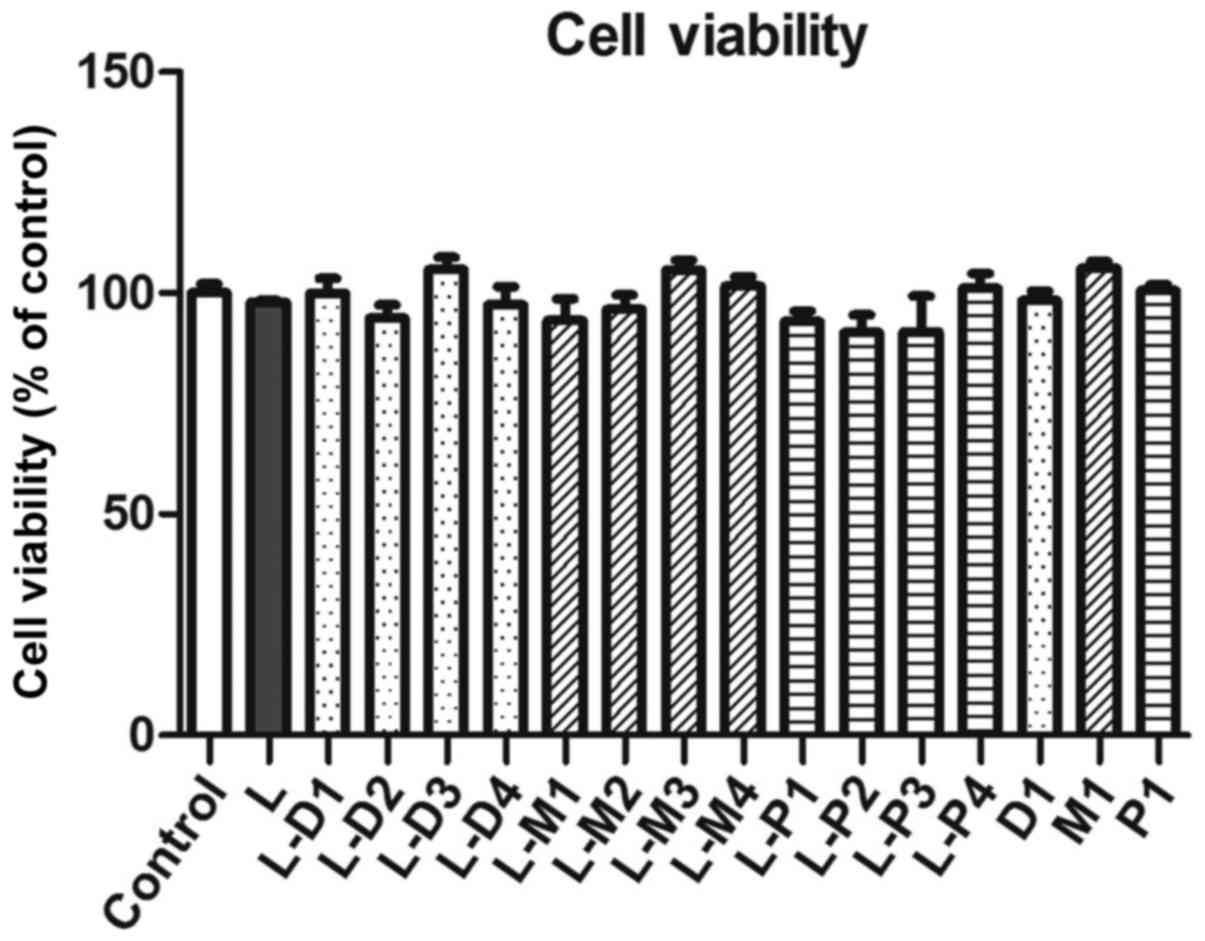
Effect of sedatives on the DCs
viability
To test the toxicity of dexmedetomidine, midazolam,
propofol, and LPS on DCs, the DCs were cultured with these drugs
and LPS, respectively, and then the cell viability was assayed
using a CCK-8 kit. Compared with the negative control group, the
DC2.4 cells viability has no significantly changes in LPS group or
in LPS-combined respectively dexmedetomidine, midazolam, and
propofol groups (Fig. 1).
Effect of sedatives on LPS-induced
cytokines
The pre-experiment showed that the TNF-α, IL-1β,
IL-6, and IL-10 mRNA expression peaked at different times in
response to treatment, but their expression levels were all
elevated after LPS treatment for 2 h. So the cell was all cultured
with drugs for 2 h. Compared with the negative control, there was a
significant increase of the TNF-α, IL-1β, IL-6, and IL-10 mRNA
expression in LPS-treated cell (all P<0.05). However, the only
dexmedetomidine, midazolam, and propofol did not affect their
expression (data not shown).
