Introduction
Metabolic syndrome is a cluster of risk factors,
including obesity, type 2 diabetes, dyslipidemia, hypertension and
cardiovascular disease (1). Dietary
intervention is a potential strategy to prevent and treat metabolic
syndrome. It has been revealed that fiber-enriched diets improve
obesity, insulin sensitivity and glucose tolerance, which is
attributed to the production of short-chain fatty acid (SCFA)
mediated by the gut microbiota (2–5). The
most abundant SCFAs in the gut include acetate, propionate and
butyrate (6). Although the major
source of butyrate is the fermentation of dietary fibers, it is
also contained in butter and cheese (7). Butyrate supplementation has been
demonstrated to ameliorate diet-induced obesity, dyslipidemia,
insulin resistance and glucose intolerance in mice (8). However, the mechanisms underlying the
beneficial effect of butyrate has remained largely elusive.
It is well accepted that butyrate acts not only as a
signaling molecule for the G-protein-coupled-receptor 41 (GPR41)
and GPR43, but also as a wide-spectrum histone deacetylase (HDAC)
inhibitor (9,10). Histone deacetylases regulate gene
transcription by deacetylation of proteins, including histone
proteins and transcription factors. In recent years, HDAC has
emerged as a novel molecular target in the treatment of type 2
diabetes. It has been demonstrated that HDACs are crucial
regulators of pancreatic cell fate determination (11). Butyrate treatment was reported to
improve β-cell proliferation, function and glucose homeostasis as
well as to reduce β-cell apoptosis through HDAC inhibition and
histone acetylation in diabetic rats (12). Li et al (13) observed that sodium
butyrate-stimulated fibroblast growth factor 21 expression and
enhanced fatty acid oxidation in the liver by inhibition of HDAC3.
In addition, sodium butyrate has been demonstrated to improve
systemic insulin sensitivity and increase energy expenditure in
mice via upregulating mitochondrial function in skeletal muscle and
brown fat through peroxisome proliferator-activated receptor γ
coactivator 1α (PGC-1α) induction and elevation of adenosine
monophosphate-activated protein kinase activity (8). However, the direct effect of butyrate
on hepatic gluconeogenesis has remained to be elucidated.
Gluconeogenesis is an important pathological
contributing factor in diabetic subjects and abnormally high during
the progression of diabetes (14).
In the liver, class IIa HDACs mediate glucogan-induced
gluconeogenic gene expression through promoting deacetylation and
activation of forkhead box O (FoxO) transcription factors. Hepatic
knockdown of Class IIa HDACs in vivo resulted in lowered
blood glucose in mice (15).
Therefore, the present study investigated the direct effect of
butyrate on gluconeogenesis in isolated mouse primary hepatocytes.
Unexpectedly, butyrate significantly increased hepatic
gluconeogenesis and the expression of gluconeogenic genes, which
was different from the actions of other HDAC inhibitors.
Materials and methods
Materials
Hepatocyte medium was purchased from ScienCell
(Carlsbad, CA, USA). Dulbecco's modified Eagle's medium (DMEM),
Hank's balanced salt solution (HBSS), PBS and collagenase type IV
(200 units/mg) were from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Sodium pyruvate, sodium L-lactate,
dexamethasone, bovine serum albumin (BSA), 8-bromo-cyclic adenosine
monophosphate (8-bromo-cAMP), acetate, propionate, sodium butyrate,
Trichostatin A (TSA), CI994, Entinostat (MS-275), PCI-34051 and
tubacin were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). All primers used for reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) were synthesized by Shanghai
Biological Engineering Technology & Services Co., Ltd.
(Shanghai, China). Anti-cAMP response element-binding protein
(CREB; cat no. 9197S) and anti-phosphorylated (p)-CREB (Ser133; cat
no. 9198) as well as anti-mouse immunoglobulin (Ig)G (cat no.
14709) and anti-rabbit IgG conjugated with horseradish peroxidase
(cat no. 7074) were from Cell Signaling Technology Inc. (Beverly,
MA, USA). The antibody to GAPDH (cat no. sc25778) was purchased
from Santa Cruz Biotechnology, Inc. (Danvers, MA, USA).
Isolation and culture of mouse primary
hepatocytes
A total of 20 Male C57BL/6 mice (age, 6–8 weeks;
weight, 16–18 g) were purchased from Shanghai Slack Experimental
Center (Shanghai, China). Primary hepatocytes were isolated from
C57BL/6 mice by a modified version of the collagenase method. In a
biosafety cabinet, mouse livers were perfused with 10 ml
calcium-free HBSS through the portal vein under anesthesia with 10%
chloral hydrate (Sigma-Aldrich; Merck, KGaA). The liver was excised
by careful dissection and transferred to a 10-cm tissue culture
dish. Mice were then sacrificed via cervical dislocation. The
isolated liver was perfused with 0.05% collagenase IV (Gibco;
Thermo Fisher Scientific, Inc.) dissolved in 20 ml
calcium-containing HBSS in a recirculating manner for 15 min.
Hepatocytes extracted from digested livers were filtered through a
100-µm cell strainer, washed 3 times with PBS and re-suspended in
hepatocyte medium containing 100 U/ml penicillin, 100 µg/ml
streptomycin, 0.1% BSA and hepatocyte growth factor (all from
ScienCell Research Laboratories, Inc., San Diego, CA, USA). The
cells were seeded on 6-well or 24-well plates and incubated in a
tissue culture incubator at 37°C (95% air and 5% CO2).
After 24 h, the cells had already attached to the plates and the
medium was replaced with DMEM containing 5.5 mM glucose and 0.25%
BSA, followed by drug treatment. All mice used in the present study
for isolation of hepatocytes were fed a normal diet under a regular
schedule and were not fasted. All of the experimental procedures
involving the use of animals were approved by the Animal Use and
Care Committee of Shanghai Jiaotong University School of Medicine
(Shanghai, China).
