Introduction
Alzheimer's disease (AD) is a neurodegenerative
disorder associated with pathological deposition of β-amyloid (Aβ)
peptides, intracellular neurofibrillary tangles and neuronal loss
caused by neuronal apoptosis. A complex contribution of several
genetic and environmental risk factors is necessary for the
initiation and progression of AD (1,2).
Currently, due to the lack of effective treatment options, the
disease ultimately results in patient mortality (3,4). Several
studies have investigated the potential association between Aβ and
the development of AD (5–7). Genetic studies have revealed abnormal
production of Aβ peptides in cell culture and animal models
(5–7). A vast body of evidence suggests that
the alterations in physicochemical properties and concentration of
Aβ1-42 may initiate the transition of neurons from the
physiological to the pathological state (8–10).
The etiology and pathogenesis of AD have remained
elusive, and the majority of the observations have confirmed that
Aβ deposition in the brain leads to the intracellular accumulation
of reactive oxygen species (ROS). Although oxidative stress does
not show specific clinical symptoms, the overproduction of ROS may
induce cell injury via lipid peroxidation, DNA damage and
regulation of apoptotic proteins (11–13).
Subsequently, apoptosis and cell cycle arrest further result in
neuronal cell death (14). In
addition, several studies have demonstrated an association between
oxidative stress and apoptosis in AD (14,15).
Therefore, it has been proposed that compounds that ameliorate
Aβ-induced oxidative stress may be used for the treatment and/or
prevention of AD. Antioxidants that prevent or delay Aβ-induced
apoptosis may be a promising therapeutic strategy against AD
(16–19).
Vanillin is a primary active component extracted
from vanilla beans, which has long been used as a component of
perfume and for food and medical applications (20). Ethyl vanillin (EVA), an analogue of
vanillin, is widely used as a food additive (Fig. 1), and the safety and long-established
properties of EVA have been previously investigated (21). Furthermore, it is reported that EVA
at doses <0.5 mM is unable to diminish viability of the
macrophage cells (22). Due to its
safety as a food additive, a number of studies have investigated
the multifunctional effects of EVA, including antioxidative,
antimutagenic, antiangiogenetic, anti-sickling and analgesic
activities (23–26). It has been reported that EVA serves a
protective role against protein oxidation and induction of
apoptosis caused by rotenone in a rotenone-induced rat model of
Parkinson's disease (27). A
previous study suggested that the antioxidative activity of EVA is
more potent compared with vanillin, as demonstrated by oxidative
hemolysis inhibition assays (28).
In addition, EVA can reduce the increase in ROS levels and
metalloproteinase-9 expression levels in lipopolysaccharide
(LPS)-stimulated macrophages, indicating that it could protect from
neurodegeneration and oxidative damage (21,29).
Based on the above evidence, the current study investigated the
pro-survival activity of EVA against the oxidative damage caused by
Aβ1-42-induced toxicity in PC12 cells. The ability of
EVA to protect against Aβ-induced neurotoxicity was assessed in
PC12 cells and initial experiments were performed to investigate
the potential mechanism of its action.
Materials and methods
Reagents
Ethyl vanillin (EVA), nerve growth factor (NGF),
Aβ1-42, Dulbecco's modified Eagle medium (DMEM), fetal
bovine serum (FBS) and horse serum (HS) were obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). TUNEL Detection Kit
(#11684817910) was purchased from Roche (Basel, Switzerland). The
kits for malondialdehyde (MDA), superoxide dismutase (SOD),
catalase (CAT), glutathione peroxidase (GSH-Px) and lactate
dehydrogenase (LDH) detection were provided from Nanjing Jiancheng
Bioengineering Institute (Nanjing, China). ROS assay kit and
mitochondrial membrane potential with JC-1 kit were purchased from
Beyotime Institute of Biotechnology (Haimen, China). Polyclonal
antibodies against cleaved caspase-3 (#9661), apoptosis regulator
Bcl-2 (#2876s) and apoptosis regulator Bax (#2772) were obtained
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Horseradish peroxidase conjugated goat anti-rabbit antibodies
(#GB23303) and bovine serum albumin (#G5001) were obtained from
Servicebio, Inc. (Woburn, MA, USA).
