Introduction
Idiopathic interstitial pneumonias (IIPs) are a
group of heterogeneous diseases with unknown etiology,
characterized by pulmonary parenchymal distortion caused by varying
amounts of fibrosis and inflammation, classified under the heading
of diffuse parenchymal lung diseases. While IIPs have been reported
since the 19th century, it was in 2002 that the American Thoracic
Society (ATS) and the European Respiratory Society (ERS) were the
first to define and categorize this condition into seven groups
considering clinical, radiological and histopathological findings
(Table I) (1), and this classification was revised by
the ATS and ERS in 2013 (2). The
current classification divides IIPs into three groups: Major IIP,
rare IIP and unclassifiable IIP (Table
II). Major IIP is further divided into three groups, namely
chronic fibrosing IIPs, smoking-associated IIPs and acute or
sub-acute IIPs (Table III).
 | Table I.Histological and clinical
classification of interstitial pneumonia. |
Table I.
Histological and clinical
classification of interstitial pneumonia.
| Histological
pattern |
Clinical-radiological-pathological
diagnosis |
|---|
| Usual interstitial
pneumonia | Idiopathic
pulmonary fibrosis/cryptogenic fibrosing alveolitis |
| Non-specific
interstitial pneumonia | Non-specific
interstitial pneumonia |
| Organized
Pneumonia | Cryptogenic
organized pneumonia |
| Diffuse alveolar
injury | Acute interstitial
pneumonia |
| Respiratory
bronchiolitis | Respiratory
bronchiolitis interstitial lung disease |
| Desquamative
interstitial pneumonia | Desquamative
interstitial pneumonia |
| Lymphoid
interstitial pneumonia | Lymphoid
interstitial pneumonia |
| Table II.American thoracic Society/European
respiratory society classification of IIP from 2013. |
Table II.
American thoracic Society/European
respiratory society classification of IIP from 2013.
| IIP
classification | Pathology |
|---|
| Major IIP | Idiopathic
pulmonary fibrosis |
|
| Idiopathic
nonspecific interstitial pneumonia |
|
| Respiratory
bronchiolitis interstitial lung disease |
|
| Desquamative
interstitial pneumonia |
|
| Cryptogenic
organized pneumonia |
|
| Acute interstitial
pneumonia |
| Rare IIP | Idiopathic
lymphocytic interstitial pneumonia |
|
| Idiopathic
pleuroparenchymal fibroelastosis |
| Unclassified
IIP | – |
| Table III.Classification of major IIP. |
Table III.
Classification of major IIP.
|
Category/clinical-radiological-pathological
diagnosis | Morphological
pattern |
|---|
| IIP with chronic
fibrosis | Usual interstitial
pneumonia |
|
Idiopathic pulmonary
fibrosis | Non-specific
interstitial pneumonia |
|
Idiopathic non-specific
interstitial pneumonia |
|
| IIP associated with
smoking | Respiratory
bronchiolitis |
|
Respiratory bronchiolitis
interstitial lung disease | Desquamative
interstitial pneumonia |
|
Desquamative interstitial
pneumonia |
|
| Acute/sub-acute
IIP | Organized
pneumonia |
|
Cryptogenic organized
pneumonia | Diffuse alveolar
injury |
| Acute
interstitial pneumonia |
|
Desquamative interstitial pneumonia (DIP) is a major
type of smoking-associated IIP, which is characterized by
accumulation of alveolar macrophages in alveolar lumens and septa
and develops secondary to mainly active or passive exposure to
cigarette smoke. It was first defined in 1965 by Liebow et
al (3), who described lesions
exhibiting extensive alveolar cell proliferation and desquamation
associated with mild thickening in distal airways, but not with
necrosis (4). The disease was
earlier named as such based on the assumption that it was caused by
desquamation of alveolar epithelium into the alveoli. Although the
ATS considered ‘alveolar macrophage pneumonia’ as a more
explanatory definition in 2002, this was renounced later (1).
The prevalence of DIP is not exactly known. Global
or national epidemiological data are usually for idiopathic
pulmonary fibrosis (IPF), which is the most common form of IIPs. No
epidemiological data are available for rare forms of IIP, such as
DIP; however, a limited number of case-based studies are available
(5).
Etiology
Despite the more common occurrence in the 4th or 5th
decade of life, pediatric cases have also been described in whom
surfactant pathology rather than smoking has been implicated. Also
smoking-associated pediatric cases have been described (1). The male-to-female ratio is 2:1.
While 100% of respiratory bronchiolitis-associated
interstitial lung disease (RB-ILD) cases are linked with cigarette
smoking, this figure is 90% for DIP. In addition to smoking, other
factors such as systemic disease, infections,
environmental/occupational exposure to hazardous agents as well as
drugs may be associated with DIP. Although RB-ILD does not occur in
children, DIP may rarely be seen in children. In contrast with the
significant male predominance (factor 2:1) among DIP cases, there
is no marked predominance in RB-ILD. The two conditions generally
occur in the 4th to 5th decades of life (6).
With regard to the link between DIP and IPF, a less
marked association with cigarette smoking exists for IPF (41–83% of
cases). IPF mostly occurs in the middle-aged or elderly population,
with a reported male-to-female ratio between 1:1 and 2:1. It may
rarely occur during childhood (6).
Risk factors
Smoking
The most important widely-recognized factor in the
etiology of DIP is the active or passive exposure to cigarette
smoke (7). It was reported in 58–91%
of smokers (4,6,8–10). Vassollo (11) described smoking as the most important
factor in DIP etiology. Craig et al (12) assessed histopathologically confirmed
interstitial pneumonia cases and detected a smoking history in 12
of 20 DIP cases, a number which they concluded to be significantly
higher compared with that among RB cases. Ryu et al
(13) emphasized smoking in the
etiology of DIP and stressed that progressive and fatal cases had a
history of heavy smoking.
While an association between smoking and DIP has
been fully demonstrated, DIP is also seen in either non-smokers or
smoking quitters. This implies the involvement of other etiological
risk factors such as environmental or occupational exposure,
accompanying diseases or medication use, with or without
smoking.
Occupational or environmental exposure
to hazardous substances
Regarding occupational exposure to hazardous agents
in 8 DIP cases with no smoking history in the study by Craig et
al (12), diesel or fire smoke,
or beryllium exposure had occurred in these cases. In their study
on a series of 10 cases, Moon et al (14) detected solder smoke and occupational
wood dust exposure in two different non-smoker cases.