Upon co-treatment with LPS, dexmedetomidine did not
significantly affect the LPS-induced TNF-α, IL-1β, IL-6 and IL-10
mRNA expression at the used four concentrations (P>0.05,
Fig. 2). However, midazolam, at all
four concentrations, markedly inhibited LPS-induced IL-6 mRNA
expression (Fig. 2C). The inhibition
ratio of midazolam was up to 84.3±7.6, 51.6±8.7, 20.6±9.2, and
31.3±8.0% at 50, 10, 1 and 0.1 µM, respectively.
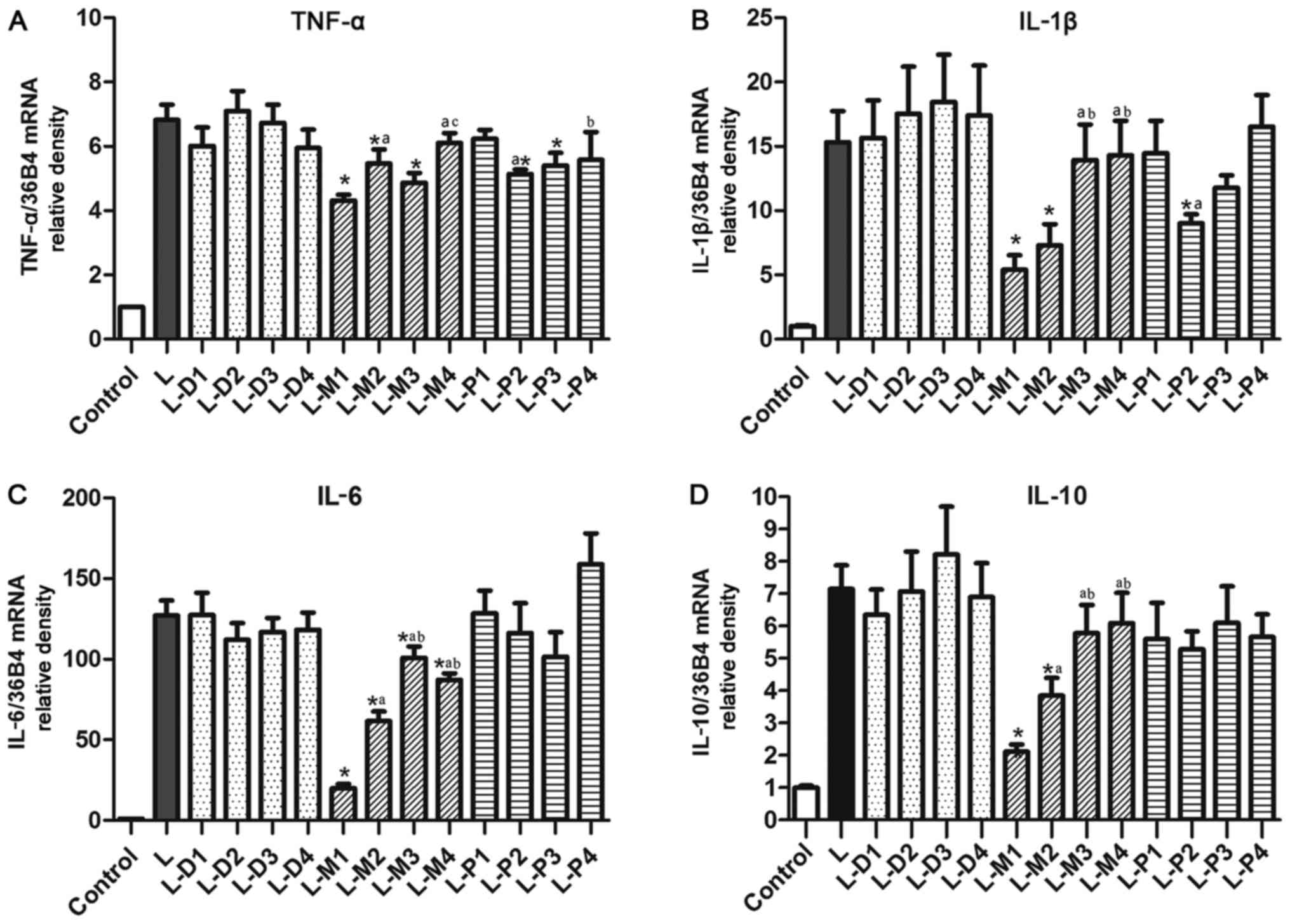 | Figure 2.Effects of dexmedetomidine,
midazolam, and propofol on LPS-enhanced cytokine mRNA levels. (A)
The TNF-α mRNA levels. (B) The IL-1β mRNA levels. (C) The IL-6 mRNA
levels. (D) The IL-10 mRNA levels. *P<0.05 vs. L group,
aP<0.05 vs. L-D1, L-M1 or L-P1 group, respectively.
bP<0.05 vs. L-D1, L-M1 or L-P1 group, respectively.
cP<0.05 vs. L-D1, L-M1 or L-P1 group, respectively.