Cell culture
Undifferentiated PC12 cells were obtained from the
American Type Culture Collection (Manassas, VA, USA). Cells were
cultured with 5 ng/ml NGF in DMEM containing 10% HS, 5% FBS and 1%
penicillin-streptomycin in a CO2 incubator (37°C and 5%
CO2). The medium was changed every other day. The cells
were seeded at a density of 1×104 cells/ml and allowed to grow for
24 h prior to experimentation.
Cell viability and LDH assay
To assess the protective effect of EVA against
Aβ1-42-induced cytotoxicity, cell viability was detected
by the MTT reduction assay, as previously described.
Aβ1-42 aggregates were prepared as previously described
(30). PC12 cells were cultured at a
density of 1×104 cells/well in poly-L-lysine-coated 96-well
microplates for 24 h at 37°C. The cells were pretreated with
various doses of EVA (0, 10, 30 and 100 µM) for 12 h and then
exposed to 20 µM of Aβ1-42 for 12 h. The EVA group was
treated with 100 µM of EVA for 24 h. At the end of the drug
treatment, 20 µl of MTT solution (5 mg/ml) was added to each well
and the cells were incubated at 37°C for 4 h. The supernatants were
subsequently replaced with 150 ml of dimethylsulfoxide. Cell
viability was determined at a wavelength of 570 nm using an ELISA
reader (Varioskan™ Flash Multimode Reader; Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
The LDH activity is an in vitro cellular
toxicity marker that was determined in the present study using a
commercial assay kit. In brief, PC12 cells were cultured at a
density of 1×104 cells/well in poly-L-lysine-coated 96-well
microplates for 24 h at 37°C. The cells were pretreated with
various doses of EVA (0, 10, 30 and 100 µM) for 12 h and then
exposed to 20 µM of Aβ1-42 for 12 h. At the end of the
drug treatment, 100 µl of the incubation medium was collected for
the extracellular LDH activity assay. The adherent cells were
washed with phosphate buffered saline (PBS) three times and
subsequently homogenized. The homogenate was centrifuged at 4,000 ×
g for 30 min at 37°C and the supernatant was collected for the
intracellular LDH activity assay. The absorbance of each sample was
measured at a wavelength of 490 nm using a microplate reader
according to the manufacturer's protocol. LDH release was
calculated as follows:
LDH release rate (%) = LDH activity in
the culture mediumLDH activity in the culture medium + LDH activity
in cell × 100%
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labelling
(TUNEL) assay
Apoptotic cells were observed by TUNEL assay.
Briefly, PC12 cells were cultured and treated on circular glass
coverslips. The cells were pretreated with 100 µM of EVA for 12 h
and then exposed to 20 µM Aβ1-42 for 12 h. At the end of
drug treatment, the cells were fixed in 4% paraformaldehyde for 15
min at 4°C and washed with PBS. The samples were further incubated
in a blocking solution (obtained from the assay kit) for 20 min at
37°C and permeabilizing solution (0.1% Triton X-100 in 0.1% sodium
acetate) for 30 min at 4°C. Subsequently, the cells were stained by
TUNEL reaction solution for 1 h at 37°C, followed by cultured with
3% H2O2 solution for 15 min and rinsed with
PBS for 10 min. The cells were finally treated with
3,3′-diaminobenzidine (DAB) substrate to produce a dark brown
precipitate. Coverslips were stained with hematoxylin for 1 min at
37°C and mounted with neutral balsam. The number of TUNEL-positive
cells was counted at 6 high-power (magnification, ×400) fields per
slide.
Lipid peroxidation assay
The MDA content, an index of lipid peroxidation, was
detected using a commercial assay kit according to the
manufacturer's protocol. Briefly, PC12 cells were cultured at a
density of 4×105 cells/well in 6-well microplates for 24 h at 37°C.
The cells were pretreated with various doses of EVA (0, 10, 30 and
100 µM) for 12 h and then exposed to 20 µM Aβ1-42 for 12
h. At the end of the drug treatment, the cells were washed with PBS
and lysed in radio immunoprecipitation assay (RIPA) lysis buffer.
The mixture was centrifuged at 12,000 × g for 30 min at 4°C. The
collected supernatant samples were used for analysis of MDA
concentration.