Another important occupation affecting DIP is the
textile industry. Lougheed et al (15) reported biopsy-based DIP cases in
textile workers employed at cotton/polyester factories. A study
performed on 88 textile workers with DIP included five cases with
no current smoking status, of which two were non-smokers and the
remaining three had a prior smoking history. The authors concluded
that a possible factor responsible for DIP development may be
aflatoxin inhalation in the workplace. They reported that DIP may
develop after exposure to substances including asbestos, talc,
graphite, silica and aluminum (15).
Liebow and Carrington (16) reported
on DIP cases associated with exposure to tungsten cobalt. In a
study on the effects of metals on the lung, Nemery (17) suggested that desquamative and
giant-cell interstitial pneumonias may occur due to cobalt
exposure. Asbestos-associated DIP cases were reported by Corrin and
Price (18) in a shipyard worker and
by Freed et al (19) in a
construction worker who was employed for 32 years. Hull and Abraham
(20) reported exposure to aluminum
welding smoke as a risk factor for DIP. A different type of
occupational exposure as a risk factor was pointed out by Nakazawa
et al (21), who presented a
radiologically and histopathologically confirmed DIP case who had
been subjected to heavy exposure to occupational waterproof spray.
Despite the presence of a smoking history, this occupational factor
was regarded to be responsible for the disease. All of these
findings, in spite of mostly being derived from case-based studies,
demonstrated that environmental or occupational exposure to
hazardous substances may have an important role in the etiology of
DIP.
Psychotropic substance or medication
use and chronic diseases
DIP cases associated with chronic drug or
psychotropic drug use have been reported. Carrington et al
(22) suggested marijuana use as an
etiological factor. Pathologically confirmed DIP cases developed
secondary to marijuana use and their specimens revealed gold-brown
particle-laden macrophages, which was reported to be a distinctive
feature of smoking-associated DIP (21). Gill (23) also presented a cannabis-associated
case. Certain drugs, including macrolides, sirolimus,
nitrofurantoin, sulfasalazine and tocainide were reported to be
associated with DIP (21,24). Sirolimus is an immunosuppressive
agent whose application is indicated in solid organ transplant
patients, amongst whom a large number of pulmonary toxicity cases
were reported after sirolimus use. Under circumstances where
post-transplant deterioration, dyspnea and hypoxia occur without
any evidence of infection, this deterioration may be associated
with DIP (25).
DIP was also demonstrated to occur during chronic
diseases. A study on 26 DIP cases by Tubbs et al (4) reported that 58% of subjects had a
history of smoking, and that rheumatologic disorders may have been
responsible for the disease in the remaining patients. Ishii et
al (26), who presented 24 cases
where DIP and rheumatologic disease were comorbid, detected
positivity of certain markers such as rheumatoid factor and
anti-nuclear antibody, and recommended that diagnosis of DIP is
established upon histopathological evidence. Kawabata et al
(27) reported 20 DIP cases emerging
during systemic lupus erythematosus.
There are also case presentations showing an
association between DIP and infectious diseases. Iskandar et
al (28) reported on the
development of DIP due to immunological response mechanisms during
hepatitis C infection. Similarly, Hasegawa et al (29) reported DIP in a 72-year-old patient
with chronic hepatitis C infection who had no history of active or
passive smoking. The condition was thought to occur secondary to an
immunological response (29).
Cytomegalovirus (CMV) infection was also reported to be involved in
the etiology of DIP. Schroten et al (30) reported a case of DIP in an
8-month-old infant, developed after CMV infection. Sung et
al (31) reported a DIP case
which occurred during CMV and aspergillus infections in a renal
transplant patient.
Rare causes of DIP have also been reported. Arai
et al (32) presented a DIP
case developed after receiving a tattoo, which was attributed to a
foreign body reaction. While the most recognized etiological factor
is exposure to cigarette smoke, DIP may also result from chronic
diseases, medication use and occupational or environmental exposure
to various substances, as mentioned above. In particular, DIP
should be considered in rheumatologic disease and in transplant
patients with new onset of radiological alterations and clinical
deterioration in the absence of infection.
Diagnosis
Laboratory and bronchoalveolar lavage
(BAL) findings
Laboratory tests usually do not reveal any abnormal
findings (3). Certain patients were
reported to have polycythemia, which was thought to be associated
with respiratory insufficiency (3).
A study reported elevated lysozyme levels (33). Another study reported increased
levels of erythrocyte sedimentation rate, lactate dehydrogenase,
immunoglobulin (Ig)G, IgE and interleukin-6 (34).
Slight elevations in eosinophil, neutrophil,
lymphocyte and macrophage counts in the BAL fluid have been
reported (27,34,35).
However, it should be kept in mind that these laboratory and BAL
findings are non-specific and to date, no specific sign has been
defined.
Radiological finding
Radiological diagnosis of DIP
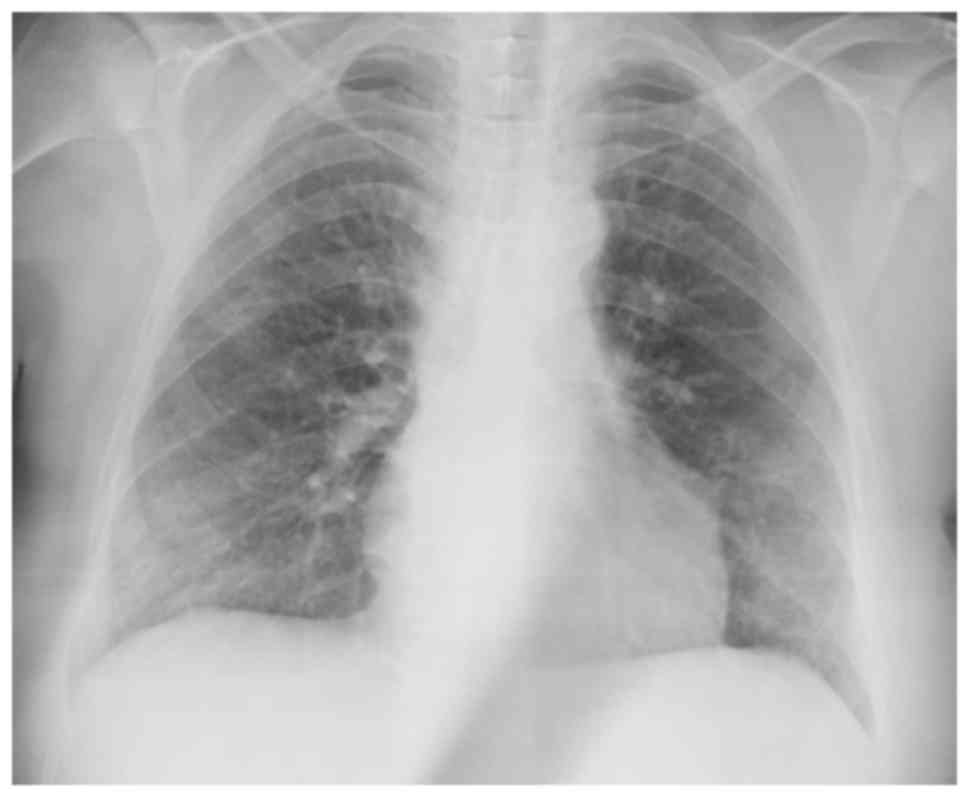
Chest X-ray findings are non-specific (Fig. 1; patient provided informed consent
for inclusion in the present study). While patchy ground-glass
opacities located peripherally in lower zones are observed in
certain cases, others have normal chest X-rays (1).