Cell culture and treatments
Mouse primary hepatocytes were cultured in DMEM
supplemented with 0.25% BSA overnight at 37°C with 5%
CO2. To evaluate the effect of butyrate on hepatic
gluconeogenesis, mouse primary hepatocytes were incubated with
sodium butyrate for 8 h at various concentrations (0, 0.1, 1, 5, or
10 mmol/l). To investigate the effect of HDAC inhibitors on hepatic
gluconeogenesis, cells were incubated in glucose-free DMEM
containing gluconeogenic substrates (10 mmol/l sodium lactate and 1
mmol/l sodium pyruvate), 8-bromo-cAMP (100 µmol/l) and with the
following HDAC inhibitors: TSA (100 nmol/l), CI994 (10 µmol/l),
MS-275 (10 µmol/l), PCI-34051 (10 µmol/l), Tubacin (10 µmol/l) or
sodium butyrate (5 mmol/l). Following 8 h incubation at 37°C, cell
culture supernatants were collected for measuring the glucose
content. Mouse primary hepatocytes were then treated with sodium
butyrate (5 mmol/l) and TSA (100 nmol/l) in the presence of
8-bromo-cAMP (100 µmol/l) for 8 h at 37°C, the mRNA expression of
gluconeogenic genes were detected using RT-qPCR. To compare the
effects of three SCFAs on hepatic glucose production, mouse
hepatocytes were treated with acetate (5 mmol/l), propionate (5
mmol/l) or sodium butyrate (5 mmol/l) for 8 h at 37°C in the
presence of gluconeogenic substrates (10 mmol/l sodium lactate and
1 mmol/l sodium pyruvate). To test whether SCFAs affect hepatic
gluconeogenesis as substrates, mouse hepatocytes were incubated
with one of three different SCFAs (acetate, propionate, or sodium
butyrate; 5 mmol/l each) and 100 µM 8-bromo-cAMP in the absence of
gluconeogenic substrates (10 mmol/l sodium lactate and 1 mmol/l
sodium pyruvate) for 8 h at 37°C. To assess the phosphorylation
levels of CREB, mouse hepatocytes were treated with butyrate (5
mmol/l) or 8-bromo-cAMP (100 µmol/l) for 1 h at 37°C.
In vitro glucose production assay
Glucose production was assayed as described
previously (16). In brief,
hepatocytes were seeded into 24-well plates at 2.5×105
cells/well. After 24 h, these hepatocytes were pre-stimulated with
DMEM containing 5.5 mM glucose, 0.25% BSA and 100 nM dexamethasone
for 16 h. Cells were then washed three times with PBS and incubated
in glucose production buffer (DMEM without glucose, serum or phenol
red, and supplemented with 1 mM sodium pyruvate and 10 mM sodium
lactate). After 8 h, the cell culture supernatants were collected
for measuring the glucose content using a glucose oxidase kit
(Applygen Co., Beijing, China).
RNA extraction and RT-qPCR
Total RNA was extracted from mouse primary
hepatocytes using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Complementary DNA was synthesized by RT using Random Primers
(Promega Corp., Madison, WI, USA) according to the manufacturer's
protocol. RT-qPCR was performed in a Roche LightCycler 480 system
(Roche Diagnostics, Basel, Switzerland) using SYBR Premix EX Taq
(Takara, Tokyo, Japan). The PCR conditions were as follows:
Denaturation at 95°C for 10 sec, followed by 40 cycles of 95°C for
5 sec. The annealing temperature was then reduced to 60°C for 31
sec with a final elongation step at 72°C for 300 sec. At the end of
the amplification, a melting curve was generated in the temperature
range of 60–95°C. The sequences of primers used are listed in
Table I. Relative gene expression
levels were quantified based on the cycle threshold (Cq) values and
normalized to the reference gene β-actin. The relative gene
expression was calculated using the 2−ΔΔCq method
(17).
 | Table I.Sequences of primers used for
polymerase chain reaction. |
Table I.
Sequences of primers used for
polymerase chain reaction.
| Gene | Sequence no. | Primer sequence
(5′-3′) | Product length
(bp) |
|---|
| PEPCK | NM_011044.2 | F,
GTGCTGGAGTGGATGTTCGG | 258 |
|
|
| R,
CTGGCTGATTCTCTGTTTCAGG |
|
| G6pase | NM_008061.4 | F,
ACTGTGGGCATCAATCTCCTC | 344 |
|
|
| R,
CGGGACAGACAGACGTTCAGC |
|
| FoxO1 | NM_019739.3 | F,
AAGAGCGTGCCCTACTTCAA | 157 |
|
|
| R,
CTCCCTCTGGATTGAGCATC |
|
| PGC-1α | NM_008904.2 | F,
ATACCGCAAAGAGCACGAGAAG | 253 |
|
|
| R,
CTCAAGAGCAGCGAAAGCGTCACAG |
|
| HNF4α | NM_008261.3 | F,
ATGCGACTCTCTAAAACCCTTG | 135 |
|
|
| R,
ACCTTCAGATGGGGACGTGT |
|
| β-actin | NM_007393.5 | F,
GGCTGTATTCCCCTCCATCG | 154 |
|
|
| R,
CCAGTTGGTAACAATGCCATGT |
|
Western blot analysis
Primary hepatocytes were treated with
radioimmunoprecipitation lysis buffer containing protease and
phosphatase inhibitors (Merck KGaA) and centrifuged at a speed of
6,000 × g for 10 min at 4°C. The supernatant was then used to
analyze the expression levels of specific proteins. Protein
concentration was determined using a BCA protein assay kit (Thermo
Fisher Scientific, Inc.). A total of 10 µg protein was loaded per
lane and separated in 15% SDS-PAGE. Samples were then transferred
onto polyvinylidene fluoride membranes (ECL Advance; Cell Signaling
Technology, Inc). Prior to western blot analysis, membranes were
blocked with 5% skimmed milk for 2 h at 37°C and subsequently
incubated at 4°C overnight with rabbit anti-mouse primary
antibodies sourced from Cell Signaling Technology Inc. These
included cAMP response element-binding protein (CREB; cat. no.
9197S; 1:1,000) and phosphorylated (p)-CREB (Ser133; cat. no. 9198;
1:1,000). GAPDH antibodies (cat. no. sc25778; 1:1,000) were
purchased from Santa Cruz Biotechnology, Inc. Membranes were then
incubated with mouse anti-rabbit horseradish peroxidase-labeled IgG
secondary antibodies (1:2,000; cat. no. 7074; Cell Signaling
Technology Inc.,) overnight at 4°C. The results were visualized
using an immobilon ECL ultra western HRP substrate (cat. no.
WBULS0100; Merck KGaA) and images were captured using an LAS-4000
Super CCD Remote Control Science Imaging System (Fuji, Tokyo,
Japan).
Adenosine triphosphate (ATP)
assay
The amount of ATP was measured by the
luciferin-luciferase method according to the protocol of the ATP
detection kit (cat. no. S0026; Beyotime Institute of Biotechnology,
Inc., Haimen, China). Primary hepatocytes were seeded onto 24-well
plates at 2.5×105 cells/well. After 24 h, these cells
received the same dexamethasone pre-stimulation as described in the
glucose production assay. The cells were then treated with
propionate (5 mmol/l) and different concentrations of sodium
butyrate (0, 0.1, 1, 5 and 10 mmol/l) for 1 h prior to lysis with
lysis buffer (200 µl/well) from the ATP detection kit. After
centrifugation at 12,000 × g for 5 min at 4°C, the supernatant was
transferred to a fresh tube for the ATP test. The luminescence of a
20-µl sample was assayed in a luminometer (Perkin Elmer, Inc.,
Waltham, MA, USA) together with 100 µl ATP detection buffer from
the ATP detection kit. The protein concentration was determined
using a bicinchoninic acid protein assay kit (Thermo Fisher
Scientific, Inc.). The concentration of ATP was normalized to that
of protein in the same cell lysate.
