Introduction
Distal myopathy with rimmed vacuoles (DMRV), also
known as GNE myopathy, Nonaka myopathy, and hereditary
inclusion body myopathy, is an autosomal recessive hereditary
distal myopathy (1). DMRV was first
described in Iranian-Jewish patients as a vacuolar myopathy
affecting the distal muscles of the lower limbs (2). Since then, more and more cases of DMRV
have been reported from all over the world, especially after the
responsible gene was identified. DMRV is caused by mutations in the
UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine
kinase (GNE) gene (3,4). The GNE gene is located on
chromosome 9p13-p12. Its major transcript consists of 13 exons,
with exons 1 and 13 being non-coding (5). The gene encodes the bifunctional enzyme
GNE, which has both epimerase and kinase activity. GNE is a
rate-limiting enzyme that catalyzes the initial two steps in the
biosynthesis of sialic acid (6).
Sialic acid is widely distributed in the tissues and plays an
important role in the formation of protein folding. GNE mutations
influence and decrease the activity of epimerase and kinase,
resulting in the declined level of sialic acid. Abnormal
aggregating of the protein, such as including tau protein and
amyloid in muscle fibers can be induced by the decrease of
intracellular sialic acid. The systems of both ubiquitin proteasome
and autophagy lysosome are then activated, which might be the
reasons of RVs formation (7).
DMRV manifests as abnormal gait, and weakness and
atrophy of the distal muscles of the lower extremities, mainly the
tibialis anterior, with little or no involvement of the quadriceps
muscles even in the advanced stages of the disease. In fact,
quadriceps sparing is a characteristic feature of DMRV (2). The initial presentation is foot drop
and abnormal gait resulting from the involvement of the ankle
dorsiflexors. DMRV is a progressive disease, and most patients will
begin to rely on wheelchairs for mobility about 12 years after the
onset of symptoms (8). DMRV does not
lead to cardiomyopathy even in the late stages (9,10).
Furthermore, no studies have reported respiratory failure in DMRV,
except for one study from Japan (11).
Magnetic resonance imaging (MRI) of the muscles is
not specific but helps to differentiate DMRV from other conditions
such as myositis. Muscle biopsy specimens from patients with DMRV
exhibit rimmed vacuoles, protein aggregation, variation in muscle
fiber size, and an absence of inflammatory cells (5,7). Serum
creatine kinase (CK) is usually within normal limits or only mildly
elevated. The identification of GNE mutations in genetic
studies is diagnostic. Thus far, more than 150 mutations in the
GNE gene have been reported worldwide (12). However, relatively few GNE
mutations have been reported from China. The first two cases were
reported in 2006, and currently, approximately 90 cases of DMRV
along with the associated GNE mutations have been reported
in China (13–15).
In this series, we retrospectively analyzed the
clinical manifestations, pathological features, and gene mutations
in seven unrelated Chinese DMRV patients. We identified eight
GNE mutations in these patients, including five known
mutations (p.D207V, p.C44S, p.G576R, p.A669P, and p.D218G) and
three mutations that had not been reported before (c.455_ 456insC,
p.P421L, and p.A287T). We hope that our findings will improve our
understanding of the pathogenic mechanisms underlying DMRV and
facilitate the diagnosis of DMRV.
Materials and methods
Clinical data
Seven patients were diagnosed with DMRV in the First
Affiliated Hospital of Jilin University, between 2013 and 2016. All
of these patients voluntarily joined this study, and provided
written informed consent before being enrolled into the study. The
experimental protocol was established according to the ethical
guidelines of the Declaration of Helsinki, and was approved by the
Human Ethics Committee of Jilin University (Changchun, China). All
patients underwent detailed clinical evaluation, and the following
data were collected from each patient: Family history, age at
onset, distribution of muscle weakness, serum CK level, and
findings of muscle biopsy, electromyography, muscle MRI, and
peripheral blood genetic testing.
Muscle biopsy
Open muscle biopsy was performed in all patients.
The muscle specimens were vertically embedded in tragacanth gum,
which was applied on a cork. The specimens were then frozen for
20–30 sec, under slight shaking, in isopentane that had been
precooled in liquid nitrogen. After this, the specimens were stored
at a temperature of −80°C. For histological examination, serial
frozen sections (8 µm) were stained using routine histochemical
methods, including hematoxylin-eosin, modified Gomori trichrome,
periodic acid Schiff, NADH-tetrazolium, succinate dehydrogenase,
cytochrome C oxidase, and oil red O.