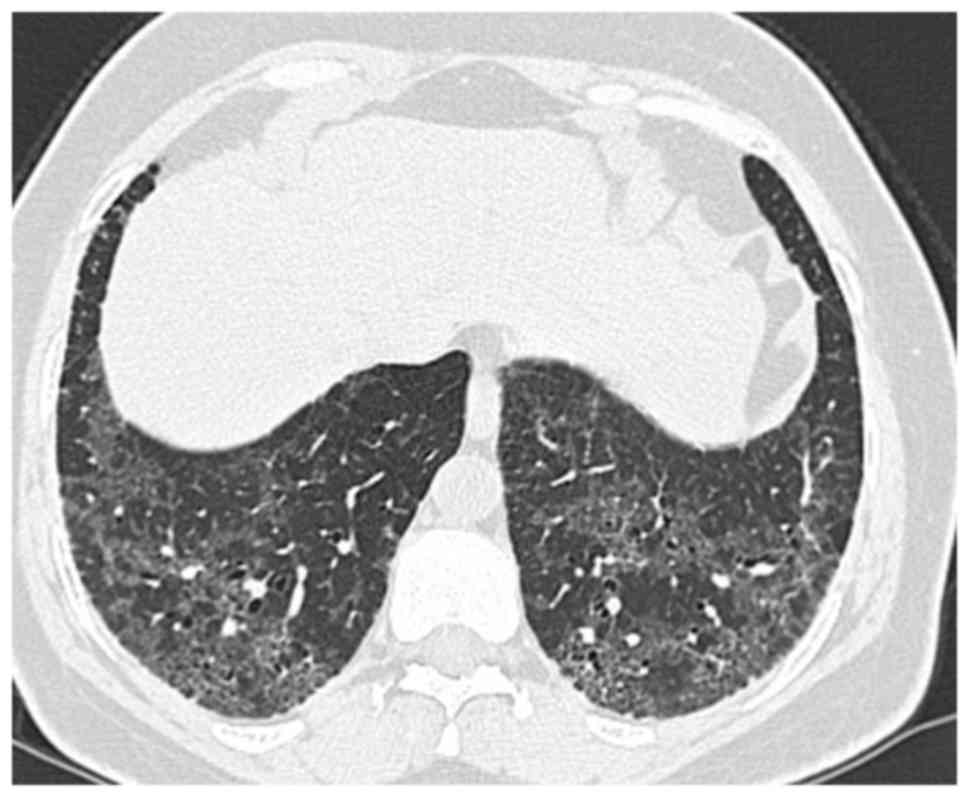
Diagnostic radiological findings are rather apparent
from thoracic high-resolution computed tomography (HRCT; Fig. 2; patient provided informed consent
for inclusion in the present study). An invariable HRCT finding is
the presence of a diffuse ground-glass appearance, usually
symmetrical and frequently involving the middle and lower zones
(6,36,37).
While involvement of upper zones is seen in most cases, their
predominant involvement is relatively rare. All regions may be
affected, although subpleural involvement is most common. Other
HRCT findings include irregular lines and traction bronchiectases
indicating parenchymal distortion (38). Peripheral microcysts implying dilated
bronchioles and alveolar ducts may be seen (39). Honeycomb appearance is not
common.
Differential diagnosis
Differential diagnoses should include RB-ILD,
hypersensitivity pneumonia and non-specific interstitial pneumonia
(NSIP). Certain features outlined below may help to rule out these
diagnoses. At times, HRCT findings of RB-ILD may include
centrilobular micronodules, ground-glass appearance, linear
reticular appearance, atelectasis and emphysema (40–43).
Centrilobular nodules and patchy ground-glass appearance tend to be
diffuse with no predominance of any zone, unlike the appearance in
DIP (44). Emphysema and patchy
hypoattenuation are mainly seen in lower zones. Radiological
findings of NSIP often consists of bilateral, symmetrical and
subpleural ground-glass appearance, which is at times superposed by
irregularly linear or reticular opacities or traction
bronchiectases (39,45,46).
Chronic hypersensitivity pneumonia displays at ground-glass
appearance, millimetric centrilobular nodules and mosaic perfusion
pattern associated with air entrapment in HRCT (47,48). In
addition, certain patients may have a honeycomb appearance
(49).
Histopathological examination
Histopathological diagnosis of
DIP
Due to non-specific laboratory, BAL and radiological
findings, surgical biopsy is indicated in cases with suspected IIP
with no classical usual interstitial pneumonia (UIP) pattern on
HRCT, as recommended by the ATS and ERS (1,50).
When first described, the predominant histological
feature of the disease was thought to be desquamation of alveolar
epithelial cells, and hence it was named DIP (3). However, electron microscopy images
indicated macrophage accumulation in alveoli rather than
desquamation of epithelial cells (4). The major characteristic of DIP is
proliferation of pneumocytes over the course of alveolar septa and
the presence of numerous macrophages, diffusely distributed along
with pulmonary acini within alveoli (1,10,51).
These macrophages are termed smoker's macrophages, and exhibit
eosinophilic cytoplasm and fine granular light brown pigments
(10). Fine granular iron may be
seen in the cytoplasm of macrophages (52), multinuclear giant cells are frequent
(3,5), and eosinophil and lymphoid aggregates
may be observed (1,3,4). The
alveolar structure is usually preserved despite mild chronic
interstitial inflammation. Albeit rare, interstitial fibrosis may
be seen. Emphysema is usually observed, but no or minimal honeycomb
appearance is present (12).