Dichloro-dihydro-fluorescein diacetate
(DCFH-DA) assay for ROS
The accumulation of intracellular ROS was monitored
using the fluorescent dye DCFH-DA as a probe. Briefly, PC12 cells
were cultured at a density of 4×105 cells/well in 6-well
microplates for 24 h at 37°C. The cells were pretreated with
various doses of EVA (0, 10, 30 and 100 µM) for 12 h and then
exposed to 20 µM of Aβ1-42 for 12 h. At the end of the
drug treatment, 1 ml of 10 µM DCFH-DA solution (part of the ROS
kit) was added to the cells and cultured at 37°C for 30 min. In the
presence of ROS, DCFH was converted to DCF, as the kit protocol
states. Subsequently, the cells were rinsed three times with PBS
and the fluorescence intensity was visualized by fluorescence
microscopy (Carl Zeiss AG, Oberkochen, Germany). The levels of
intracellular ROS of each group were normalized to those of the
control group (untreated PC12 cells).
Antioxidant system assay
PC12 cells were cultured at a density of 4×105
cells/well in 6-well microplates for 24 h at 37°C. The cells were
pretreated with various doses of EVA (0, 10, 30 and 100 µM) for 12
h and then exposed to 20 µM Aβ1-42 for 12 h consecutive.
At the end of the drug treatment, the cells were washed with PBS
and lysed in RIPA lysis buffer. The mixture was centrifuged at
12,000 × g for 30 min at 4°C. The collected supernatants were used
for the following analyses.
SOD, CAT and GSH-Px activity levels in PC12 cells
were measured by commercial assay kits according to the
instructions provided by the manufacturer. SOD activity was
detected using the xanthine oxidase method at a wavelength of 550
nm using a spectrophotometer. CAT activity was determined using by
measuring the absorbance at a wavelength of 405 nm with a
spectrophotometer. GSH-Px activity was measured by quantifying the
rate of oxidation of reduced glutathione to oxidized glutathione by
H2O2 at a wavelength of 412 nm. The activity
levels were normalized to the protein concentration of each
sample.
Measurement of mitochondrial membrane
potential
Mitochondrial membrane potential was detected using
the fluorescent JC-1 dye probe. PC12 cells were cultured at a
density of 4×105 cells/well in 6-well microplates for 24 h at 37°C.
The cells were pretreated with various doses of EVA (0, 10, 30 and
100 µM) for 12 h and then exposed to 20 µM of Aβ1-42 for
12 h. At the end of the drug treatment, the cells were treated with
1 ml of JC-1 for 30 min at room temperature. Fluorescence intensity
alterations were determined using flow cytometry (CXP 2.1, Beckman
Coulter, Inc., Brea, CA, USA). The data are presented as the ratio
of red to green signal. The changes in mitochondrial membrane
potential were calculated as the integral of the decrease in the
ratio of JC-1 fluorescence.
Immunohistochemistry (IHC) assay
IHC analysis was used to evaluate the levels of
cleaved caspase-3, Bax and Bcl-2 proteins. Briefly, PC12 cells were
cultured and pretreated with 100 µM of EVA for 12 h at 37°C and
subsequently exposed to 20 µM of Aβ1-42 for 12 h at
37°C. At the end of the drug treatment, the cells were fixed in 4%
paraformaldehyde for 15 min at 4°C and washed with PBS. Cells were
then heated for 10 min in citrate buffer (pH 6.0) for antigen
retrieval. The slides were subsequently exposed to 3%
H2O2 solution for 25 min and blocked in 5%
bovine serum albumin at room temperature for 30 min. Fixed cells
were incubated with primary antibodies diluted in PBS (anti-cleaved
caspase-3, 1:1,000; anti-Bax, 1:1,000; anti-Bcl-2, 1:500) overnight
at 4°C. The samples were washed and treated with secondary antibody
(goat anti-rabbit, 1:3,000) diluted in PBS at 37°C for 45 min. DAB
was used to produce a dark brown precipitate at 37°C for 30 sec and
hematoxylin was used to stain the nuclei of the cells at 37°C for 1
min. The cells were observed and images were captured with a
fluorescence microscope (magnification, ×400; Carl Zeiss AG). The
ratio of brown to total area was analyzed with Image-Pro Plus in 6
fields of view (Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
The differences between two groups were assessed with one-way
analysis of variance followed by Dunnett's post hoc test. Analyses
were performed using SPSS software (version 17.0; SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of EVA on
Aβ1-42-induced cytotoxicity in PC12 cells
Aβ1-42 can induce both oxidative stress
and apoptosis in PC12 cells (13).