Genetic analysis
GNE mutation analysis was performed in all
patients. Genomic DNA was extracted from the peripheral blood by
the method of Qiagen FlexiGene DNA kit (Qiagen, AB, Sollentuna,
Sweden). Genomic libraries were built after DNA fragmented, and PCR
amplification using designed primers (Table I) was applied to screen out a
successful built segments of genomic libraries, following by hybrid
capture to obtain needed sequencing fragments. The target fragments
were enriched by PCR amplification and the products were purified
and examined successively after above procedures. Then the NEXTSEQ
500 sequencer (Illumina, Inc., San Diego, CA, USA) was applied to
analyze these PCR products to obtain original data. CASAVA software
(1.8.2; Illumina, Inc.) was used to convert original data into
recognizable base sequences, which were analyzed by biological
information analysis system to select mutations that in accordance
with clinical features. At last, mutations were further confirmed
via Sanger sequencing pedigree validation. NM_001128227 was chosen
as a reference sequence in this study (16). In addition, all mutations were
further confirmed using the Human Gene Mutation Database, Database
of Single Nucleotide Polymorphisms and 1,000 Genomes Project.
 | Table I.Primer sequences for polymerase chain
reaction. |
Table I.
Primer sequences for polymerase chain
reaction.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Exon no. | Forward | Reverse |
|---|
| 3 |
CATAAGTGGAGGTGCAAAAAAAGAT |
ACATAAAAACTAAGCAGCAGAACAG |
| 4 |
TTGCAACTCGGAGGTTCGTC |
ACTGTGCACGGCAGGAAGAT |
| 7 |
TGCCCGACCATCTTTCACTT |
GCACCCTGTGACCACTGACA |
| 10 |
TATTTCCTTGCAGTCCTTGGTAAC |
ATGTGGCTCACCTTCAGGCTCTAG |
| 12 |
CAAAGGCTTTAGGGGCAGTG |
GACACTGCAAAGCACCTGTC |
Statistical analysis
Data are expressed as the mean ± standard deviation.
Average age and mean duration of the disease were analyzed using
Excel (version 2013; Microsoft Corp. Redmond, WA, USA). Descriptive
analysis of these data was carried out in results discussion. There
were no relevant variance analysis of groups because of the small
sample size.
Results
Clinical features
This study involved two male and five female
patients. The patients were aged 26–46 years old, and their disease
course ranged from 2–10 years. The average age at onset was
29.8±4.5 years (range, 23–36 years) and the mean duration of the
disease was 6.7±3.6 years (range, 2–10 years). Lower-limb weakness
was present in four patients, upper-limb weakness in one patient,
and both upper- and lower-limb weakness in two patients. None of
the patients had a family history of DMRV (Table II). All patients were found to have
normal cranial nerve function on nervous system examination.
According to the Medical Research Council scale, the muscle
strength of the upper and lower extremities was >3 in all
patients, except for patient #2, in whom the tibialis anterior
muscle strength was 2. This patient had difficulty in standing and
walking, and had the gait associated with foot drop when she first
presented at our hospital. The main clinical feature in all
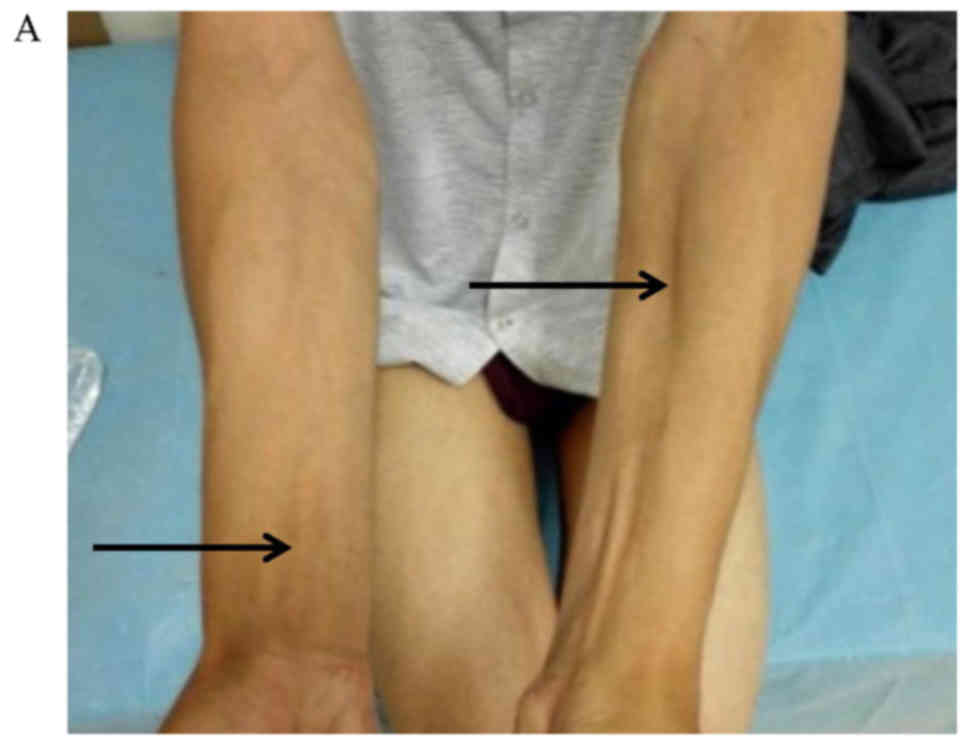
patients was weakness and atrophy of the distal limbs, with visible
involvement of both the legs and hands (Fig. 1). Four patients had foot drop and
duck step. At the time of presentation, all patients were ambulant
and did not need to be supported (except for patient #2). The
tendon reflexes were weak or absent, but cognition, cranial nerve
function, and coordination were unaffected. No patient complained
of paresthesia, and none had cardiac or respiratory problems.