Statistical analysis
All values are expressed as the mean ± standard
error of the mean from at least three independent experiments. Two
group comparisons were performed using a Student's t-test. All
statistical analyses were performed using SPSS 19.0 (IBM Corp.,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
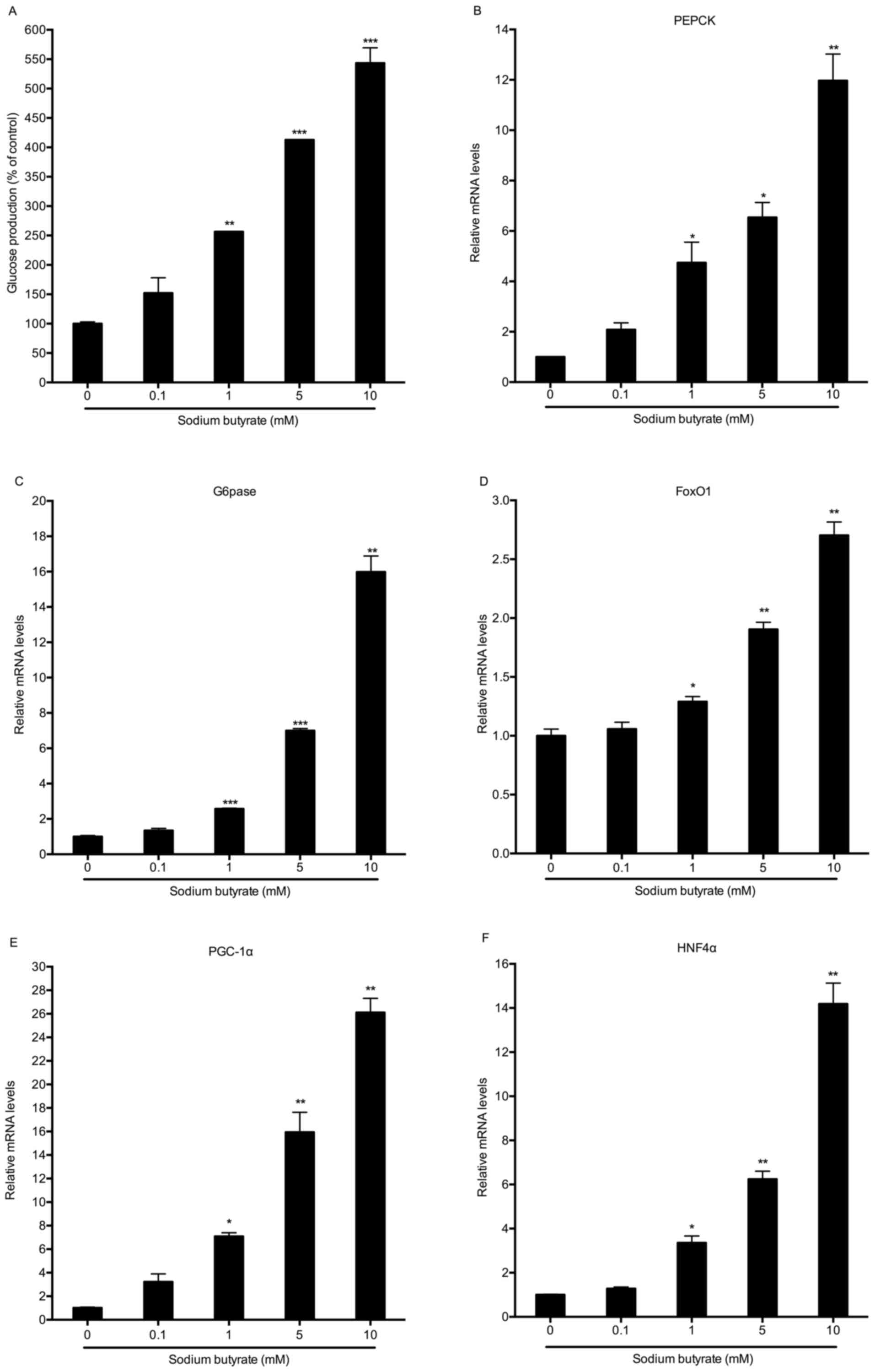
Sodium butyrate stimulates hepatic
glucose production and gluconeogenic gene expression
To determine the effects of butyrate on hepatic
gluconeogenesis, mouse primary hepatocytes were incubated with
various concentrations of sodium butyrate for 8 h. Unexpectedly,
sodium butyrate stimulated hepatic glucose production in a
dose-dependent manner, causing a significant increase at the
concentration of 1 mM (P<0.01; Fig.
1A). Under the same conditions, the expression of genes
associated with hepatic gluconeogenesis, including
phosphoenolpyruvate carboxykinase (PEPCK),
glucose-6-phosphatase (G6Pase), FoxO1, PGC-1α and hepatocyte
nuclear factor 4α (HNF4α) was also increased. Consistent with the
results on gluconeogenesis, sodium butyrate dose-dependently
increased the mRNA expression of these gluconeogenic genes, with
the maximum efficacy at the concentration of 10 mM (Fig. 1B-F).
Effects of various HDAC inhibitors on
hepatic gluconeogenesis
To investigate whether sodium butyrate-stimulated
gluconeogenesis is involved in the inhibition of HDACs, the effects
of other HDAC inhibitors, including TSA, CI994, MS-275, PCI-34051
and tubacin, on hepatic glucose production in mouse primary
hepatocytes were detected. After mouse hepatocytes were incubated
with 100 µM 8-bromo-cAMP for 8 h, glucose production was markedly
increased. In the presence of 100 nM TSA, 10 µM CI994 or 10 µM
PCI-34051, 8-bromo-cAMP-stimulated gluconeogenesis was
significantly decreased. However, sodium butyrate treatment further
enhanced glucose production induced by 8-bromo-cAMP. The other two
HDAC inhibitors had no significant effect (Fig. 2A). TSA is a potent inhibitor of class
I and II HDAC (18). The present
study further compared the effects of TSA and sodium butyrate on
the expression of the five gluconeogenic genes. As expected, the
expression of all of these gluconeogenic genes was strongly induced
by 8-bromo-cAMP. Contrary to the action of sodium butyrate, TSA
suppressed cAMP-stimulated gluconeogenic gene expression (Fig. 2B-F). It is therefore unlikely that
sodium butyrate promotes gluconeogenesis via inhibiting HDAC
activity.