The viability of Aβ1-42-treated PC12 cells was assessed
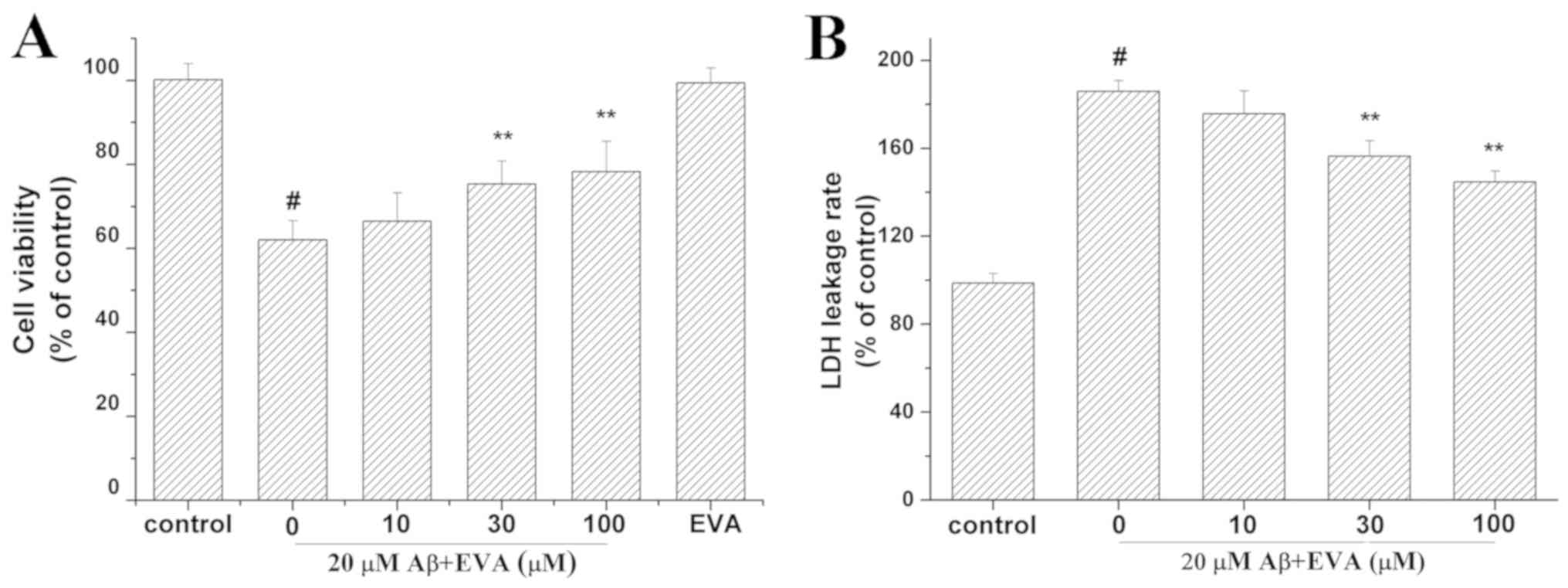
in the presence of EVA using the MTT assay (Fig. 2A). Cell viability was reduced to
62.07% in PC12 cells treated with 20 µM Aβ1-42 compared
with the control group in the absence of Aβ1-42. In
contrast to the concentration of 20 µM Aβ1-42 alone,
pretreatment with different doses (10, 30 and 100 µM) of EVA
increased cell viability to 66.47, 75.38 and 78.29% of the control
value, respectively, in a dose-dependent manner. Furthermore, PC12
cells cultured with 100 µM of EVA exhibited a cell viability of
99.49% compared with that noted in the control group in the absence
of Aβ1-42 at 24 h after the treatment. This result
suggested that 100 µM of EVA alone did not reduce the viability of
PC12 cells.
The LDH release assay was used to evaluate the
effect of treatment with EVA on Aβ1-42-induced
cytotoxicity (Fig. 2B). The LDH
release rate was increased to 185.84% in PC12 cells incubated with
20 µM of Aβ1-42 compared with the control group
untreated with Aβ1-42. Compared with the group treated
with 20 µM Aβ1-42 only, pretreatment with different
doses (10, 30 and 100 µM) of EVA decreased the LDH release rate to
175.66, 156.43 and 144.67% of the control value, respectively, in a
dose-dependent manner.