LPS, Lipopolysaccharides; TNF, tumor necrosis factor; IL,
interleukin. |
Midazolam at a concentration of 10 and 50 µM also
significantly inhibited the LPS-induced IL-1β, IL-6 and IL-10
expression. The LPS-induced IL-6 and IL-10 expression were not
markedly affected by propofol at any of the tested concentrations
(Fig. 2C and D). Moreover, as
Fig. 2A and B showed that propofol
did not affect the LPS-enhanced TNF-α and IL-1β expression at both
of the highest (100 µM) and lowest concentrations (1 µM), while
propofol at 50 and 30 µM could inhibit the induced TNF-α expression
by 24.7±7.2 and 20.7±9.1%, respectively. Additionally, the enhanced
IL-1β expression was significantly suppressed by propofol at 50 and
15 µM with the inhibition rate of 41.1±16.5 and by 28.9±17.5%,
respectively, but this did not reach statistical significance.
Effect of sedatives on LPS-induced
cytokines levels
As shown in Fig. 3,
LPS could enhance significantly TNF-α, IL-1β, and IL-6 levels in
DCs (P<0.05). In addition to the 1 µM dexmedetomidine that could
inhibit LPS-induced IL-1β and IL-6 levels with the inhibition ratio
of 24.7±7.2 and 17.5±5% (P<0.0), respectively, the other used
concentrations did not affect TNF-α, IL-1β, and IL-6 levels.
Midazolam could inhibit LPS-enhanced IL-6 protein expression at
four used concentration (50 µM: 90.7±4.9%, 10 µM: 45.9±12.3%, 1 µM:
21.7±7.4%, 0.1 µM: 25.5±4.42%). A similar negative effect on
LPS-induced IL-6 expression was seen after propofol treatment at 15
µM (L-P3) and 1 µM (L-P4), with a reduction by 7.6±2.5 and
21.5±3.6%, respectively. Propofol did not significantly affect the
LPS-induced TNF-α levels, while midazolam could significantly
suppress it by 49.9±13.3% at 50 µM and by 28.2±10.3% at 10 µM, and
non-significantly by 13.6±7.6% at 1 µM and 17.0±7.8% at 0.1 µM. A
similar inhibitory effect could also been seen in midazolam
treatment on LPS-induced IL-1β levels by 87.3±6.7% at 50 µM and by
17.2±6.8% at 10 µM, and non-significantly by 14.3±7.1% at 1 µM and
by 11.4±6.9% at 0.1 µM. LPS-enhanced IL-1β levels could also be
suppressed by propofol at 50, 15 and 1 µM by 47.4±10.9, 33.7±11.2,
and 51.1±16.8%, respectively. Propofol at 100 µM also showed an
inhibitory effect on reducing LPS-induced IL-1β by 25.3±11.6%, but
it was not statistically significant. Additionally, we found that
IL-10 levels in all treatment groups were below the detection limit
(data not shown).
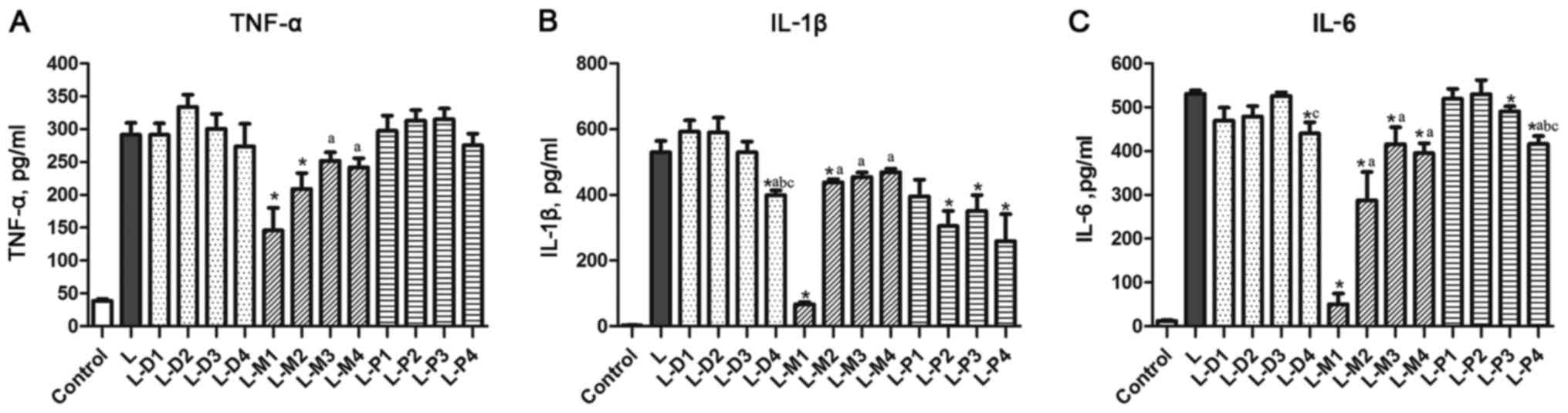 | Figure 3.Effects of dexmedetomidine,
midazolam, and propofol on LPS-enhanced cytokine levels. (A) The
TNF-α mRNA levels. (B) The IL-1β mRNA levels. (C) The IL-6 mRNA
levels. *P<0.05 vs. L group, aP<0.05 vs. L-D1,
L-M1 or L-P1 group, respectively. bP<0.05 vs. L-D1,
L-M1 or L-P1 group, respectively. cP<0.05 vs. L-D1,
L-M1 or L-P1 group, respectively. LPS, Lipopolysaccharides; TNF,
tumor necrosis factor; IL, interleukin. |
Effect of sedatives on the
LPS-stimulated NF-κB and MAPK pathways
Western blotting was used to assess the protein
expression of COX2, IκB-α, ERK-MAPK, p38-MAPK, and JNK-MAPK in
DC2.4 cells. Based on a pre-experiment, 15 min was chosen as the
treatment time for all proteins, except for COX2, whose treatment
time was 10 h. COX2, phospho-P38, phospho-ERK, and phospho-JNK
expression was low in the control group, but was clearly enhanced
by LPS treatment (Fig. 4A).