Differential diagnosis based on
histopathology
DIP is listed among smoking-associated IIPs along
with RB-ILD and pulmonary Langerhans cell histiocytosis (PLCH)
(12,35). RB is a commonly incidental
histopathological lesion of smokers, being mainly asymptomatic
(53). RB-ILD is a type of RB
characterized by respiratory symptoms as well as radiological and
spirometric abnormalities observed in certain smokers. Based on
histological findings, of RB could not be differentiated from
RB-ILD (54). Although these two
clinical entities have distinct diagnostic criteria,
histopathological differentiation is compelling (2). The major histological finding in RB-ILD
is accumulation of alveolar macrophages in respiratory bronchioles.
The most important difference between DIP and RB-ILD is the diffuse
and uniform character of this accumulation and lesions in DIP, as
compared with the bronchiolocentric character of RB-ILD (40). Unlike in DIP, biopsy of RB-ILD
reveals less interstitial fibrosis, and less eosinophil and
lymphoid follicles in the interstitium are seen (12). Giant cells are not identified in
RB-ILD patients. Since RB-ILD as well as DIP are associated with
smoking and smokers with other ILDs have common histopathological
features with RB-ILD and DIP, the diagnosis of the latter two
conditions may only be established in the absence of signs of ILD
(55–58).
NSIP should also be kept in mind for the
differential diagnosis of DIP. Varying degrees of fibrosis are seen
during histopathological examination of fibrotic NSIP (59). While certain patients have patchy
fibrosis due to remodeling of the lung (60), a more diffuse involvement with
preservation of the alveolar structure may also be observed.
Airspace macrophages are not present. In certain smoking patients,
differential diagnosis of DIP and fibrotic NSIP based on
radiological or histopathological findings is compelling. It has
been suggested that DIP may transform into fibrotic NSIP and
certain NSIP cases may be associated with smoking (26), which still remains controversial.
The major histological characteristics of early
PLCH, a smoking-associated ILD, is the presence of peribronchial
nodules containing Langerhans and inflammatory cells (61). Not grouped among IIPs, airspace
enlargement with fibrosis [also termed smoking-associated
interstitial fibrosis (SRIF)] and combined pulmonary fibrosis and
emphysema (CPFE) are the two conditions histopathologically
observed in smokers, and which should be distinguished from DIP.
SRIF is a recently described histological pattern (62). It was defined after observation of
certain alterations in non-neoplastic pulmonary areas of smoking
lung cancer patients who underwent lobectomy (57,62,63).
Histopathological observations are incidentally made in these
asymptomatic patients. They have mainly sub-pleural emphysematous
areas and smoker's macrophages within alveoli. Thickened alveolar
septa are seen due to hyperplastic smooth-muscle fibers and
eosinophilic collagen structures. While SRIF is an incidentally
detected radiological and histopathological condition, CPFE, which
is rather a comorbid pathology of IPF, has clinical signs
associated with emphysema and interstitial fibrosis (2).
Clinical features
DIP, a chronic disease, mostly occurs in male
smokers in association with non-specific symptoms responsive to
steroid therapy and has a better prognosis than UIP. To date, no
large-scale clinical studies have been performed in DIP patients.
Factors responsible for the scarcity of data on the clinical course
of this condition include the retrospective nature of the available
information as well as its rare occurrence. Despite this, a general
consensus exists as to the nature of its symptoms, association with
smoking, age and gender distribution, findings of respiratory
function tests, steroid responsivity and mortality (Table IV).
| Table IV.Clinical and functional findings and
the disease course in patients with desquamative interstitial
pneumonia. |
Table IV.
Clinical and functional findings and
the disease course in patients with desquamative interstitial
pneumonia.
| Parameter | Ryu et al
(13) | Kawabata et
al (34) | Craig et al
(12) | Yousem et al
(10) |
|---|
| N | 23 | 31 | 20 | 36 |
| Male sex | 11/23 (48) | 29/31 (93.5) | 12/20 (60) | 26/36 (72.2) |
| Age (years) | 46±10 | 55±13 | 43 | 42 (17–67) |
| Smoking status
(current) | 18/23 (78) | 28/30 (93) | 12/20 (60) | 33/36 (91.6) |
| Smoking status
(previous) | 2 (8) |
|
|
|
| Smoking history
(pack years) | 38±21 | 52±41 |
| 36 (10–71) |
| Symptoms |
|
|
|
|
| None | 1 (4) |
|
| 5/34 (15) |
| Dyspnea | 20 (87) |
|
| 29/34 (85) |
| Cough | 10 (43) |
|
| 26/32 (81) |
| Sputum |
|
|
| 17/33 (52) |
| Chest pain | 4 (17) |
|
|
|
| Physical signs |
|
|
|
|
| Inspiratory
crackles | 13 (57) |
|
| 5/9 (56) |
| Digital
clubbing | 6 (26) |
|
| 15/36 (42) |
| Pulmonary
function |
|
|
|
|
| Restrictive | 6 (30) |
|
|
|
| Obstructive | 3 (15) |
|
|
|
| Low diffusion
capacity only | 7 (35) |
|
|
|
| Normal | 4 (20) |
|
|
|
| Total lung
capacity, PP | 84.8±18.8 |
|
| 94 (43–133) |
| FVC, PP | 74.1±16.7 | 84±23 (VC) |
| 68 (33–124) |
| DLCO, PP | 52.8±16.7 |
|
| 45 (32–78) |
| Oxygen saturation
at rest, % | 93.8±3.6 |
|
|
|
| Oxygen saturation
with exercise, % | 89.4±5.1 |
|
|
|
| PaO2,
mmHg |
| 79±11 |
|
|
| Mortality, % | 26% | 78% (10 years) |
| 8/36 (32) |
In their retrospective study, Ryu et al
(13) examined 33 patients with DIP
and 12 patients with RB-ILD. The patient characteristics in these
subjects are summarized in Table
IV. In that study, a total of 21 DIP patients (91%) received
corticosteroids and 4 quit smoking (27%). Improvement was found in
1 patient (5%), while 12 patients (63%) had stable disease, while
the disease deteriorated in 1 patient (5%) and 5 patients succumbed
to their disease (26%). The causes of death included respiratory
failure in 3, lung cancer in 1 and hepatic carcinoma in
another.
In the study by Kawabata et al (34), a total of 31 DIP patients were
followed-up for a duration of 99 months. Their patient
characteristics are depicted in Table
IV. Patients received pulsed or oral steroids at a daily dose
of 15–80 mg, with clinical improvement in 90% of the cases. Seventy
patients quit smoking prior to or after treatment. Deaths occurred
due to DIP progression, lung cancer, fulminant pulmonary disease
after lobectomy and diffuse alveolar disease in one patient each
and due to causes not associated with lung disease in another four
patients. Lobectomy was performed in three patients due to lung
cancer. The 10-year survival rate was 78%.