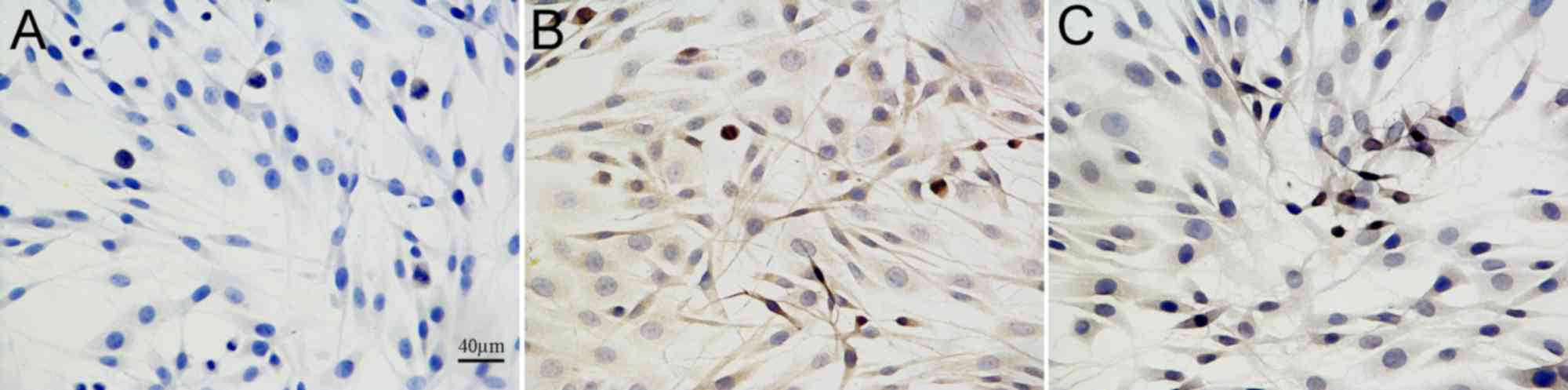
To further investigate the protective effect of EVA
on the Aβ1-42-induced PC12 cell damage, the rate of
apoptosis of PC12 cells was observed by the TUNEL assay (Fig. 3). The apoptotic cells were
characterized by the appearance of intense brown staining. Control
PC12 cells were not treated with Aβ1-42 or EVA.
Treatment of PC12 cells with 20 µM of Aβ1-42 markedly
increased the levels of apoptosis compared with those noted in the
control cells. This effect was ameliorated by pretreatment of the
cells with 100 µM of EVA. The results indicated that
Aβ1-42 inducedPC12 cell injury, and that EVA possessed
the ability to inhibit this change induced by
Aβ1-42.
Effect of EVA on oxidative stress
induced in PC12 cells
Oxidative stress serves an important role in the
induction of PC12 cell apoptosis by Aβ1-42 (13). The current study investigated the
effects of EVA on the MDA levels and the levels of intracellular
ROS (Fig. 4) in PC12 cells following
treatment with Aβ1-42. The production of ROS and the MDA
levels were increased to 203.91% and 1.79 nmol/mg (control MDA
value, 0.87 nmol/mg), respectively in PC12 cells induced with 20 µM
of Aβ1-42, compared with the control group. Pretreatment
of Aβ1-42-treated cells with different doses (10, 30 and
100 µM) of EVA decreased the intracellular ROS levels to 174.93,
160.86 and 139.51%, respectively, and the MDA levels to 1.61, 1.51
and 1.34 nmol/mg/protein, respectively, in a dose-dependent
manner.