Dexmedetomidine, midazolam, and propofol could not change the basal
expression levels of these proteins (data not shown).
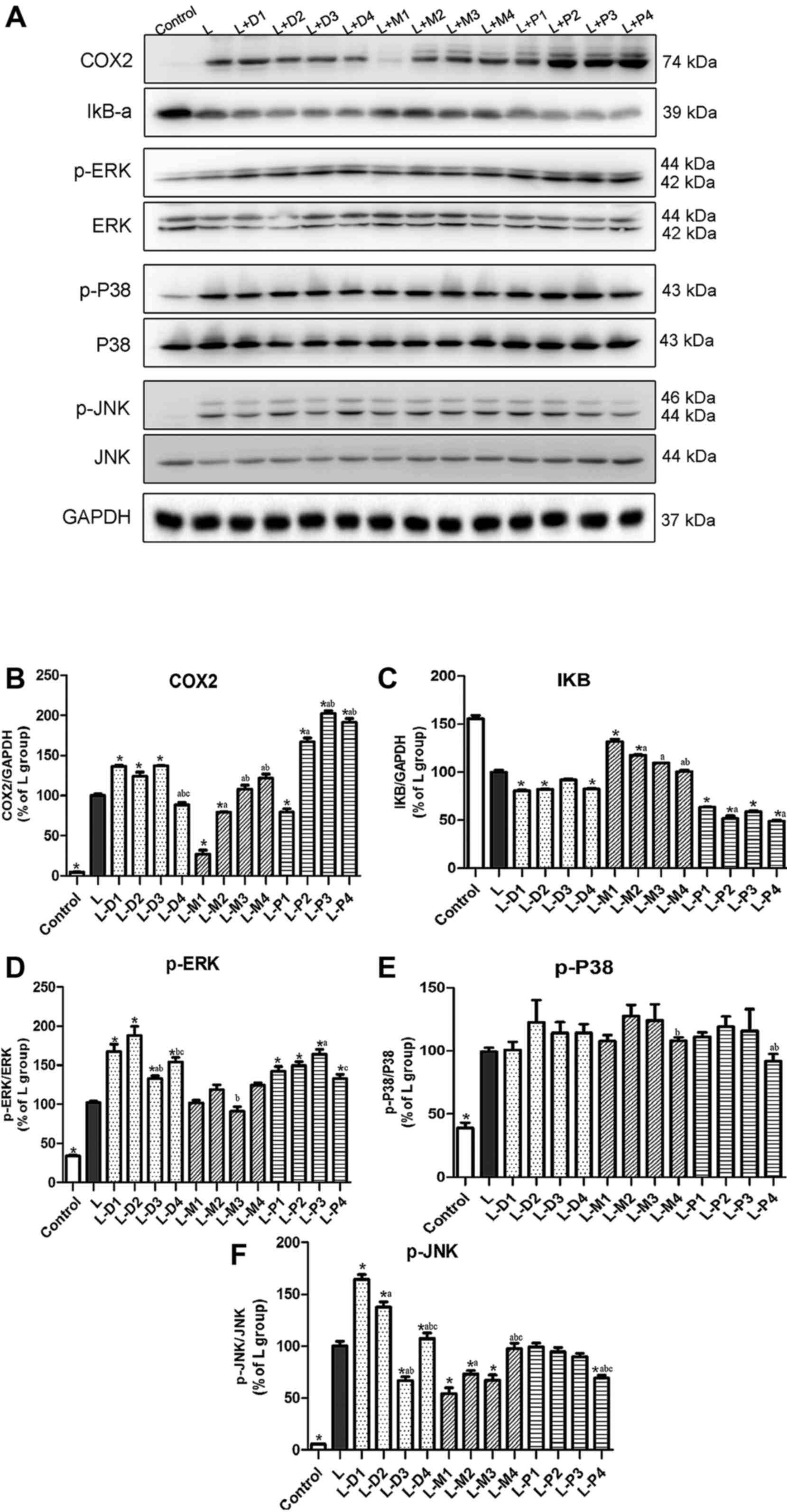 | Figure 4.Effect of dexmedetomidine, midazolam,
and propofol on LPS-stimulated NF-κB and MAPK pathways. (A) The
western blot of COX2, IκB-α, p-ERK, p-p38 and p-JNK. (B-F) The
graphics with the data from COX2, IκB-α, p-ERK, p-p38 and p-JNK
protein level. *P<0.05 vs. L group, aP<0.05 vs.