| Table II.Clinical features of seven Chinese
patients with DMRV. |
Table II.
Clinical features of seven Chinese
patients with DMRV.
|
|
|
|
|
|
|
| U-Ex strength | L-Ex strength |
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Patient no. | Sex | Age (years) | Age at onset
(years) | Disease course
(years) | Onset symptoms | Family history | Distal | Proximal | TA | GC | QF | IP | Muscle atrophy | Tendon
reflexes | EMG | CK (U/l) |
|---|
| 1 | F | 35 | 27 | 8 | Weakness of both
hands | – | 3 | 4 | 3 | 3 | 5 | 3 | Both hands | Absent | M | Normal |
| 2 | F | 29 | 27 | 2 | Weakness of both
legs | – | 5 | 5 | 2 | 3 | 5 | 4 | Not found | Weakened | M | 258 |
| 3 | Ma | 26 | 23 | 3 | Weakness of all
four limbs | – | 4 | 5 |
| 4 | 5 | 5 | Both hands and
calves | Weakened | M | 678 |
| 4 | Ma | 43 | 33 | 10 | Weakness of all
four limbs | – | 4 | 5 | 3 | 4 | 5 | 5 | Both forearms and
calves | Normal | M | 578 |
| 5 | F | 34 | 30 | 4 | Weakness of both
legs | – | 4 | 5 | 3 | 3 | 4 | 4 | Both forearms and
calves | Weakened | M | 254 |
| 6 | F | 46 | 36 | 10 | Weakness of both
legs | – | 4 | 5 | 3 | 4 | 5 | 4 | Both forearms and
hands | Weakened | M | 583 |
| 7 | F | 43 | 33 | 10 | Weakness of both
legs | – | 5 | 5 | 3 | 4 | 4 | 4 | Both calves | Weakened | M | 777 |
The peak serum CK level was 777 U/l (normal, 40–200
U/l); thus, the CK level was mildly elevated (no more than three
times the normal level). Electromyography of the upper and lower
limb muscles showed myopathic changes. Three patients (#2, #4, and
#7) underwent muscle MRI. Bilateral MRI examination of the calf
muscles (Fig. 2) showed obvious
involvement of the tibialis anterior, tibialis posterior, extensor
digitorum longus, peroneus longus, peroneus brevis, and
gastrocnemius. In patient #4, the muscles in the medial and
posterior compartments of the thighs were mainly involved. The main
changes observed on muscle MRI were muscle atrophy and steatosis.
No obvious inflammatory exudation was observed, and the quadriceps
femoris muscles were not involved.