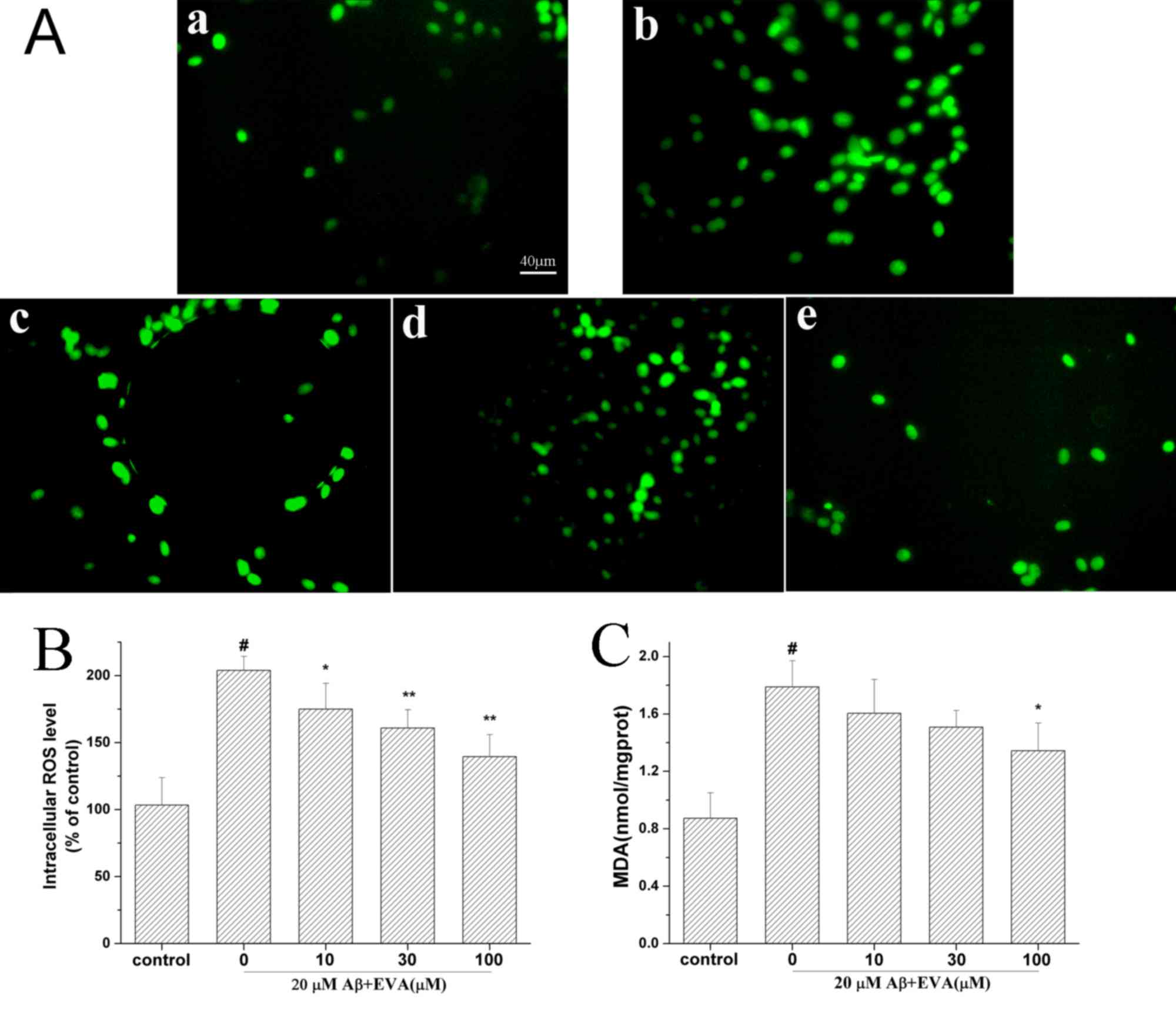 | Figure 4.Protective effect of EVA on oxidative
stress in Aβ1-42-treated PC12 cells. Cells were
incubated in the absence of Aβ1-42 (control) or in the
presence of 20 µM of Aβ1-42 following pretreatment with
different concentrations of EVA. (A) ROS production was measured
with a fluorescence microscope (magnification, ×200). a, Control
group. b, 20 µM Aβ1-42 single treatment. c, 20 µM
Aβ1-42+10 µM EVA. d, 20 µM Aβ1-42+30 µM EVA.
e, 20 µM Aβ1-42+100 µM EVA. (B) ROS levels were measured
using a standard assay. Data are presented as the percentage of the
control group and expressed as the mean ± standard deviation (n=3).
(C) MDA content was measured with an MDA assay. The results are
expressed as the mean ± standard deviation (n=5). #P<0.01
compared with the control group untreated with Aβ1-42.