L-D1, L-M1 or L-P1 group, respectively. bP<0.05 vs.
L-D1, L-M1 or L-P1 group, respectively. cP<0.05 vs.
L-D1, L-M1 or L-P1 group, respectively. LPS, Lipopolysaccharides;
NF-κB, nuclear factor-κB; TNF, tumor necrosis factor; IL,
interleukin; COX2, cyclooxygenase-2; p-, phospho; ERK,
extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase. |
As showed in Fig. 4A,
midazolam inhibited LPS-enhanced COX2 protein expression at
concentrations of 10 and 50 µM. Co-treatment with LPS and
dexmedetomidine at 0.01, 0.1, and 1 µM promoted COX2 protein
expression. Similarly, co-treatment with LPS and propofol at
concentrations of 1, 15, and 30 µM promoted COX2 expression.
However, the effect of propofol at 50 µM actually had an inhibitory
effect. IκB-α, a crucial NF-κB component, has a suppressive effect
on inflammation. In particular, midazolam at 10 and 50 µM
suppressed the LPS-mediated reduction in the expression of IκB-α
protein, but the LPS-induced decreased in IκB-α protein was
enhanced by both dexmedetomidine and propofol at all tested
concentrations.
In order to explore whether MAPK signaling
participates in the inflammatory process, the three main proteins
involved in MAPK signaling, viz., phospho-p38, phospho-ERK, and
phospho-JNK, as well as their reference proteins, were
investigated. LPS could enhance the expression of phospho-ERK,
phospho-p38, and phospho-JNK, but none of the three anesthetic
drugs changed the basal protein secretion.
While midazolam did not significantly affect the
expression of LPS enhanced phospho-ERK, both dexmedetomidine and
propofol promoted this expression at the used four concentrations.
Phospho-JNK expression, induced by LPS, could be inhibited by
midazolam (at 50, 10 and 1 µM) and propofol (1 µM). and was
enhanced by dexmedetomidine (at 1, 0.1 and 0.001 µM). However, this
enhancement by dexmedetomidine was reduced at a low concentration,
and at 0.01 µM, dexmedetomidine rather had an inhibitory effect.
Furthermore, none of the three drugs affected the LPS-enhanced
expression of phospho-p38.
Discussion
Recently, the effects of sedative drugs on
inflammation have become a matter of interest (1,2).
Propofol is an intravenous anesthetic that is widely used for
anesthesia and sedation, and Inada et al reoprted that
intravenous anesthetic propofol suppresses prostaglandin E2
production in murine DC (25).
Dexmedetomidine has an effect on the migratory capacity of DCs both
in vitro and in vivo (17). Generally, TNF-α, IL-1β, and IL-6 are
regarded as important pro-inflammatory cytokines, while IL-10 is
considered as an anti-inflammatory cytokine (26–28).
This study aimed to investigate the effect of dexmedetomidine,
propofol and midazolam on pro-inflammatory cytokines production in
LPS-induced DCs are lacking.
Our study demonstrated that dexmedetomidine has a
biphasic effect that enhanced inflammation at high clinical
concentrations (10, 1, and 0.1 µM) and inhibited inflammation at
the lowest clinical concentration (0.001 µM), and this mechanism
was related to NF-κB and JNK-MAPK signalling pathway. However,
beyond this concentration, dexmedetomidine did not significantly
influence pro-inflammatory or anti-inflammatory cytokine expression
that had been enhanced by LPS stimulation of DCs. Midazolam at the
highest clinical dosage and 5 times this dose
concentration-dependently inhibited LPS-induced TNF-α, IL-1β, and
IL-6 expression at mRNA and protein levels. Interestingly, IL-10
expression was also inhibited by midazolam. This indicated that
midazolam-mediated inhibition of the maturation of DCs may play a
role in its anti-inflammatory action (18). Propofol can partly inhibit
LPS-induced expression of pro-inflammatory cytokines, such as IL-1β
and IL-6, and its anti-inflammatory effect may be achieved by
inhibiting JNK-MAPK signalling, and enhancing NF-κB and ERK-MAPK
signaling at clinical concentrations.
The numbers of reports about the effects of propofol
on inflammation outnumber those of the other two sedatives
(29). Though the inhibit effect of
propofol on cytokine were similar seen in many previous study, our
study shows that propofol could enhanced LPS-induced COX2, that may
be because of the difference cells. As antigen-processing and
presenting cells, activation of DCs in inflammation was earlier
than others in their study, like macrophage and microglia (30–32).
Moreover, the concentration of propofol based on clinical blood
concentration in our study was lower than the study on DC (33).