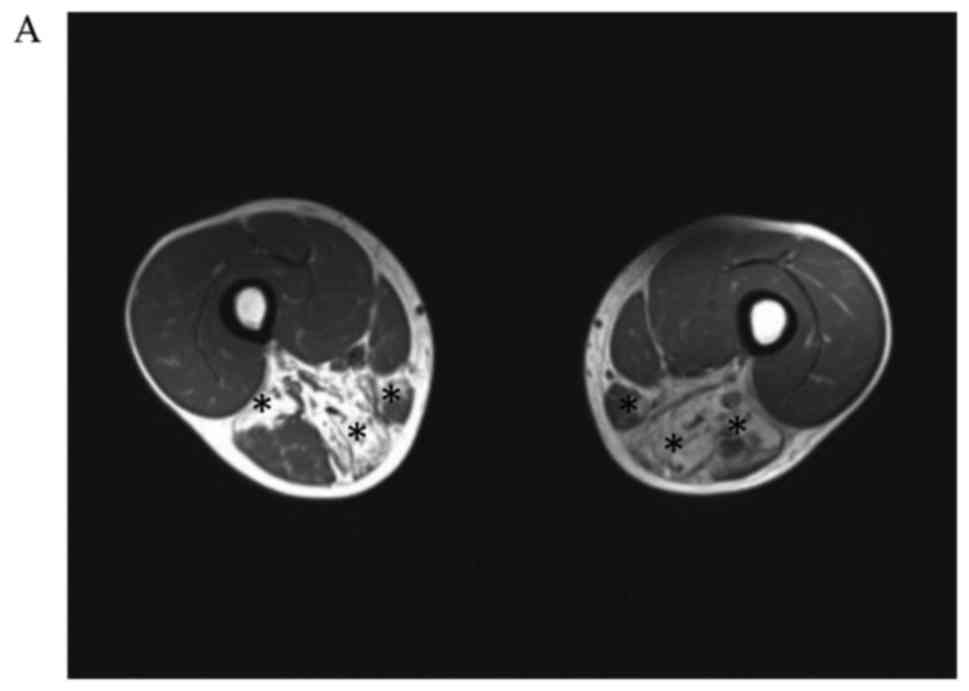 | Figure 2.Axial T1-weighted MR images in patient
#4. (A) An MR image was taken in the transverse plane through the
thighs shows severe fatty replacement of the muscles in the medial
and posterior compartments. (B) An MR image taken in the transverse
plane through the calves, shows marked involvement of the tibialis
anterior, tibialis posterior, extensor digitorum longus, peroneus
longus, peroneus brevis and gastrocnemius. (C) Axial T1-weighted MR
images in patient #2. An MR image taken in the transverse plane
through the calves shows marked involvement of the tibialis
anterior, tibialis posterior, extensor digitorum longus, peroneus
longus, peroneus brevis and gastrocnemius. (D) Axial T1-weighted MR
images in patient #7. An MR image taken in the transverse plane
through the calves shows marked fatty replacement of the muscles in
the anterolateral and anteromedial areas. The asterisks indicate
muscles with fatty replacement. MR, magnetic resonance. |
Pathological features
Hematoxylin and eosin staining of muscle biopsy
specimens showed rimmed vacuoles, fiber size variation, and
basophilic granule deposition around the vacuoles (Fig. 3, picture A, B-1, C-G). Small empty
spaces surrounded by tiny red granules were observed in the
cytoplasm of muscle fibers on modified Gomori trichrome staining
(Fig. 3, picture Bb). Inflammatory
cell infiltration was not seen, but degeneration and necrosis of
the muscle fibers were found. Internal nuclei were discovered in
patients #3, #4, and #7. Among all the patients, patient #2
(Fig. 3, picture Ba and Bb) had the
most atrophic muscle fibers, which is consistent with the severe
muscle weakness observed on clinical examination in this patient.
Periodic acid Schiff, NADH-tetrazolium, succinate dehydrogenase,
cytochrome C oxidase, and oil red O staining showed no obvious
abnormalities.
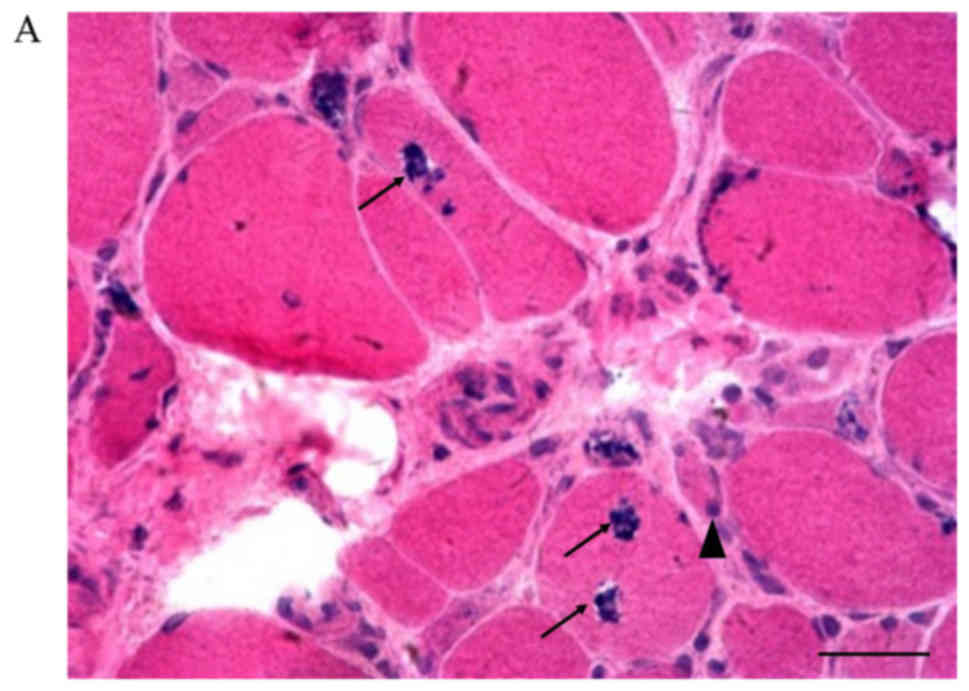 | Figure 3.Muscle biopsy examination in all the
study patients. (A) Patient #1, c.455_456insC. Some rimmed vacuoles
with basophilic granule deposition and inclusion bodies in angular
or round atrophic fibers are seen. Degeneration and necrotic muscle
fibers can also be found (hematoxylin and eosin staining;
magnification, ×200). (Ba) Patient #2, p.D207V. Variation in muscle
fiber size is observed. Some necrotic muscle fibers can be seen
(hematoxylin and eosin staining; magnification, ×200). (Bb) Patient
#2, p.D207V. Small empty spaces surrounded by tiny red granules in
the cytoplasm of muscle fibers (modified Gomori trichrome staining;
magnification, ×200). (C) Patient #3, p.C44S/p.G576R. Internal
nuclei and atrophic fibers are present (hematoxylin and eosin
staining; magnification, ×400). (D) Patient #4, p.D207V/p.A669P.
Rimmed vacuoles in two angular fibers and internal nuclei are
present (hematoxylin and eosin staining; magnification, ×400). (E)