*P<0.05 and **P<0.01 compared with the
Aβ1-42-treated group. ROS, reactive oxygen species; EVA,
ethyl vanillin; Aβ, β-amyloid; MDA, malondialdehyde. |
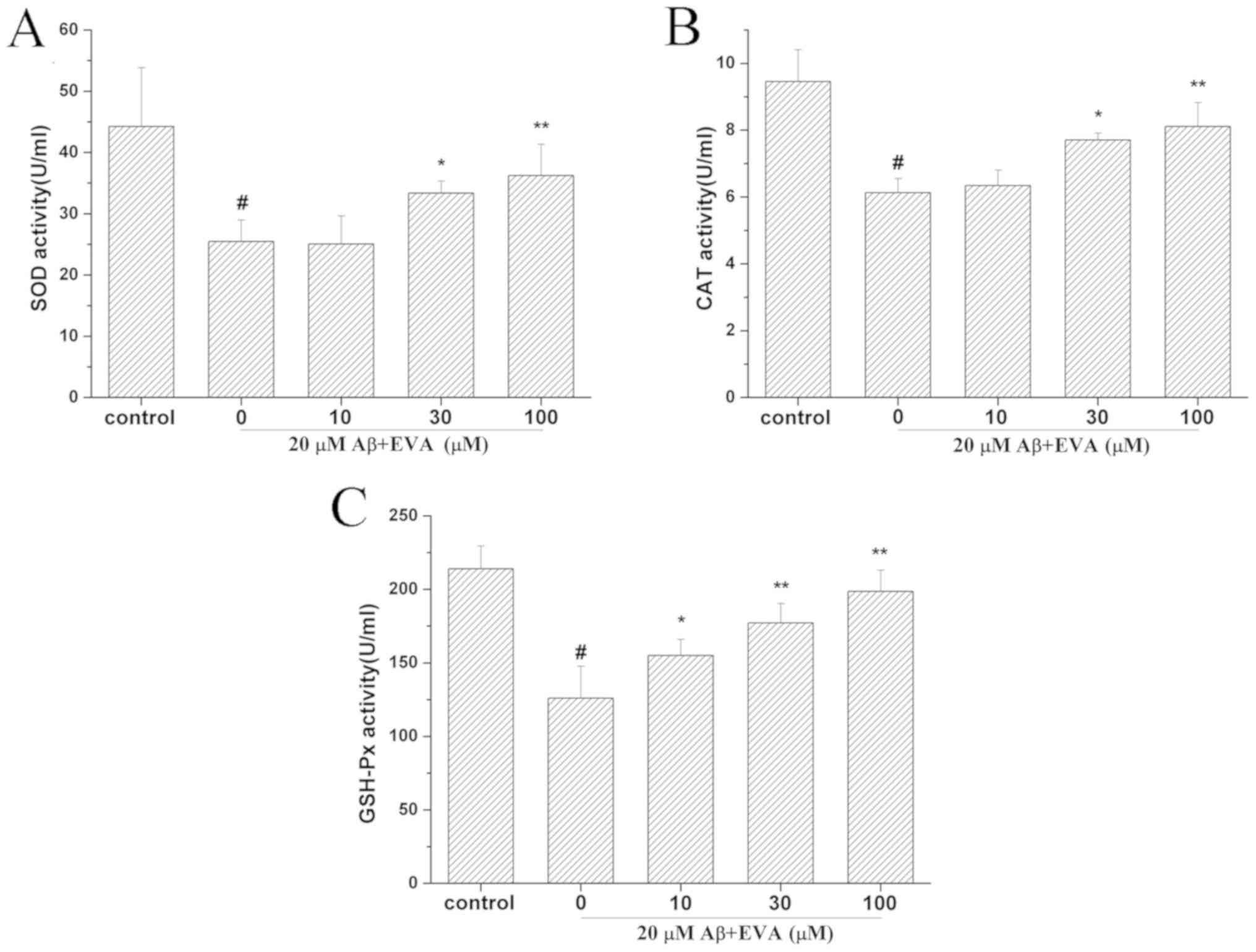
Effects of EVA on the antioxidative
enzyme activities in PC12 cells treated with Aβ1-42
To determine the effect of EVA on the activities of
antioxidative enzymes in Aβ1-42-treated PC12 cells, the
activity levels of SOD (Fig. 5A),
CAT (Fig. 5B) and GSH-Px (Fig. 5C) were detected. The results
indicated that treatment with Aβ1-42 reduced the
activities of SOD, CAT and GSH-Px to 25.47 U/ml (control SOD value,
44.25 U/ml), 6.13 U/ml (control CAT value, 9.46 U/ml) and 126.01
U/ml (control GSH-Px value, 213.91 U/ml), respectively. However,
administration of different doses of EVA (10, 30 and 100 µM)
ameliorated the activity levels of SOD (25.07, 33.37 and 36.24
U/ml, respectively), CAT (6.34, 7.71 and 8.11 U/ml, respectively)
and GSH-Px (155.13, 177.26 and 198.63 U/ml, respectively). These
activity levels of SOD, CAT and GSH-Px following pretreatment with
EVA were close to those of the control cells and the protective
effect was dose-dependent.