Inflammatory cytokines are closely associated with
inflammatory signaling, particularly with signalling via the NF-κB
and MAPK pathways. We investigated whether the drugs studied
influenced either or both pathways. In order to explore NF-κB
signaling, we detected IκB-α, an important component of the NF-κB
complex that can be degraded from the complex, by western blotting.
Transcription factors such as NF-κB also mediate the expression of
some inducible genes, such as the gene encoding COX-2. When LPS
binds to its target protein on the surface of cells, it induces
signaling via the CD14 receptor and activation of IκB kinase. Once
IκB is phosphorylated in the IκB-NF-κB complex, IκB is degraded,
and the free NF-κB translocates from the cytoplasm into the nucleus
where it induces COX2 expression (12,34). We
showed that dexmedetomidine in high concentration could increase
IκB-α degradation and enhance LPS-induced COX-2 expression. Similar
effects were seen after co-treatment of cells with propofol at
clinically relevant concentrations and LPS, but the suppressive
effect of propofol was much more marked than that of
dexmedetomidine. In contrast, co-treatment of cells with LPS and
midazolam markedly inhibited the NF-κB signaling, by suppressing
IκB-α degradation, and thereby decreasing the expression of
COX2.
ERK, p38, and JNK protein are three different but
important proteins in the MAPK signaling pathway (35). Our study corroborated data from a
previous study, which showed that LPS could enhance the expression
of ERK-MAPK, p38-MAPK, and JNK-MAPK markedly in DCs (13). However, we show for the first time
that dexmedetomidine could enhance the LPS-induced ERK and JNK
signaling at a high clinical concentration. JNK-MAPK also
contributed to the anti-inflammatory effect of midazolam. In our
study, propofol enhanced ERK-MAPK, but inhibited JNK-MAPK
signaling. LPS-stimulated p38 MAPK signaling was not markedly
changed by an of the three drugs.
Interestingly, after stimulation of DCs with LPS, a
significant enhancement of COX-2 protein expression and NF-κB
signalling could be seen with treatment with high clinical
concentrations of dexmedetomidine, but the cytokine levels were not
markedly affected. Two reasons may account for this phenomenon.
First, the expression of cytokines is regulated by many factors
besides the NF-κB pathway, such as their inducer genes, or their
levels per se (11,36). Second, beside binding to the highly
selective a2-adrenergic receptors, dexmedetomidine can also bind to
other kinds of receptors that could affect inflammation in the
opposite direction, as may be supported by the biphasic effect
noted in this and a previous study (6). Furthermore, as also noted in previous
studies, propofol had a partial and slightly anti-inflammatory
effect on DCs. For the first time, our study found that this effect
may be realized through suppression of the JNK pathway, which
perhaps inhibits maturation of DCs (37). However, it should be mentioned that
COX-2, NF-κB, and ERK-MAPK were enhanced significantly at the same
time. Previous studies had reported different effects of ERK-,
Ep38-, and JNK-MAPKs (13). It is
possible that, as with dexmedetomidine, other kinds of receptors
could be involved in the inflammatory effects. The difference may
also be related Eto the specific function of DCs, viz., their
antigen-processing and -presenting capacity. It may be beneficial
to have a weak response to LPS-induced inflammation at first. This
may account for the anti-inflammatory response in individuals with
infection who are receiving dexmedetomidine or propofol, as
compared with midazolam and other sedatives (3,38,39).
The study had some limitations. Firstly, this study
employed a transformed DC line of murine origin (DC2.4 cells)
rather than fresh murine or human DCs. Secondly, although the
mechanism underlying the effect of the sedative on inflammation was
investigated, the mechanism was not fully elucidated, as specific
antagonists or inhibitors of the relevant factors were not
employed. In the future, studies should make use of selective
antagonists against the sedatives, or the NF-κB and MAPK pathways.
Moreover, as antigen-processing and -presenting cells, the
migratory capacity of DCs and cell-surface molecules related to
their maturity were not investigated, and should be studied in the
future.
In conclusion, this study yielded a number of novel
insights. i) Midazolam can markedly inhibit LPS-induced
inflammatory responses of DCs, and the NF-κB and JNK-MAPK pathways
are suppressed during this process. ii) Dexmedetomidine has a
biphasic effect that enhanced inflammation at high clinical
concentrations (10, 1, and 0.1 µM) and inhibited inflammation at
the lowest clinical concentration (0.001 µM), and this mechanism
was related to NF-κB and JNK-MAPK signalling. iii) Propofol can
partly inhibit LPS-induced expression of pro-inflammatory
cytokines, such as IL-1β and IL-6, and its anti-inflammatory effect
may be achieved by inhibition of JNK-MAPK, and enhanced NF-κB and
ERK-MAPK signaling, at clinical concentrations. iv) Although both
midazolam and propofol at clinical dosages show anti-inflammatory
properties in LPS-induced inflammation, which of the former was
more marked. Thus, this study helped to elucidate the function of
sedatives in inflammation. In addition, it facilitates rational
implementation of these three sedatives in patients undergoing
tracheal intubation with sepsis or multiple organ dysfunction
syndrome.