Craig et al (12) reported an average survival of 8.8
years for non-smokers and 7 years for smokers during the follow-up
of their patients. Due to the presence of occupational exposure to
hazardous substances in their patient group, the authors concluded
that it was not possible to definitely determine the causative role
of DIP in the development of clinical signs.
Bressieux-Degueldre et al (64) reported on a 30-month old female
patient who presented with respiratory failure. Physical
examination revealed the presence of respiratory rales on
auscultation. After a diagnosis of DIP was established,
corticosteroid treatment was commenced and the patient was reported
to be oxygen-dependent at 2 years of follow-up. DIP represents the
most common form of pediatric interstitial lung disease, which has
been linked with a genetic defect in surfactant proteins in
children. A possible diagnosis of interstitial lung disease should
be suspected in children with cough, dyspnea or tachypnea lasting
>3 months. Aggressive nutritional support as well as preventive
measures against infections are important in the management of
children with DIP. Although corticosteroids and immunosuppressive
agents are recommended, the prognosis is poor. Lung transplantation
represents a therapeutic option during terminal disease.
A 44-year-old patient presenting with shortness of
breath, right-sided pleuritic chest pain and orthopnea was
described by Behnia and Cummings (65). The patient was found to be
sub-febrile and tachycardic, and also had rales in the lower zones
of the lung on auscultation. Pulmonary angiography revealed no
filling defects. Clinically, the patient was hypotensive and died
at 5 days after admission; the diagnosis was established on
autopsy.
In addition to those presenting with cough, dyspnea
or chest pain, patients presenting with non-specific symptoms such
as slightly increased body temperature, myalgia, weight loss,
fatigue or respiratory failure have also been reported (66,67).
Yousem et al (10) assessed 36 DIP patients with chronic
symptoms. The clinical characteristics of these patients are
summarized in Table IV. In this
retrospective evaluation, 14 patients (56%) exhibited improvement
during follow-up, while 3 (12%) were stable and 8 patients (32%)
died. Comparison between RB-ILD and DIP patients revealed an
average diffusion capacity of 62 and 45% in these groups,
respectively.
The most common symptoms of DIP include dyspnea and
dry cough of insidious onset that may last for weeks or months. In
addition, patients with respiratory failure, fever, fatigue and
weakness have been reported. Digital clubbing is present in nearly
half of the cases (1). Chest pain
and weight loss may also occur. Hemoptysis is rare, as are
asymptomatic cases. Cyanosis may be seen. DIP may occur in
association with connective tissue disorders such as scleroderma,
lupus or rheumatoid arthritis, and their symptoms may accompany
those of DIP (68).
In patients with DIP, the lung volume is normal or
mild restrictive abnormalities may be detected, with moderately
reduced diffusing capacity of the lungs for carbon monoxide, which
is an indicator of the severity of the underlying disorder. In the
assessment of the severity of the functional impairment in DIP,
emphysema, which is a frequent co-morbidity, may be a confounding
factor (69). Cor pulmonale is rare
and hypoxia occurs in the advanced stages of the disease (70).
In each of the two forms of smoking-associated IIPs,
DIP and RB-ILD, cough and shortness of breath represent the most
common symptoms with an insidious onset (6). Symptoms and clinical features are
non-specific in DIP and RB-ILD. The nature of dyspnea in DIP and
RB-ILD is slowly progressing exercise dyspnea (69). While rales on auscultation may be
heard in each of the two conditions, digital clubbing is more
common in DIP. Approximately 60% of DIP patients are found to have
rales on auscultation on physical examination. Respiratory function
tests reveal a restrictive pattern in DIP, while a mixed defect or
normal result may be observed in RB-ILD (6). RB-ILD and DIP respond favorably to
steroids with good prognosis, and a complete response may
occur.
In IPF, the onset is insidious. Dry cough and
shortness of breath comprise the most common symptoms. Almost all
patients have rales on auscultation and digital clubbing is more
common (50–70%). Compared to IPF, a less marked effect is observed
in respiratory function tests in DIP (6).
Treatment and prognosis
Prognosis in DIP is usually favorable and the
majority of the patients improve with quitting smoking and
corticosteroid therapy. The 10-year survival rate is ~70% (1), with a mortality rate between 6 and 28%
(71). Of the untreated patients,
almost two thirds have a poor prognosis (6). Despite an insidious onset, the disease
may exhibit a rapidly progressive course. Progression to severe
fibrosis is rare (70).
Compared to IPF, DIP has a more moderate prognosis
with a better response to anti-inflammatory treatment. The mainstay
of treatment is quitting smoking, which may, on its own, be
sufficient in certain cases (72).
Environmental factors and drugs are also implicated in the
development of DIP (15,18,73,74).
Therefore, termination of any probable environmental risk factors
is reasonable. In a study assessing a series of 5 cases of
occupational DIP, Lougheed et al (15) reported improvement in two cases after
moving off the workplace requiring no steroid treatment, and
chronic respiratory insufficiency despite steroid treatment in the
three remaining cases.
Systemic steroid treatment is recommended in
moderately or severely symptomatic patients who progressed despite
quitting smoking. While certain patients respond to this treatment
based on clinical and radiological findings, others remain stable
or the disease progresses (10,75–77). Ryu
et al (13) followed 23 DIP
patients for 12 years and reported symptomatic improvement in 24%
of cases that were initiated with steroid treatment. However, they
also reported recurrence in certain patients after termination of
steroid therapy, even in smoking quitters, where recurrence rates
were higher in those resuming smoking or with passive cigarette
smoking. It remains to be elucidated whether improvement after
steroid initiation depends on steroid treatment or termination of
smoking, or results from the natural course of the disease.
However, observational studies indicated progression in most of the
patients who did not receive any treatment (22). Although no randomized controlled
clinical trial proving its efficacy is available, initiation of
steroid treatment in all patients with diagnosis of DIP appears to
be a reasonable approach with continuance of the treatment upon
clinical, radiological and functional improvement and
discontinuance in case of no response. Initial steroid treatment
consists of 40–60 mg/day for 6 weeks, followed by tapering and
cessation of the treatment within 6–9 months (33).
Based on the potential anti-inflammatory effects of
macrolides in steroid-refractory cases, Knyazhitskiy et al
(78) reported rapid and dramatic
improvement in all clinical and radiological parameters with
clarithromycin treatment in a patient who was refractory to steroid
therapy. Similarly, in a case where no response was obtained after
initiation of oral steroids, clinical and radiological improvement
was observed at one month after reduction of the steroid dose and
addition of clarithromycin (79).