Patient #5, p.D207V/p.D218G. Several small, angular, atrophic
fibers are seen. Rimmed vacuoles can be observed in one angular
fiber (hematoxylin and eosin staining; magnification, ×200). (F)
Patient #6, p.C44S. Rimmed vacuoles are seen in a round fiber, and
internal nuclei are present (hematoxylin and eosin staining;
magnification, ×200). (G) Patient #4, p.D207V/p.A669P. Necrotic
fibers with rimmed vacuoles are present (hematoxylin and eosin
staining; magnification, ×400). Inflammatory cell infiltration is
not seen in any panel. Scale bars, 100 µm. The arrowheads indicate
atrophic fibers and the arrows indicate rimmed vacuoles. |
Genetic features
The GNE genes of the seven patients were
analyzed, and the results are shown in Table III. A total of eight different
mutations, including one frameshift mutation and seven missense
mutations, were detected. Six mutations sites spanned the epimerase
region, while two sites were located in the kinase domain. Of the
eight mutations, five were known mutations, namely, p.D207V,
p.C44S, p.G576R, p.A669P, and p.D218G, while three had never been
reported before, namely, c.455_ 456insC, p.P421L, and p.A287T.
Patients #2, #4, and #5 had the same mutation in exon 4
(c.620A>T), which resulted in a p.D207V amino acid change, and
patients #3 and #6 had an identical mutation in exon 3
(c.131G>C), which led to the p.C44S amino acid transformation.
Patient #1 had a frameshift mutation, c.455_456insC, which resulted
in the early termination of the codon coding for the amino acid.
Patient #7 had two different mutations in exons 7 and 4 in the
epimerase region, resulting in p.P421L and p.A287T amino acid
changes.
| Table III.Genetic analysis results of seven
Chinese patients with DMRV. |
Table III.
Genetic analysis results of seven
Chinese patients with DMRV.
| Patient no. | Mutation type | GNE mutation | Exon | Domain |
|---|
| 1 | Frameshift | c.455_456InsC | 4 | Epimerase |
| 2 | Missense | c.620A>T
(p.D207V) | 4 | Epimerase |
| 3 | Missense | c.131G>C
(p.C44S) | 3 | Epimerase |
|
| Missense | c.1726G>C
(p.G576R) | 10 | Kinase |
| 4 | Missense | c.620A>T
(p.D207V) | 4 | Epimerase |
|
| Missense | c.2005G>C
(p.A669P) | 12 | Kinase |
| 5 | Missense | c.620A>T
(p.D207V) | 4 | Epimerase |
|
| Missense | c.653A>G
(p.D218G) | 4 | Epimerase |
| 6 | Missense | c.131G>C
(p.C44S) | 3 | Epimerase |
| 7 | Missense | c.1262C>T
(p.P421L) | 7 | Epimerase |
|
| Missense | c.859G>A
(p.A287T) | 4 | Epimerase |
All of our patients began to have symptoms such as
progressive distal and proximal muscle weakness in adulthood, and
none of the patients had a family history of DMRV. These clinical
features are consistent with previous reports (5). Muscle atrophy was present in five
patients; the observation of rimmed vacuoles on muscle biopsy and
the identification of GNE mutations on gene analysis
confirmed the diagnosis in these patients.