Acknowledgements
The authors would like to thank Wang Qi, Zheng Libin
and Luo Jingfeng (Department of Central Laboratory, Sir Run Run
Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou,
China) for their technical support.
Funding
The present study was supported by The Science
Technology Department of Zhejiang Province, China (grant no.
2012C33039).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FG conceived and designed the study, and drafted the
manuscript. YD collected the data and XY analyzed the data. XC
performed the experiments, reviewed and edited the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yuki K, Soriano SG and Shimaoka M:
Sedative drug modulates T-cell and lymphocyte function-associated
antigen-1 function. Anesth Analg. 112:830–838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pandharipande PP, Sanders RD, Girard TD,
McGrane S, Thompson JL, Shintani AK, Herr DL, Maze M and Ely EW:
MENDS investigators: Effect of dexmedetomidine versus lorazepam on
outcome in patients with sepsis: An a priori-designed analysis of
the MENDS randomized controlled trial. Crit Care. 14:R382010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ruokonen E, Parviainen I, Jakob SM, Nunes
S, Kaukonen M, Shepherd ST, Sarapohja T, Bratty JR and Takala J:
‘Dexmedetomidine for Continuous Sedation’ Investigators:
Dexmedetomidine versus propofol/midazolam for long-term sedation
during mechanical ventilation. Int Care Med. 35:282–290. 2009.
View Article : Google Scholar
|
|
4
|
Jung HS, Joo JD, Jeon YS, Lee JA, Kim DW,
In JH, Rhee HY and Choi JW: Comparison of an intraoperative
infusion of dexmedetomidine or remifentanil on perioperative
haemodynamics, hypnosis and sedation and postoperative pain
control. J Int Med Res. 39:1890–1899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roquilly A, Josien R and Asehnoune K:
Midazolam impairs immune functions: It's time to take care of
dendritic cells. Anesthesiology. 114:237–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lai YC, Tsai PS and Huang CJ: Effects of
dexmedetomidine on regulating endotoxin-induced up-regulation of
inflammatory molecules in murine macrophages. J Surg Res.
154:212–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lipscomb MF and Masten BJ: Dendritic
cells: Immune regulators in health and disease. Physiol Rev.
82:97–130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blanco P, Palucka AK, Pascual V and
Banchereau J: Dendritic cells and cytokines in human inflammatory
and autoimmune diseases. Cytokine Growth Factor Rev. 19:41–52.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dowling D, Hamilton CM and O'Neill SM: A
comparative analysis of cytokine responses, cell surface marker
expression and MAPKs in DCs matured with LPS compared with a panel
of TLR ligands. Cytokine. 41:254–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Neves BM and Cruz MV: Differential roles
of PI3-Kinase, MAPKs and NF-kappaB on the manipulation of dendritic
cell T(h)1/T(h)2 cytokine/chemokine polarizing profile. Mol
Immunol. 46:2481–2492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim SF: The nitric oxide-mediated
regulation of prostaglandin signaling in medicine. Vitam Horm.
96:211–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aktan F: iNOS-mediated nitric oxide
production and its regulation. Life Sci. 75:639–653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakahara T, Moroi Y, Uchi H and Furue M:
Differential role of MAPK signaling in human dendritic cell
maturation and Th1/Th2 engagement. J Dermatol Sci. 42:1–11. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen RM, Chen TG, Chen TL, Lin LL, Chang
CC, Chang HC and Wu CH: Anti-inflammatory and antioxidative effects
of propofol on lipopolysaccharide-activated macrophages. Ann N Y
Acad Sci. 1042:262–271. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SN, Son SC, Lee SM, Kim CS, Yoo DG,
Lee SK, Hur GM, Park JB and Jeon BH: Midazolam inhibits
proinflammatory mediators in the lipopolysaccharide-activated
macrophage. Anesthesiology. 105:105–110. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng M, Wang YL, Wang CY and Chen C:
Dexmedetomidine attenuates lipopolysaccharide-induced
proinflammatory response in primary microglia. J Surg Res.