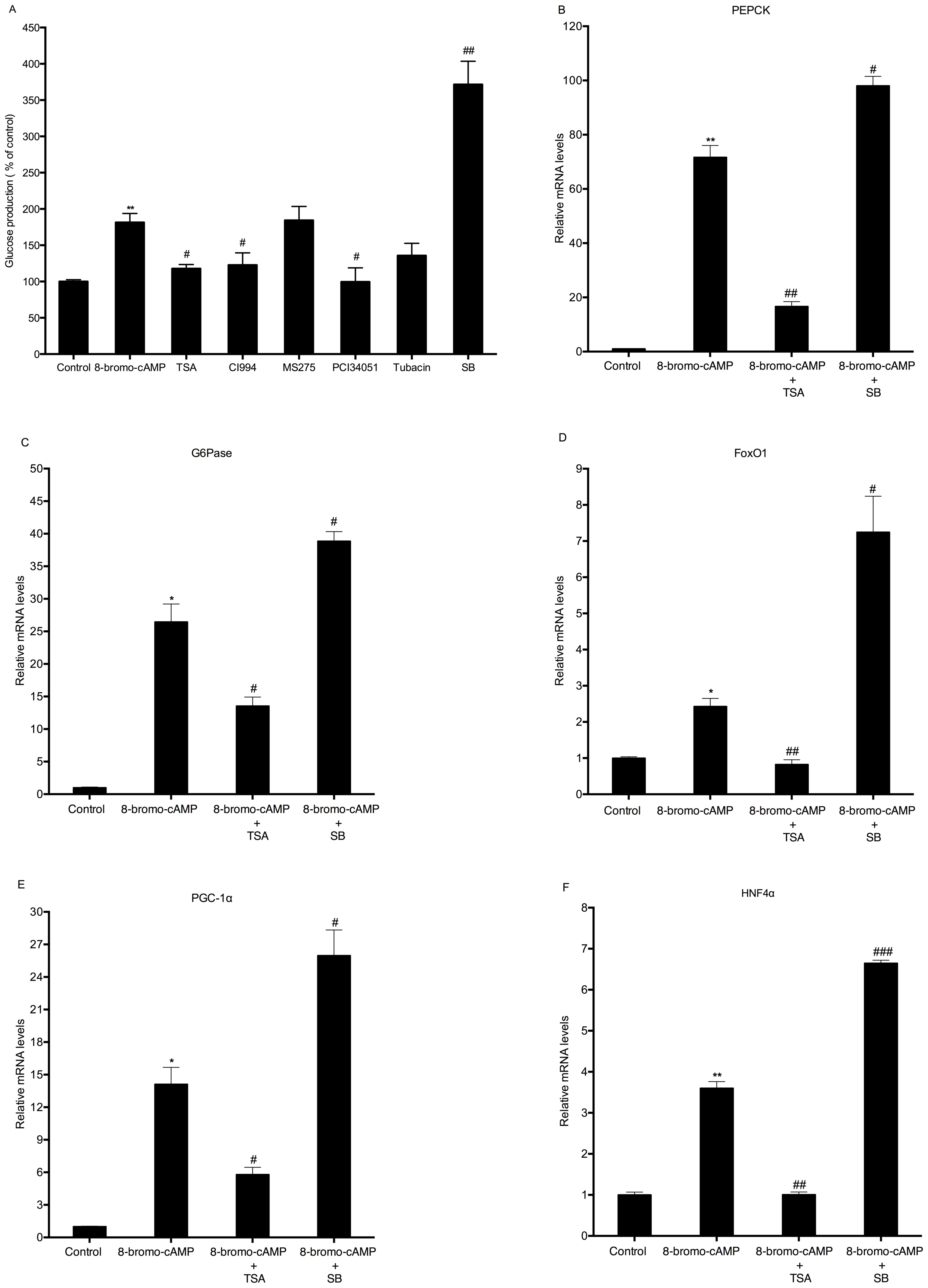 | Figure 2.Effect of various HDAC inhibitors on
hepatic glucose production. (A) Mouse hepatocytes were incubated
with different HDAC inhibitors (100 nM TSA, 10 µM CI994, 10 µM
MS-275, 10 µM PCI-34051, 10 µM Tubacin or 5 mM SB) in glucose-free
Dulbecco's modified Eagle's medium containing gluconeogenic
substrates (10 mM sodium lactate and 1 mM sodium pyruvate) and 100
µM 8-bromo-cAMP for 8 h. The cell culture supernatants were
collected for measuring the glucose content. (B-F) After mouse
hepatocytes were treated with 5 mM sodium butyrate and 100 nM TSA
in the presence of 100 µM 8-bromo-cAMP for 8 h, the mRNA
expressions of gluconeogenic genes were detected by
reverse-transcription quantitative polymerase chain reaction
analysis. Values are expressed as the mean ± standard error of the
mean of three separate experiments. *P<0.05, **P<0.01
compared with control group; #P<0.05,
##P<0.01, ###P<0.001 compared with
8-bromo-cAMP group. 8-bromo-cAMP, 8-bromo-cyclic adenosine
monophosphate; TSA, Trichostatin A; MS-275, Entinostat; HDAC,
histone deacetylase; SB, sodium butyrate; FOX, forkhead box; PEPCK,
phosphoenolpyruvate carboxykinase; G6pase, glucose 6-phosphatase;
PGC1α, peroxisome proliferator-activated receptor γ coactivator 1α;
HNF4α, hepatocyte nuclear factor 4α. |
Effects of various SCFAs on hepatic
glucose production and gluconeogenic gene expression
To investigate whether sodium butyrate stimulates
gluconeogenesis as an energy substrate, the present study compared
the effects of three SCFAs on hepatic glucose production. Mouse
hepatocytes were treated with 5 mM acetate, propionate or sodium
butyrate for 8 h in the presence of gluconeogenic substrates
(sodium lactate and sodium pyruvate). Similar to sodium butyrate,
propionate also promoted hepatic glucose production (Fig. 3A). Sodium butyrate supplementation
led to increases in the expression of all of the selected
gluconeogenic genes. Propionate significantly triggered the mRNA
expression of PEPCK, G6Pase, FoxO1 and PGC-1α, but not that of
HNF4α (Fig. 3B-F). Although acetate
had no significant effects on glucose production, it significantly
increased the expression of FoxO1 and PGC-1α (Fig. 3A, D and E).