There is no definite connection between phenotype
and genotype in DMRV, but it has been reported that a homozygous or
heterozygous p.V572L mutation could cause early pathological
changes and more obvious clinical symptoms (17). The p.V572L/p.D207V heterozygous
mutation has been associated with mild symptoms, while the
homozygous p.D207V mutation causes few or even no clinical
symptoms. In our sample, the p.V572L mutation was not observed, but
one patient had a homozygous p.D207V mutation. This patient had an
early onset at the age of 27 years, leading to severe muscle
weakness, which is not in accordance with previous reports. We also
found two compound heterozygous mutations of p.D207V/p.A669P and
p.D207V/p.D218; in both these patients, the muscle strength was
>3, and symptom onset was relatively late, at the ages of 33 and
30 years, respectively. The patient with the compound heterozygous
mutation p.C44S/p.G576R and the one with a homozygous p.C44S
mutation had similar muscle strengths and atrophy in the hand
muscles, despite having different ages at symptom onset, i.e., 23
and 36 years. The two patients with later onset shared the same
missense mutation, p.D207V. Patients #1 and #7 had mutations
c.455_456insC and p.P421L/p.A287T, respectively, which had never
been reported before. Thus, the clinical symptoms in these patients
could not be statistically analyzed (12). The age at onset in these patients was
27 and 33 years, respectively (Table
II). Patient #1 had a muscle strength score of >3 and a
normal CK level, while patient #7 had a muscle strength score of
>4 (except in the tibialis anterior) and the highest CK level in
our sample. It is reported that CK results of this disease are
ranging from normal or mildly increased level (8,18) to the
high level of above 1,000 IU/l in 23% of patients (19). DMRV is caused by abnormal proteins
depositing in the cytoplasm with the relative integrity of muscle
cell membranes. Thus, CK release is prohibited by the cell
membranes and there is no amounts of CK released into the blood to
cause a high level of CK (7).
Therefore, CK level of DMRV is closely related to the pathogenesis
of this disease. Overall, the above results indicate that different
mutations lead to different clinical symptoms. However, there is a
need enlarge the sample size to draw definitive conclusions about
the connection between phenotype and genotype. A 2012 study
described the clinical features of DMRV according to the domain
affected by the mutation, i.e., whether the mutation was within the
UDP-GlcNAc 2-epimerase domain (ED) or the
N-acetylmannosamine kinase domain (KD) (10). The authors reported that KD/KD
mutations are associated with a more severe phenotype than ED/KD
mutations, and that ED/ED mutations appear to be associated with a
disease severity intermediate between ED/KD and KD/KD mutations
(10). In our series, patients #5
and #7 had ED/ED mutations, and had weaker quadriceps femoris and
iliopsoas muscles than patients #3 and #4, who harbored ED/KD
mutations. This finding is consistent with the above report. The
connection between phenotype and genotype should be investigated in
greater detail in future studies.
Discussion
DMRV has been reported worldwide in populations of
diverse ethnicities. GNE mutations have been reported in
Persian Jews, people from the Caucasus region, and the Indian,
Thai, Japanese, African, South Korean, and Chinese populations
(1,15,17,1).
These populations show much genetic heterogeneity and at least
three instances of the founder effect (12). The first described instance of the
founder effect refers to the M712T mutation, which is predominantly
found in patients of Middle Eastern descent and also occurs in
Muslim people of Bedouin and Palestinian descent as well as among
Italian and Japanese people (23–25). The
second variant consists of mutations that are common among Japanese
patients, namely, p.V572L, p.C44S, and p.D207V (1). The last variant is p.I618T, which is
found in the Romani population (10). Korean patients show a high rate of
p.V572L, followed by p.C44S (22).
With the growing popularity of genetic testing, an increasing
number of patients have been diagnosed with DMRV in China. On the
basis of relevant papers published by Chinese researchers, we
conclude that the mutations p.L508S, p.A631V, p.D207V, and p.A524V
have a high incidence in China, and the mutation p.L508S has only
been found in China (13,15). The mutation p.V572L, which is
relatively common among Japanese patients, was not found in our
series, and neither was the mutation p.L508S, which has a high
incidence in China. The most common mutation in our study was
p.D207V, which is the second most common mutation among Japanese
DMRV patients (1,17) and is also very common among Chinese
patients (15). Thus, our findings
are consistent with the high incidence rates of the p.D207V
mutation in these two countries. Two of our patients had the p.C44S
mutation, which is seldom reported among Chinese patients but is
the third and second most common GNE mutation in Japan and
South Korea (17,26), with mutation frequencies of 3.5 and
26.2%, respectively. The geographic relationship among the three
countries indicates a possible founder effect of this GNE
myopathy. However, the incidences are various, indicating the
genotype patterns are not completely consistent though some common
mutations are shared among them (15). The mutations p.G576R, p.A669P, and
p.D218G, which were reported in 2015 and 2012 (10,14),
were also found in three of our patients. All but two mutations in
our study were located in the epimerase region, and the majority of
the mutations in our series were missense mutations (Table III); these results are in
accordance with previous reports (17). The three previously unknown mutations
that we found, namely c.455_456insC, p.P421L, and p.A287T might be
beneficial to analyze the hot spot mutations in the future though
our observation is limited to a small cohort of patients.