179:e219–e225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueshima H, Inada T and Shingu K:
Suppression of phagosome proteolysis and Matrigel migration with
the α2-adrenergic receptor agonist dexmedetomidine in murine
dendritic cells. Immunopharmacol Immunotoxicol. 35:558–566. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohta N, Ohashi Y, Takayama C, Mashimo T
and Fujino Y: Midazolam suppresses maturation of murine dendritic
cells and priming of lipopolysaccharide-induced t helper 1-type
immune response. Anesthesiology. 114:355–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brown M, Zhang Y, Dermine S, de Wynter EA,
Hart C, Kitchener H, Stern PL, Skinner MA and Stacey SN: Dendritic
cells infected with recombinant fowlpox virus vectors are potent
and long-acting stimulators of transgene-specific class I
restricted T lymphocyte activity. Gene Ther. 7:1680–1689. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dyck JB, Maze M, Haack C, Azarnoff DL,
Vuorilehto L and Shafer SL: Computer-controlled infusion of
intravenous dexmedetomidine hydrochloride in adult human
volunteers. Anesthesiology. 78:821–828. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Swart EL, Zuideveld KP, de Jongh J, Danhof
M, Thijs LG and van Schijndel Strack RM: Population pharmacodynamic
modelling of lorazepam- and midazolam-induced sedation upon
long-term continuous infusion in critically ill patients. Eur J
Clin Pharmacol. 62:185–194. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gepts E, Camu F, Cockshott ID and Douglas
EJ: Disposition of propofol administered as constant rate
intravenous infusions in humans. Anesth Analg. 66:1256–1263. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meng ZX, Li S, Wang L, Ko HJ, Lee Y, Jung
DY, Okutsu M, Yan Z, Kim JK and Lin JD: Baf60c drives glycolytic
metabolism in the muscle and improves systemic glucose homeostasis
through Deptor-mediated Akt activation. Nat Med. 19:640–645. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arend WP, Palmer G and Gabay C: IL-1,
IL-18 and IL-33 families of cytokines. Immunol Rev. 223:20–38.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Inada T, Kubo K, Ueshima H and Shingu K:
Intravenous anesthetic propofol suppresses prostaglandin E2
production in murine dendritic cells. J Immunotoxicol. 8:359–366.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ren K and Torres R: Role of
interleukin-1beta during pain and inflammation. Brain Res Rev.
60:57–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zelová H and Hošek J: TNF-α signalling and
inflammation: Interactions between old acquaintances. Inflamm Res.
62:641–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scheller J, Garbers C and Rose-John S:
Interleukin-6: From basic biology to selective blockade of
pro-inflammatory activities. Semin Immunol. 26:2–12. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
González-Correa JA, Cruz-Andreotti E,
Arrebola MM, López-Villodres JA, Jódar M and De La Cruz JP: Effects
of propofol on the leukocyte nitric oxide pathway: In vitro and ex
vivo studies in surgical patients. Naunyn Schmiedebergs Arch
Pharmacol. 376:331–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen RM, Wu GJ, Tai YT, Sun WZ, Lin YL,
Jean WC and Chen TL: Propofol reduces nitric oxide biosynthesis in
lipopolysaccharide-activated macrophages by downregulating the
expression of inducible nitric oxide synthase. Arch Toxicol.
77:418–423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng M, Ye JS, Wang YL, Chen C and Wang
CY: Posttreatment with propofol attenuates
lipopolysaccharide-induced up-regulation of inflammatory molecules
in primary microglia. Inflamm Res. 63:411–418. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiu WT, Lin YL, Chou CW and Chen RM:
Propofol inhibits lipoteichoic acid-induced iNOS gene expression in
macrophages possibly through downregulation of toll-like receptor
2-mediated activation of Raf-MEK1/2-ERK1/2-IKK-NFκB. Chem Biol
Interact. 181:430–439. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee CJ, Tai YT, Lin YL and Chen RM:
Molecular mechanisms of propofol-involved suppression of no
biosynthesis and inducible iNOS gene expression in LPS-stimulated
macrophage-like raw 264.7 cells. Shock. 33:93–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faugaret D, Lemoine R, Baron C, Lebranchu
Y and Velge-Roussel F: Mycophenolic acid differentially affects
dendritic cell maturation induced by tumor necrosis factor-alpha
and lipopolysaccharide through a different modulation of MAPK
signaling. Mol Immunol. 47:1848–1859. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nakahara T, Uchi H, Urabe K, Chen Q, Furue
M and Moroi Y: Role of c-Jun N-terminal kinase on
lipopolysaccharide induced maturation of human monocyte-derived
dendritic cells. Int Immunol. 16:1701–1709. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jin Y, Wi HJ, Choi MH, Hong ST and Bae YM:
Regulation of anti-inflammatory cytokines IL-10 and TGF-β in mouse
dendritic cells through treatment with Clonorchis sinensis crude
antigen. Exp Mol Med. 46:e742014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Handley ME, Thakker M, Pollara G, Chain BM
and Katz DR: JNK activation limits dendritic cell maturation in
response to reactive oxygen species by the induction of apoptosis.
Free Radic Biol Med. 38:1637–1652. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kadoi Y, Saito S, Kawauchi C, Hinohara H
and Kunimoto F: Comparative effects of propofol vs. dexmedetomidine
on cerebrovascular carbon dioxide reactivity in patients with
septic shock. Br J Anaesth. 100:224–229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jakob S, Ruokonen E, Grounds RM, Sarapohja
T, Garratt C, Pocock SJ, Bratty JR and Takala J: Dexmedetomidine
for Long-Term Sedation Investigators: Dexmedetomidine vs. midazolam
or propofol for sedation during prolonged mechanical ventilation:
Two randomized controlled trials. JAMA. 307:1151–1160. 2012.
View Article : Google Scholar : PubMed/NCBI
|