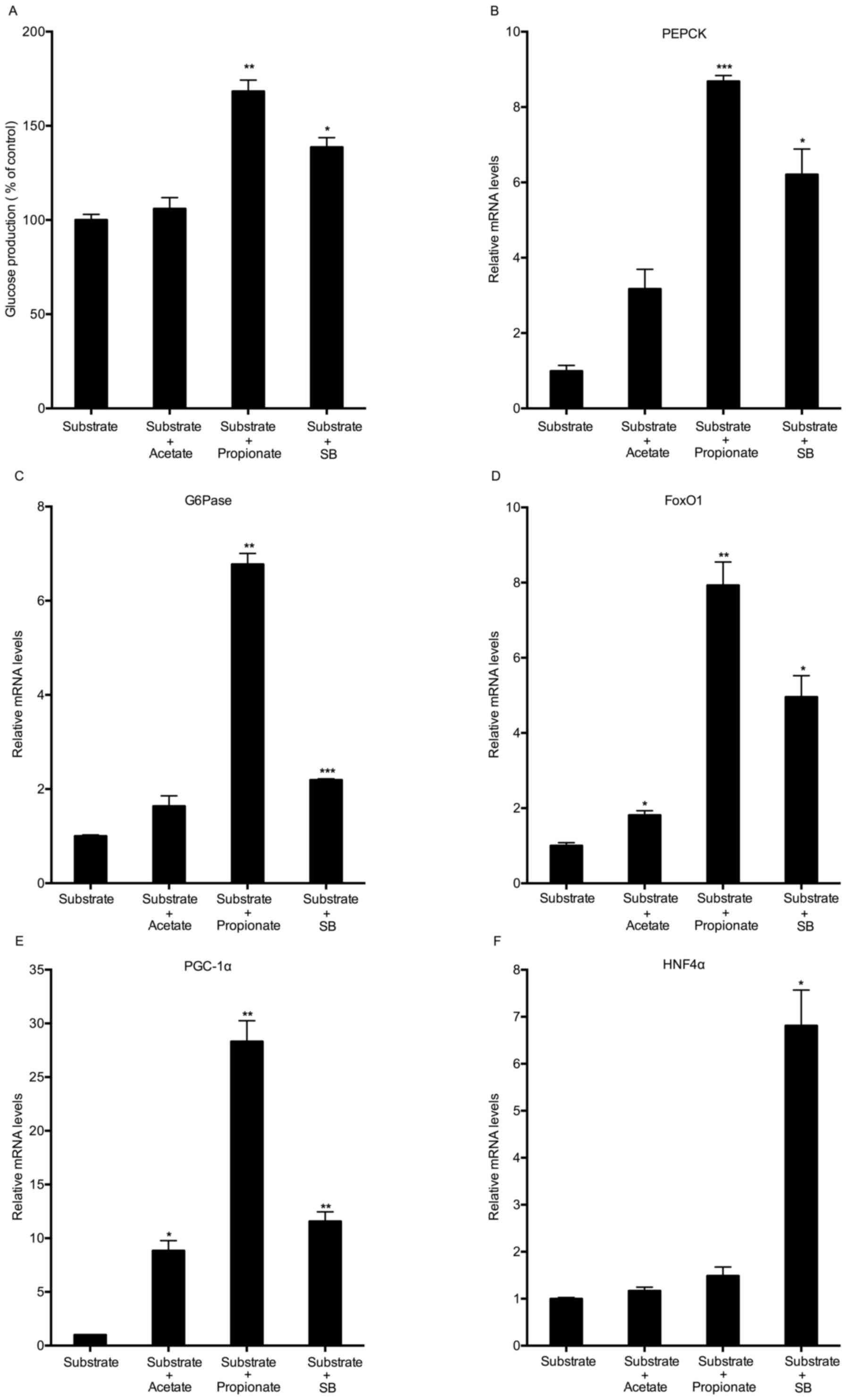 | Figure 3.Effect of various short-chain fatty
acids on hepatic glucose production and gluconeogenic gene
expression. (A) Mouse hepatocytes were incubated with 5 mM acetate,
propionate or SB in glucose-free Dulbecco's modified Eagle's medium
containing gluconeogenic substrates (10 mM sodium lactate and 1 mM
sodium pyruvate) for 8 h. The cell culture supernatants were
collected for measuring glucose content. (B-F) Under the same
conditions, the mRNA expression of gluconeogenic genes was detected
by reverse-transcription quantitative polymerase chain reaction
analysis. Values are expressed as the mean ± standard error of the
mean of three separate experiments. *P<0.05, **P<0.01,
***P<0.001 compared with substrate group. SB, sodium butyrate;
FOX, forkhead box; PEPCK, phosphoenolpyruvate carboxykinase;
G6pase, glucose 6-phosphatase; PGC1α, peroxisome
proliferator-activated receptor γ coactivator 1α; HNF4α, hepatocyte
nuclear factor 4α. |
Sodium butyrate stimulates hepatic
glucose production as a substrate
To test whether sodium butyrate induces glucose
production as a type of gluconeogenic substrate, mouse primary
hepatocytes were incubated with one of three SCFAs in the absence
of gluconeogenic substrates. Acetate, propionate and sodium
butyrate all increased hepatic gluconeogenesis. Propionate had the
most noticeable effect. In the presence of three SCFAs, addition of
8-bromo-cAMP further enhanced the glucose production (Fig. 4A). In agreement with the results on
gluconeogenesis, 8-bromo-cAMP significantly enhanced butyrate and
propionate-stimulated expression of the five gluconeogenic genes
(Fig. 4B-F). Acetate supplementation
only led to mild increases in PEPCK and PGC-1α expression (Fig. 4B and E). It is well accepted that
propionate is a substrate for hepatic glucose production (19). Therefore, it is reasonable to presume
that butyrate also stimulates hepatic gluconeogenesis as a
substrate.