There is still a lack of analysis of hot spot
mutations in China probably because of the low incidence rate of
DMRV and the lack of recognition of this disease. DMRV is usually
diagnosed using clinical and pathological features, though genetic
analysis is diagnostic. With increasing knowledge about the
mechanisms of this disease, a number of therapeutic methods have
been developed and applied, such as supplementation with
N-acetylmannosamine, sialic acid, and intravenous
immunoglobulin (as a source of sialic acid) (7). However, it remains unclear whether
metabolic supplementation can correct the defect or modify the
course of the disease.
In conclusion, GNE mutations were detected in
all seven patients in this series, confirming the diagnosis of
DMRV. This research reported five known mutations and three novel
GNE mutations. These results show that there still exist
unknown mutations in DMRV, which could be used to enrich the
GNE gene spectrum of DMRV myopathy in Chinese patients. Most
clinical neurologists are not well aware of DMRV because of the low
incidence of the disease, whose diagnosis depends on muscle biopsy
and genetic testing. With increasing awareness of DMRV among
physicians, we believe that more DMRV cases will be reported in the
future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81601088) and the
Natural Science Foundation of Jilin Province (grant no.
20160520164JH).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FS and JM contributed equally to this work. FS was a
major contributor in writing the manuscript and interpreting the
patients' data. JM designed the genetic test experiments,
interpreted the experimental results and critically revised the
manuscript for important intellectual content. XY assisted with the
writing and editing of the manuscript and interpreted the gene
analysis. XL collected the patient data. XW helped to collect and
process the data. All authors agreed to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Human Ethics
Committee of Jilin University and all patients provided written
informed consent prior to their inclusion.
Patient consent for publication
All patients provided written informed consent for
the publication of their associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nishino I, Noguchi S, Murayama K, Driss A,
Sugie K, Oya Y, Nagata T, Chida K, Takahashi T, Takusa Y, et al:
Distal myopathy with rimmed vacuoles is allelic to hereditary
inclusion body myopathy. Neurology. 59:1689–1693. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Argov Z and Yarom R: ‘Rimmed vacuole
myopathy’ sparing the quadriceps. A unique disorder in Iranian
Jews. J Neurol Sci. 64:33–43. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kayashima T, Matsuo H, Satoh A, Ohta T,
Yoshiura K, Matsumoto N, Nakane Y, Niikawa N and Kishino T: Nonaka
myopathy is caused by mutations in the
UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene
(GNE). J Hum Genet. 47:77–79. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eisenberg I, Avidan N, Potikha T, Hochner
H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini
G, et al: The UDP-N-acetylglucosamine
2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive
hereditary inclusion body myopathy. Nat Genet. 29:83–87. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huizing M and Krasnewich DM: Hereditary
inclusion body myopathy: A decade of progress. Biochim Biophys
Acta. 1792:881–887. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh R and Arya R: GNE myopathy and cell
apoptosis: A comparative mutation analysis. Mol Neurobiol.