Data on cytotoxic and immunosuppressive agents for
treating DIP are inconclusive. A limited number of studies reported
favorable outcomes after azathioprine and methotrexate treatment
(66). Lung transplantation may be
the treatment of choice in cases with severe and persistent
disease. However, recurrence may also be seen after transplantation
(80–82).
Acknowledgements
The authors are grateful for the support of
Professor Arzu Mirici (Department of Pulmonary Diseases, Çanakkale
University School of Medicine, Çanakkale, Turkey) and President of
the Young Academicians Group (GEAK) of the Turkish Respiratory
Society, who contributed to the planning of this review. All of the
authors of the present study are also members of GEAK.
References
|
1
|
American Thoracic Society; European
Respiratory Society: American Thoracic Society/European Respiratory
Society International Multidisciplinary Consensus Classification of
the Idiopathic Interstitial Pneumonias. This joint statement of the
American Thoracic Society (ATS), and the European Respiratory
Society (ERS) was adopted by the ATS board of directors, June 2001
and by the ERS Executive Committee, June 2001. Am J Respir Crit
Care Med. 165:277–304. 2002.PubMed/NCBI
|
|
2
|
Travis WD, Costabel U, Hansell DM, King TE
Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU,
et al: An official American Thoracic Society/European Respiratory
Society Statement: Update of the international multidisciplinary
classification of the idiopathic interstitial pneumonias. Am J
Respir Crit Care Med. 188:733–748. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liebow AA, Steer A and Billingsley JG:
Desquamative interstitial pneumonia. Am J Med. 39:369–440. 1965.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tubbs RR, Benjamin SP, Reich NE, McCormack
LJ and Van Ordstrand HS: Desquamative interstitial pneumonitis.
Cellular phase of fibrosing alveolitis. Chest. 72:159–165. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Okutan O and Çalışkan T: Smoking-related
Interstitial Lung Diseases. Eurasian J Pulmonol. 13:131–139.
2011.
|
|
6
|
Ryu JH, Colby TV, Hartman TE and Vassallo
R: Smoking-related interstitial lung diseases: A concise review.
Eur Respir J. 17:122–132. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ischander M, Fan LL, Farahmand V, Langston
C and Yazdani S: Desquamative ınterstitial pneumonia in a child
related to cigarette smoke. Pediatr Pulmonol. 49:E56–E58. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Well AU, Nicholson AG and Hansell DM:
Challenges in pulmonary fibrosis. 4: Smoking induced diffuse
interstitial lung disease. Thorax. 62:904–910. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bradley B, Branley HM, Egan JJ, Greaves
MS, Hansell DM, Harrison NK, Hirani N, Hubbard R, Lake F, Millar
AB, et al: Interstitial lung disease guidelines: The British
Thoracic Society in collaboration with the Thoracic Society of
Australia and New Zealand and the Irish Thoracic Society. Thorax.
63 Suppl 5:v1–v58. 2008.PubMed/NCBI
|
|
10
|
Yousem SA, Colby TV and Gaensler EA:
Respiratory bronchiolitis-associated interstitial lung disease and
its relationship to desquamative interstitial pneumonia. Mayo Clin
Proc. 64:1373–1380. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vassallo R: Diffuse lung diseases in
cigarette smokers. Semin Respir Crit Care Med. 33:533–542. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Craig PJ, Wells AU, Doffman S, Rassl D,
Colby TV, Hansell DM, Du Bois RM and Nicholson AG: Desquamative
interstitial pneumonia, respiratory bronchiolitis and their
relationship to smoking. Histopathology. 45:275–282. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ryu JH, Myers JL, Capizzi SA, Douglas WW,
Vassallo R and Decker PA: Desquamative interstitial pneumonia and
respiratory bronchiolitis-associated interstitial lung disease.
Chest. 127:178–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moon J, du Bois RM, Colby TV, Hansell DM
and Nicholson AG: Clinical significance of respiratory
bronchiolitis on open lung biopsy and its relationship to smoking
related interstitial lung disease. Thorax. 54:1009–1014. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lougheed MD, Roos JO, Waddell WR and Munt
PW: Desquamative interstitial pneumonitis and diffuse alveolar
damage in textile workers. Potential Role of Mycotoxins Chest.
108:1196–1200. 1995.PubMed/NCBI
|
|
16
|
Liebow AA and Carrington CB: The
interstitial pneumonias. Simon M, Potchen EJ and LeMay M: Frontiers
of pulmonary radiology. 1st. New York: Grune and Stratton; pp.
102–141. 1969
|
|
17
|
Nemery B: Metal toxicity and the
respiratory tract. Eur Respir J. 3:202–219. 1990.PubMed/NCBI
|
|
18
|
Corrin B and Price AB: Electron
microscopic studies in desquamative interstitial pneumonia
associated with asbestos. Thorax. 27:324–331. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Freed JA, Miller A, Gordon RE, Fischbein
A, Kleinerman J and Langer AM: Desquamative interstitial pneumonia
associated with chrysotile asbestos fibres. Br J Ind Med.
48:332–337. 1991.PubMed/NCBI
|
|
20
|
Hull MJ and Abraham JL: Aluminum welding
fume-induced pneumoconiosis. Hum Pathol. 33:819–825. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakazawa A, Hagiwara E, Harada S, Yoshida
M, Baba T, Okudela K, Takemura T and Ogura T: Surgically proven
desquamative interstitial pneumonia induced by waterproofing spray.
Intern Med. 53:2107–2110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carrington CB, Gaensler EA, Coutu RE,
FitzGerald MX and Gupta RG: Natural history and treated course of
usual and desquamative interstitial pneumonia. N Engl J Med.
298:801–809. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gill A: Bong lung: Regular smokers of
cannabis show relatively distinctive histologic changes that
predispose to pneumothorax. Am J Surg Pathol. 29:980–982. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hage P and El Hajje MJ:
Nitrofurantoin-induced desquamative ınterstitial pneumonitis in a
7-year-old child. Pediatr Infect Dis J. 30:3632011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feagans J, Victor D, Moehlen M, Florman
SS, Regenstein F, Balart LA, Joshi S, Killackey MT, Slakey DP and
Paramesh AS: Interstitial pneumonitis in the transplant patient:
Consider sirolimus-associated pulmonary toxicity. J La State Med
Soc. 161:168–172. 2009.
|
|
26
|
Ishii H, Iwata A, Sakamoto N, Mizunoe S,
Mukae H and Kadota J: Desquamative interstitial pneumonia (DIP) in
a patient with rheumatoid arthritis: Is DIP associated with
autoimmune disorders? Intern Med. 48:827–830. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawabata Y, Takemura T, Hebisawa A, Ogura
T, Yamaguchi T, Kuriyama T, Nagai S, Sakatani M, Chida K, Sakai F,
et al: Eosinophilia in bronchoalveolar lavage fluid and
architectural destruction are features of desquamative interstitial
pneumonia. Histopathology. 52:194–202. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iskandar SB, McKinney LA, Shah L, Roy TM
and Byrd RP Jr: Desquamative interstitial pneumonia and hepatitis C
virus infection: A rare association. South Med J. 97:890–893. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hasegawa H, Nakamura Y, Kaida Y, Enomoto
N, Hashimoto D, Inui N, Suda T and Chida K: A case of desquamative
interstitial pneumonia associated with hepatitis C virus infection.