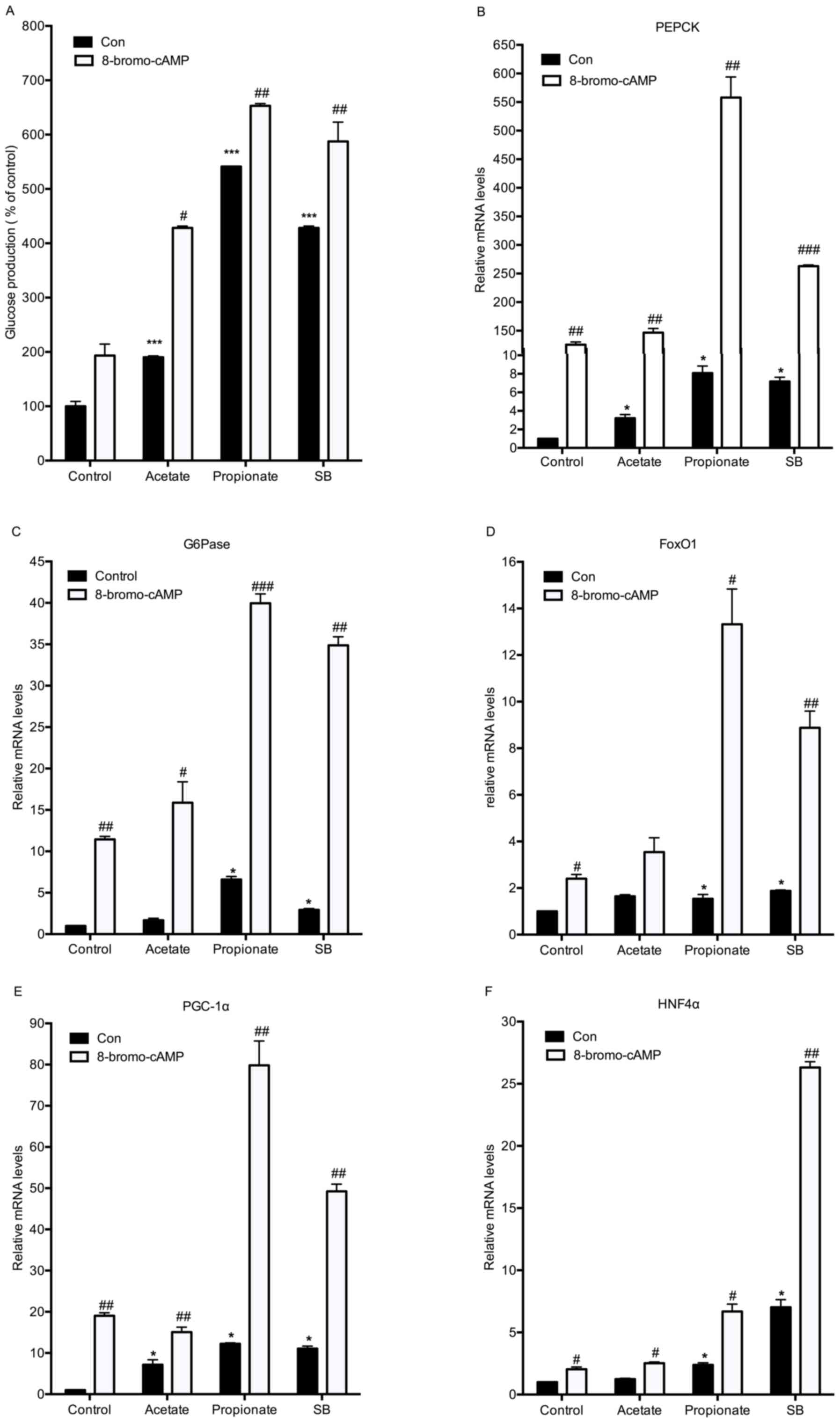 | Figure 4.Sodium butyrate stimulates hepatic
glucose production as a substrate. (A) Mouse hepatocytes were
incubated with different short-chain fatty acids (5 mM) and 100 µM
8-bromo-cAMP in the absence of gluconeogenic substrates (sodium
lactate and sodium pyruvate) for 8 h. The cell culture supernatants
were collected for measuring the glucose content. (B-F) Under the
same conditions, the mRNA expression of gluconeogenic genes was
detected by reverse-transcription quantitative polymerase chain
reaction analysis. Values are expressed as the mean ± standard
error of the mean of three separate experiments. *P<0.05,
***P<0.001 compared with control group; #P<0.05,
##P<0.01, ###P<0.001 compared with CON
group (without 8-bromo-cAMP). CON, control; 8-bromo-cAMP,
8-bromo-cyclic adenosine monophosphate; SB, sodium butyrate; FOX,
forkhead box; PEPCK, phosphoenolpyruvate carboxykinase; G6pase,
glucose 6-phosphatase; PGC1α, peroxisome proliferator-activated
receptor γ coactivator 1α; HNF4α, hepatocyte nuclear factor 4α. |
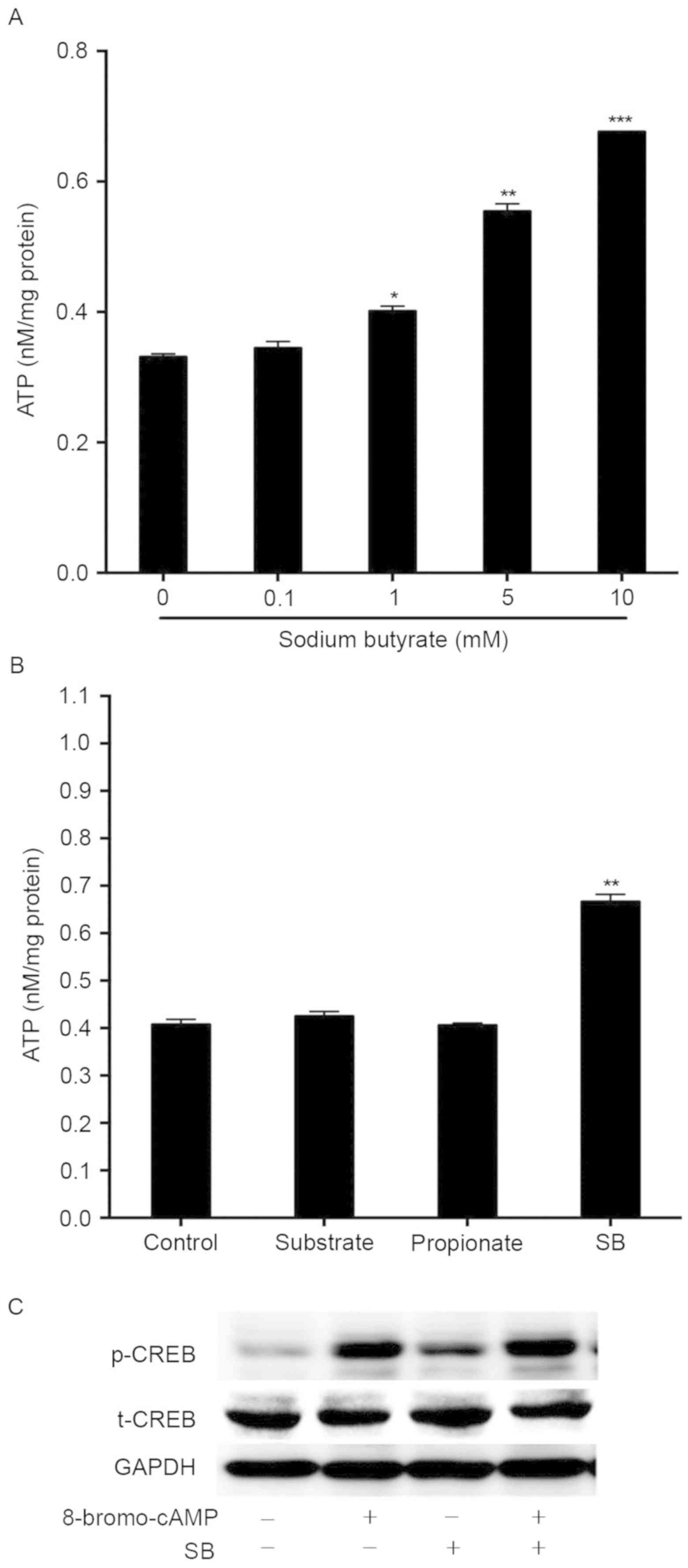
Sodium butyrate activates CREB in
mouse hepatocytes
It is well-known that cAMP/CREB is an important
signaling pathway for hepatic gluconeogenesis (20,21). The
present study investigated whether the cAMP/CREB signaling pathway
is involved in butyrate-mediated induction of gluconeogenesis. As
ATP is the substrate for cAMP production, the present study first
detected the intracellular ATP concentration after mouse primary
hepatocytes were incubated with various concentrations of sodium
butyrate for 1 h. In parallel with glucose production, sodium
butyrate treatment resulted in the accumulation of intracellular
ATP in a dose-dependent manner, exhibiting a significant effect at
the concentration of 1 mM (P<0.05; Fig. 5A). However, gluconeogenic substrates
(sodium lactate and sodium pyruvate) as well as propionate did not
alter the intracellular ATP concentration (Fig. 5B). Next, western blot analysis was
performed to evaluate the phosphorylation state of CREB in
butyrate-treated hepatocytes. Similar to 8-bromo-cAMP, sodium
butyrate also stimulated the phosphorylation of CREB (Fig. 5C). Overall, these results indicated
that butyrate activates cAMP/CREB signaling and stimulates the
expression of hepatic gluconeogenic genes, at least in part via
increasing the accumulation of intracellular ATP.