53:3088–3101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishino I, Carrillo-Carrasco N and Argov
Z: GNE myopathy: Current update and future therapy. J Neurol
Neurosurg Psychiatry. 86:385–392. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nonaka I, Noguchi S and Nishino I: Distal
myopathy with rimmed vacuoles and hereditary inclusion body
myopathy. Curr Neurol Neurosci Rep. 5:61–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mori-Yoshimura M, Oya Y, Yajima H,
Yonemoto N, Kobayashi Y, Hayashi YK, Noguchi S, Nishino I and
Murata M: GNE myopathy: A prospective natural history study of
disease progression. Neuromuscul Disord. 24:380–386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mori-Yoshimura M, Monma K, Suzuki N, Aoki
M, Kumamoto T, Tanaka K, Tomimitsu H, Nakano S, Sonoo M, Shimizu J,
et al: Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine
kinase domain mutations in the GNE gene result in a less severe GNE
myopathy phenotype compared to homozygous N-acetylmannosamine
kinase domain mutations. J Neurol Sci. 318:100–105. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mori-Yoshimura M, Oya Y, Hayashi YK,
Noguchi S, Nishino I and Murata M: Respiratory dysfunction in
patients severely affected by GNE myopathy (distal myopathy with
rimmed vacuoles). Neuromuscul Disord. 23:84–88. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Celeste FV, Vilboux T, Ciccone C, de Dios
JK, Malicdan MC, Leoyklang P, McKew JC, Gahl WA, Carrillo-Carrasco
N and Huizing M: Mutation update for GNE gene variants associated
with GNE myopathy. Hum Mutat. 35:915–926. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu X, Pu C, Huang X, Liu J and Mao Y:
Distal myopathy with rimmed vacuoles: Clinical and muscle
morphological characteristics and spectrum of GNE gene mutations in
53 Chinese patients. Neurol Res. 33:1025–1031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu N, Wang ZK, Wang HX, Li Y, Niu ZH and
Yu XF: Muscle biopsy and UDP-N-acetylglucosamine
2-epimerase/N-acetylmannosamine kinase gene mutation analysis in
two Chinese patients with distal myopathy with rimmed vacuoles.
Neuroreport. 26:598–601. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao J, Wang Z, Hong D, Lv H, Zhang W,
Chen J and Yuan Y: Mutational spectrum and clinical features in 35
unrelated mainland Chinese patients with GNE myopathy. J Neurol
Sci. 354:21–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huizing M, Carrillo-Carrasco N, Malicdan
MC, Noguchi S, Gahl WA, Mitrani-Rosenbaum S, Argov Z and Nishino I:
GNE myopathy: New name and new mutation nomenclature. Neuromuscul
Disord. 24:387–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho A, Hayashi YK, Monma K, Oya Y, Noguchi
S, Nonaka I and Nishino I: Mutation profile of the GNE gene in
Japanese patients with distal myopathy with rimmed vacuoles (GNE
myopathy). J Neurol Neurosurg Psychiatry. 85:914–917. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jay CM, Levonyak N, Nemunaitis G, Maples
PB and Nemunaitis J: Hereditary inclusion body myopathy (HIBM2).
Gene Regul Syst Bio. 3:181–190. 2009.PubMed/NCBI
|
|
19
|
Tomimitsu H, Shimizu J, Ishikawa K,
Ohkoshi N, Kanazawa I and Mizusawa H: Distal myopathy with rimmed
vacuoles (DMRV): New GNE mutations and splice variant. Neurology.
62:1607–1610. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sivakumar K and Dalakas MC: The spectrum
of familial inclusion body myopathies in 13 families and a
description of a quadriceps-sparing phenotype in non-Iranian Jews.
Neurology. 47:977–984. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liewluck T, Pho-Iam T, Limwongse C,
Thongnoppakhun W, Boonyapisit K, Raksadawan N, Murayama K, Hayashi
YK, Nishino I and Sangruchi T: Mutation analysis of the GNE gene in
distal myopathy with rimmed vacuoles (DMRV) patients in Thailand.
Muscle Nerve. 34:775–778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim BJ, Ki CS, Kim JW, Sung DH, Choi YC
and Kim SH: Mutation analysis of the GNE gene in Korean patients
with distal myopathy with rimmed vacuoles. J Hum Genet. 51:137–140.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Broccolini A, Pescatori M, D'Amico A,
Sabino A, Silvestri G, Ricci E, Servidei S, Tonali PA and Mirabella
M: An Italian family with autosomal recessive inclusion-body
myopathy and mutations in the GNE gene. Neurology. 59:1808–1809.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kalaydjieva L, Lochmüller H, Tournev I,
Baas F, Beres J, Colomer J, Guergueltcheva V, Herrmann R, Karcagi
V, King R, et al: 125th ENMC international workshop: Neuromuscular
disorders in the Roma (Gypsy) population, 23–25 April 2004,
naarden, the Netherlands. Neuromuscul Disord. 15:65–71. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eisenberg I, Grabov-Nardini G, Hochner H,
Korner M, Sadeh M, Bertorini T, Bushby K, Castellan C, Felice K,
Mendell J, et al: Mutations spectrum of GNE in hereditary inclusion
body myopathy sparing the quadriceps. Hum Mutat. 21:992003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sim JE, Park HJ, Shin HY, Nam TS, Kim SM
and Choi YC: Clinical characteristics and molecular genetic
analysis of Korean patients with GNE myopathy. Yonsei Med J.
54:578–582. 2013. View Article : Google Scholar : PubMed/NCBI
|