Nihon Kokyuki Gakka Zasshi. 47:698–703. 2009.(In Japanese).
|
|
30
|
Schroten H, Manz S, Köhler H, Wolf U,
Brockmann M and Riedel F: Fatal desquamative interstitial pneumonia
associated with proven CMV infection in an 8 month old-boy. Pediatr
Pulmonol. 25:345–347. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sung SA, Ko GJ, Kim JY, Kim MG, Lee JE,
Kim GI, Jo SK, Cho WY and Kim HK: Desquamative interstitial
pneumonia associated with concurrent cytomegalovirus and
Aspergillus pneumonia in a renal transplant recipient. Nephrol Dial
Transplant. 20:635–638. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arai T, Inoue Y, Hayashi S, Akira M,
Satoru Y, Travis WD and Sakatani M: Intractable desquamative
ınterstitial pneumonia in a tattooed man. Intern Med. 45:1055–1058.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Davies G, Wells AU and du Bois RM:
Respiratory bronchiolitis associated with interstitial lung disease
and desquamative interstitial pneumonia. Clin Chest Med. 25717–726.
(vi)2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kawabata Y, Takemura T, Hebisawa A, Sugita
Y, Ogura T, Nagai S, Sakai F, Kanauchi T and Colby TV: Desquamative
Interstitial Pneumonia Study Group: Desquamative interstitial
pneumonia may progress to lung fibrosis as characterized
radiologically. Respirology. 17:1214–1221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hidalgo A, Franquet T, Giménez A, Bordes
R, Pineda R and Madrid M: Smoking-related interstitial lung
diseases: Radiologic-pathologic correlation. Eur Radiol.
16:2463–2470. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hartman TE, Primack SL, Swensen SJ,
Hansell D, McGuinness G and Müller NL: Desquamative interstitial
pneumonia: Thin-section CT findings in 22 patients. Radiology.
187:787–790. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Akira M, Yamamoto S, Hara H, Sakatani M
and Ueda E: Serial computed tomographic evaluation in desquamative
interstitial pneumonia. Thorax. 52:333–337. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Desai SR, Ryan SM and Colby TV:
Smoking-related interstitial lung diseases: Histopathological and
imaging perspectives. Clin Radiol. 58:259–268. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mueller-Mang C, Grosse C, Schmid K,
Stiebellehner L and Bankier AA: What every radiologist should know
about idiopathic interstitial pneumonias. Radiographics.
27:595–615. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heynemann LE, Ward S, Lynch DA,
Remy-Jardin M, Johkoh T and Müller NL: Respiratory bronchiolitis,
respiratory bronchiolitis-associated interstitial lung disease, and
desquamative interstitial pneumonia: Different entities or part of
the spectrum of the same disease process? AJR Am J Roentgenol.
173:1617–1622. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Holt RM, Schmidt RA, Godwin JD and Raghu
G: High-resolution CT in respiratory bronchiolitis-associated
interstitial lung disease. J Comput Assist Tomogr. 17:46–50. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Essadki O, Chartrand-Lefebvre C, Briere J
and Grenier P: Respiratory bronchiolitis: Radiographic and CT
findings in a pathologically proven case. Eur Radiol. 8:1674–1676.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kurumagawa T, Kobayashi H, Kanoh S, Nagata
N, Aoki T, Aida S and Tamai S: Respiratory bronchiolitis-associated
interstitial lung disease. Nihon Kokyuki Gakkai Zasshi. 36:881–885.
1998.(In Japanese). PubMed/NCBI
|
|
44
|
Park JS, Brown KK, Tuder RM, Hale VA, King
Jr TE and Lynch DA: Respiratory bronchiolitis-associated
interstitial lung disease: Radiologic features with clinical and
pathologic correlation. J Comput Assist Tomogr. 26:13–20. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim DS, Collard HR and King TE Jr:
Classification and natural history of the idiopathic interstitial
pneumonias. Proc Am Thorac Soc. 3:285–292. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Akira M, Inoue G, Yamamoto S and Sakatani
M: Non-specific interstitial pneumonia: Findings on sequential CT
scans of nine patients. Thorax. 55:854–859. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Silva CI, Churg A and Müller NL:
Hypersensitivity pneumonitis: Spectrum of high-resolution CT and
pathologic findings. AJR Am J Roentgenol. 188:334–344. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Glazer CS, Rose CS and Lynch DA: Clinical
and radiologic manifestations of hypersensitivity pneumonitis. J
Thorac Imaging. 17:261–272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Silva CI, Müller NL, Lynch DA,
Curran-Everett D, Brown KK, Lee KS, Chung MP and Churg A: Chronic
hypersensitivity pneumonitis: Differentiation from idiopathic
pulmonary fibrosis and nonspecific interstitial pneumonia by using
thin-section CT. Radiology. 246:288–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bedrossian CW, Kuhn C III, Luna MA,
Conklin RH, Byrd RB and Kaplan PD: Desquamative interstitial
pneumonia-like reaction accompanying pulmonary lesion. Chest.
72:166–169. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nicholson AG: Desquamative interstitial
pneumonia. Tomashefski JF: Dail and Hammar's Pulmonary Pathology.
Berlin: Springer; pp. 710–712. 2008
|
|
52
|
Travis WD, Colby TV, Lombard CM and
Carpenter HA: A clinicopathologic study of 34 cases of diffuse
pulmonary hemorrhage with lung biopsy confirmation. Am J Surg
Pathol. 14:1112–1125. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Niewoehner DE, Kleinerman J and Rice DB:
Pathologic changes in the peripheral airways of young cigarette
smokers. N Engl J Med. 291:755–758. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Myers JL, Veal CF Jr, Shin MS and
Katzenstein AL: Respiratory bronchiolitis causing interstitial lung
disease: A clinicopathologic study of six cases. Am Rev Respir Dis.
135:880–884. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Churg A, Müller NL and Wright JL:
Respiratory bronchiolitis/interstitial lung disease. Fibrosis,
pulmonary functions, and evolving concepts. Arch Pathol Lab Med.