Discussion
The liver is the major site of gluconeogenesis from
red blood cell-derived pyruvate and lactate and from amino acid
precursors. Hepatic metabolism has a key role in the regulation of
the energy status of the whole body. Increased glucose production
through abnormally elevated hepatic gluconeogenesis is central to
the manifestation of hyperglycaemia in type 2 diabetes (22). The present study demonstrated that
butyrate promoted gluconeogenesis in mouse primary hepatocytes as a
substrate and signaling molecule, which appears paradoxical to its
beneficial effect on the whole-body energy metabolism in rodent
animals. To the best of our knowledge, there is little evidence
demonstrating that butyrate directly improves blood glucose levels
in human studies.
Gluconeogenesis is tightly controlled through the
transcriptional regulation of PEPCK and G6Pase. Numerous
transcription factors and co-activators, such as FoxO1, PGC-1α and
HNF4α, are involved in the induction of the two hepatic
gluconeogenic genes (23). Class IIa
HDAC, together with HDAC3, was reported to regulate hepatic
gluconeogenesis via deacetylation of FoxO1 (15). Butyrate is a well-characterized HDAC
inhibitor. Similar to TSA, butyrate was reported to inhibit HDAC
enzymes and increase the overall level of histone H3 acetylation
(24). A previous study demonstrated
that butyrate functioned as an HDAC inhibitor to stimulate the
proliferation of colonocytes through changing their glucose
metabolism (25). Therefore, the
present study first investigated whether butyrate affected hepatic
glucose production by acting as an HDAC inhibitor. Among six HDAC
inhibitors, only sodium butyrate increased gluconeogenesis and TSA
exerted the opposite effect. TSA is typically considered a
broad-spectrum HDAC inhibitor. The present study further detected
the expression of gluconeogenic genes in isolated mouse primary
hepatocytes treated with sodium butyrate and TSA. In consistency
with the results on gluconeogenesis, sodium butyrate enhanced
8-bromo-cAMP-stimulated hepatic gluconeogenic gene expression,
while TSA had the opposite effect. These results indicated that
butyrate stimulates gluconeogenesis and upregulates gluconeogenic
gene expression independent of HDAC.
Besides being a broad-spectrum HDAC inhibitor,
butyrate is an SCFA (6). In the
present study, the effects of three SCFAs on hepatic glucose
production and gluconeogenic gene expression were compared at the
same concentration. Similar to sodium butyrate, propionate also
stimulated gluconeogenesis and the expression of associated genes
in mouse primary hepatocytes. Propionate has long been described as
a hepatic gluconeogenic substrate (26). Infused propionate as a tracer was
reported to markedly increase hepatic tricarboxylic acid metabolism
and drive hepatic glucose production in a dose-dependent manner
(19). Dietary supplementation with
SCFAs, including propionate and butyrate, induced intestinal
glucose production. Butyrate directly stimulated PEPCK and G6Pase
expression in enterocytes in vitro (6). Therefore, it is likely that butyrate,
as propionate, is also a gluconeogenic substrate. As expected, the
present study demonstrated that sodium butyrate alone induced
hepatic glucose production and gluconeogenic gene expression in
mouse hepatocytes cultured with media void of other gluconeogenic
substrates (sodium lactate and pyruvate).
cAMP has a pivotal role in the signaling pathways of
hepatic gluconeogenesis (20). Since
intracellular ATP is the substrate of cAMP production, the present
study detected the intracellular ATP content in mouse hepatocytes
incubated with butyrate and propionate. In agreement with the
results of a previous study (27),
propionate had no effect on the ATP concentration in the present
study. However, butyrate dose-dependently induced the accumulation
of intracellular ATP, demonstrating a similar increase to that
during gluconeogenesis. Another mechanism underlying
butyrate-stimulated gluconeogenesis except as a substrate has been
reported; butyrate increased cAMP levels and protein kinase A
activity in T-cells (28) and
regulated the proliferation of porcine peripheral blood mononuclear
cells in a cAMP-dependent manner (29). The present study revealed that
butyrate stimulated the phosphorylation of CREB in mouse primary
hepatocytes. These results suggested that butyrate stimulates
gluconeogenesis via activating cAMP/CREB signaling.
In conclusion, the present study demonstrated that
butyrate has a dual role in stimulating hepatic glucose production
as a substrate and signaling molecule. cAMP/CREB signaling is
involved in butyrate-induced hepatic gluconeogenic gene expression.
Apparently, the beneficial effects of butyrate on other tissues
counterbalances its action on hepatic gluconeogenesis, eventually
improving the whole-body energy metabolism in rodent animals.
However, additional studies should be performed before sodium
butyrate is used in the treatment of type 2 diabetes mellitus in
the clinic.
Acknowledgements
Not applicable.
Funding
This work was funded by grants from the National
Natural Science Foundation of China (grant nos. 81270910, 81370876,
81471030, 81570693 and 30600294), the Natural Science Foundation of
Shanghai (grant no. 15ZR1434000), and the Academic Leaders Training
Program of Pudong Health Bureau of Shanghai (grant no.