134:27–32. 2010.PubMed/NCBI
|
|
56
|
Fraig M, Shreesha U, Savici D and
Katzenstein AL: Respiratory bronchiolitis: A clinicopathologic
study in current smokers, ex-smokers, and never-smokers. Am J Surg
Pathol. 26:647–653. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kawabata Y, Hoshi E, Murai K, Ikeya T,
Takahashi N, Saitou Y, Kurashima K, Ubukata M, Takayanagi N, Sugita
H, et al: Smoking-related changes in the background lung of
specimens resected for lung cancer: A semiquantitative study with
correlation to postoperative course. Histopathology. 53:707–714.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tazelaar HD, Wright JL and Churg A:
Desquamative interstitial pneumonia. Histopathology. 58:509–516.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Travis WD, Matsui K, Moss J and Ferrans
VJ: Idiopathic nonspecific interstitial pneumonia: Prognostic
significance of cellular and fibrosing patterns: Survival
comparison with usual interstitial pneumonia and desquamative
interstitial pneumonia. Am J Surg Pathol. 24:19–33. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nagai S, Kitaichi M, Itoh H, Nishimura K,
Izumi T and Colby TV: Idiopathic nonspecific interstitial
pneumonia/fibrosis: Comparison with idiopathic pulmonary fibrosis
and BOOP. Eur Respir J. 12:1010–1019. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Vassallo R, Ryu JH, Colby TV, Hartman T
and Limper AH: Pulmonary Langerhans'-cell histiocytosis. N Engl J
Med. 342:1969–1978. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Katzenstein AL: Smoking-related
interstitial fibrosis (SRIF), pathogenesis and treatment of usual
interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod
Pathol. 25 Suppl 1:S68–S78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Katzenstein AL, Mukhopadhyay S, Zanardi C
and Dexter E: Clinically occult interstitial fibrosis in smokers:
Classification and significance of a surprisingly common finding in
lobectomy specimens. Hum Pathol. 41:316–325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bressieux-Degueldre S, Rotman S, Hafen G,
Aubert JD and Rochat I: Idiopathic desquamative interstitial
pneumonia in a child: A case report. BMC Res Notes. 7:3832014.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Behnia MM and Cummings OW: Desquamative
interstitial pneumonia masquerading as acute life-threatening
pulmonary embolism. Am J Crit Care. 13:199–201. 2004.PubMed/NCBI
|
|
66
|
Gould TH, Buist MD, Meredith D and Thomas
PD: Fulminant desquamative interstitial pneumonitis. Anaesth
Intensive Care. 26:677–679. 1998.PubMed/NCBI
|
|
67
|
Flusser G, Gurman G, Zirkin H, Prinslo I
and Heimer D: Desquamative interstitial pneumonitis causing acute
respiratory failure, responsive only to immunosuppressants.
Respiration. 58:324–326. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Palmucci S, Roccasalva F, Puglisi S,
Torrisi SE, Vindigni V, Mauro LA, Ettorre GC, Piccoli M and
Vancheri C: Clinical and radiological features of idiopathic
interstitial pneumonias (IIPs): A pictorial review. Insights
Imaging. 5:347–364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Caminati A, Cavazza A, Sverzellati N and
Harari S: An integrated approach in the diagnosis of
smoking-related interstitial lung diseases. Eur Respir Rev.
21:207–217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Godbert B, Wissler MP and Vignaud JM:
Desquamative interstitial pneumonia: An analytic review with an
emphasis on aetiology. Eur Respir Rev. 22:117–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hagmeyer L and Randerath W:
Smoking-related interstitial lung disease. Dtsch Arztebl Int.
112:43–50. 2015.PubMed/NCBI
|
|
72
|
Matsuo K, Tada S, Kataoka M, Okahara M,
Hiramatsu J, Horiba M, Fujimori Y, Takehara H, Okamoto M, Yamadori
I and Harada M: Spontaeous remission of desquamative interstitial
pneumonia. Intern Med. 36:728–731. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Abraham JL and Hertzberg MA: Inorganic
particles associated with desquamative interstitial pneumonia.
Chest. 80 (1 Suppl):S67–S70. 1981. View Article : Google Scholar
|
|
74
|
Bone RC, Wolfe J, Sobonya RE, Kerby GR,
Stechschulte D, Ruth WE and Welch M: Desquamative interstitial
pneumonia following long-term nitrofurantoin therapy. Am J Med.
60:697–701. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Portnoy J, Veraldi KL, Schwarz MI, Cool
CD, Curran-Everett D, Cherniack RM, King TE Jr and Brown KK:
Respiratory bronchiolitis-interstitial lung disease: Long-term
outcome. Chest. 131:664–671. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hartman TE, Primack SL, Kang EY, Swensen
SJ, Hansell DM, McGuinness G and Müller NL: Disease progression in
usual interstitial pneumonia compared with desquamative
interstitial pneumonia. Assessment with serial CT. Chest.
110:378–382. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Baloira A, Xaubet A, Rodríguez Becerra E,
Romero AD, Casanova A and Ancochea J: Desquamative interstitial
pneumonia and respiratory bronchiolitis-associated interstitial
lung disease: Data from the Spanish patient registry. Arch
Bronconeumol. 44:499–503. 2008.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Knyazhitskiy A, Masson RG, Corkey R and
Joiner J: Beneficial response to macrolide antibiotic in a patient
with desquamative interstitial pneumonia refractory to
corticosteroid therapy. Chest. 134:185–187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Park DW, Kim TH, Shon JW, Yoon HJ, Park
SS, Jeon SC, Choi YW, Paik SS and Kim H: Near fatal desquamative
interstitial pneumonia with bilateral recurrent tension
pneumothorax. Sarcoidosis Vasc Diffuse Lung Dis. 32:167–171.
2015.PubMed/NCBI
|
|
80
|
Barberis M, Harari S, Tironi A and
Lampertico P: Recurrence of primary disease in a single lung
transplant recipient. Transplant Proc. 24:2660–2662.
1992.PubMed/NCBI
|
|
81
|
Verleden GM, Sels F, Van Raemdonck D,
Verbeken EK, Lerut T and Demedts M: Possible recurrence of
desquamative interstitial pneumonitis in a single lung transplant
recipient. Eur Respir J. 11:971–974. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
King MB, Jessurun J and Hertz MI:
Recurrence of desquamative interstitial pneumonia after lung
transplantation. Am J Respir Crit Care Med. 156:2003–2005. 1997.
View Article : Google Scholar : PubMed/NCBI
|