PWRd2011-01).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XJ and LZ designed the study. XJ and FZ conducted
the experiments. YZ and WX analysed the data. XJ wrote the
manuscript. LZ had primary responsibility for final content. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were reviewed and approved by
the Animal Use and Care Committee of Shanghai Jiao Tong University
(Shanghai, China) and the China Experimental Animal Protection
Association. All efforts were made to minimize the suffering of the
experimental mice.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ning G: Decade in review-type 2 diabetes
mellitus: At the centre of things. Nat Rev Endocrinol. 11:636–638.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ley RE, Turnbaugh PJ, Klein S and Gordon
JI: Microbial ecology: Human gut microbes associated with obesity.
Nature. 444:1022–1023. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Papathanasopoulos A and Camilleri M:
Dietary fiber supplements: Effects in obesity and metabolic
syndrome and relationship to gastrointestinal functions.
Gastroenterology. 138:65–72.e1-e2. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Robertson MD, Currie JM, Morgan LM, Jewell
DP and Frayn KN: Prior short-term consumption of resistant starch
enhances postprandial insulin sensitivity in healthy subjects.
Diabetologia. 46:659–665. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Robertson MD, Bickerton AS, Dennis AL,
Vidal H and Frayn KN: Insulin-sensitizing effects of dietary
resistant starch and effects on skeletal muscle and adipose tissue
metabolism. Am J Clin Nutr. 82:559–567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Vadder F, Kovatcheva-Datchary P,
Goncalves D, Vinera J, Zitoun C, Duchampt A, Bäckhed F and Mithieux
G: Microbiota-generated metabolites promote metabolic benefits via
gut-brain neural circuits. Cell. 156:84–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Robertson MD, Currie JM, Morgan LM, Jewell
DP and Frayn KN: Prior short-term consumption of resistant starch
enhances postprandial insulin sensitivity in healthy subjects.
Diabetologia. 46:659–665. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ,
Lefevre M, Cefalu WT and Ye J: Butyrate improves insulin
sensitivity and increases energy expenditure in mice. Diabetes.
58:1509–1517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin HV, Frassetto A, Kowalik EJ Jr,
Nawrocki AR, Lu MM, Kosinski JR, Hubert JA, Szeto D, Yao X, Forrest
G and Marsh DJ: Butyrate and propionate protectagainst diet-induced
obesity and regulate gut hormones via free fatty acid receptor
3-independent mechanisms. PLoS One. 7:e352402012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ellis L, Hammers H and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors. Cancer
Lett. 280:145–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Haumaitre C, Lenoir O and Scharfmann R:
Histone deacetylase inhibitors modify pancreatic cell fate
determination and amplify endocrine progenitors. Mol Cell Biol.
28:6373–6383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khan S and Jena GB: Protective role of
sodium butyrate, a HDAC inhibitor on beta-cell proliferation,
function and glucose homeostasis through modulation of p38/ERK MAPK
and apoptotic pathways: Study in juvenile diabetic rat. Chem Biol
Interact. 213:1–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H, Gao Z, Zhang J, Ye X, Xu A, Ye J and
Jia W: Sodium butyrate stimulates expression of fibroblast growth
factor 21 in liver by inhibition of histone deacetylase 3.
Diabetes. 61:797–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hundal RS, Krssak M, Dufour S, Laurent D,
Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF,
Landau BR and Shulman GI: Mechanism by which metformin reduces
glucose production in type 2 diabetes. Diabetes. 49:2063–2069.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mihaylova MM, Vasquez DS, Ravnskjaer K,
Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M and
Shaw RJ: Class IIa histone deacetylases are hormone-activated
regulators of FOXO and mammalian glucose homeostasis. Cell.
145:607–621. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ip W, Shao W, Chiang YT and Jin T: The Wnt
signaling pathway effector TCF7L2 is upregulated by insulin and
represses hepatic gluconeogenesis. Am J Physiol Endocrinol Metab.
303:E1166–E1176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taddei A, Roche D, Bickmore WA and
Almouzni G: The effects of histone deacetylase inhibitors on
heterochromatin: Implications for anticancer therapy? EMBO Rep.
6:520–524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perry RJ, Borders CB, Cline GW, Zhang XM,
Alves TC, Petersen KF, Rothman DL, Kibbey RG and Shulman GI:
Propionate increases hepatic pyruvate cycling, anaplerosis and
alters mitochondrial metabolism. J Biol Chem. 291:12161–12170.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Altarejos JY and Montminy M: CREB and the
CRTC co-activators: Sensors for hormonal and metabolic signals. Nat
Rev Mol Cell Biol. 12:141–151. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Screaton RA, Conkright MD, Katoh Y, Best
JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR III,
Takemori H, et al: The CREB coactivator TORC2 functions as a
calcium- and cAMP-sensitive coincidence detector. Cell. 119:61–74.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perriello G, Pampanelli S, Del Sindaco P,
Lalli C, Ciofetta M, Volpi E, Santeusanio F, Brunetti P and Bolli
GB: Evidence of increased systemic glucose production and
gluconeogenesis in an early stage of NIDDM. Diabetes. 46:1010–1016.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang W, Patil S, Chauhan B, Guo S, Powell
DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, et al: FoxO1
regulates multiple metabolic pathways in the liver: Effects on
gluconeogenic, glycolytic, and lipogenic gene expression. J Biol
Chem. 281:10105–10117. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiong H, Guo B, Gan Z, Song D, Lu Z, Yi H,
Wu Y, Wang Y and Du H: Butyrate upregulates endogenous host defense
peptides to enhance disease resistance in piglets via histone
deacetylase inhibition. Sci Rep. 6:270702016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Donohoe DR, Collins LB, Wali A, Bigler R,
Sun W and Bultman SJ: The Warburg effect dictates the mechanism of
butyrate-mediated histone acetylation and cell proliferation. Mol
Cell. 48:612–626. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Anderson JW and Bridges SR: Short-chain
fatty acid fermentation products of plant fiber affect glucose
metabolism of isolated rat hepatocytes. Proc Soc Exp Biol Med.
177:372–376. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Massillon D, Arinze IJ, Xu C and Bone F:
Regulation of glucose-6-phosphatase gene expression in cultured
hepatocytes and H4IIE cells by short-chain fatty acids: Role of
hepatic nuclear factor-4alpha. J Biol Chem. 278:40694–40701. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diakos C, Prieschl EE, Saemann M, Novotny
V, Bohmig G, Csonga R, Baumruker T and Zlabinger GJ: Novel mode of
interference with nuclear factor of activated T-cells regulation in
T-cells by the bacterial metabolite n-butyrate. J Biol Chem.
277:24243–24251. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weber TE and Kerr BJ: Butyrate
differentially regulates cytokines and proliferation in porcine
peripheral blood mononuclear cells. Vet Immunol Immunopathol.
113:139–147. 2006. View Article : Google Scholar : PubMed/NCBI
|