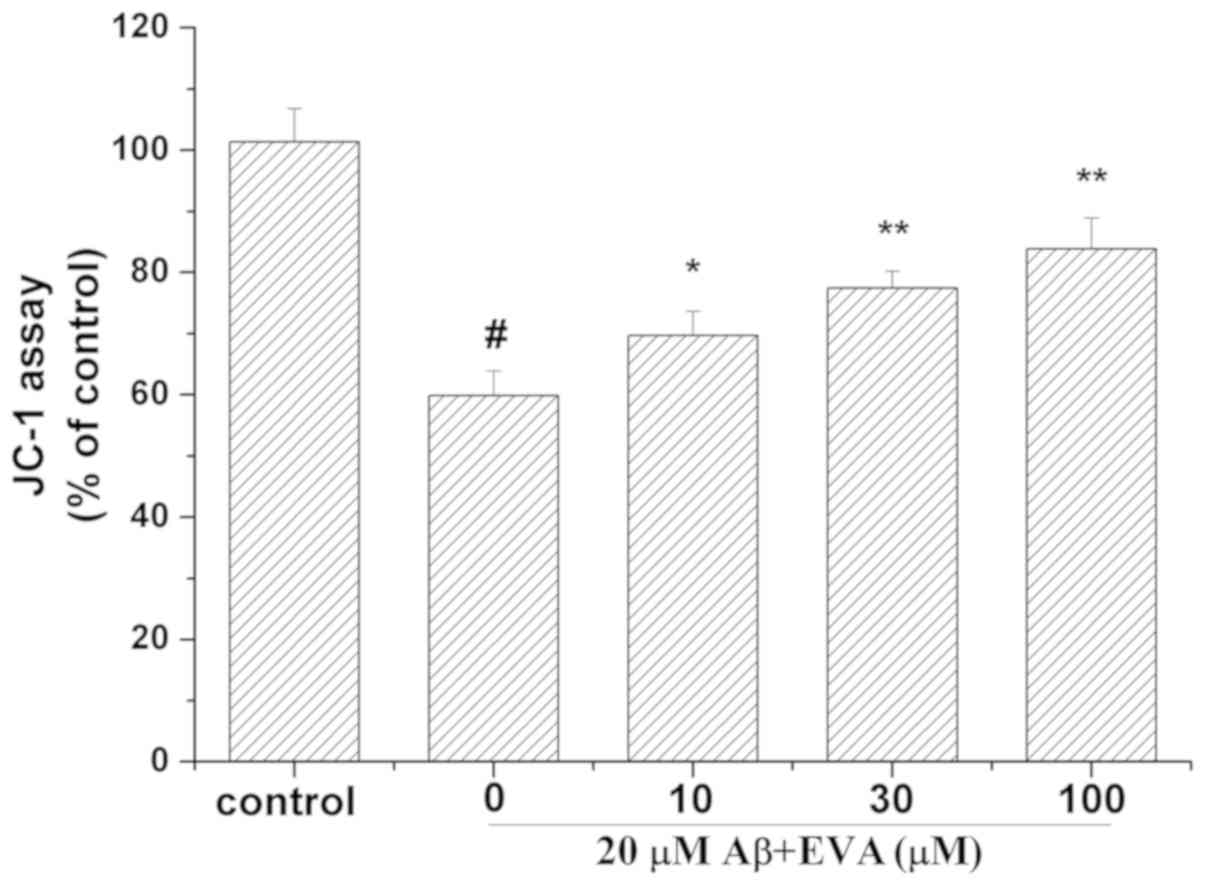
Effect of EVA on the mitochondrial
membrane potential in PC12 cells following treatment with
Aβ1-42
To determine whether EVA could stabilize the
mitochondrial function, the mitochondrial membrane potential was
measured using the JC-1 assay (Fig.
6). Treatment of PC12 cells with 20 µM Aβ1-42 for 12
h caused a reduction in the mitochondrial membrane potential to
59.91% compared with the control cells. Pretreatment of PC12 cells
with different doses of EVA (10, 30 and 100 µM), caused a
significant increase in the mitochondrial membrane potential
(69.72, 77.45 and 83.85% compared with the control value,
respectively) in a dose-dependent manner.
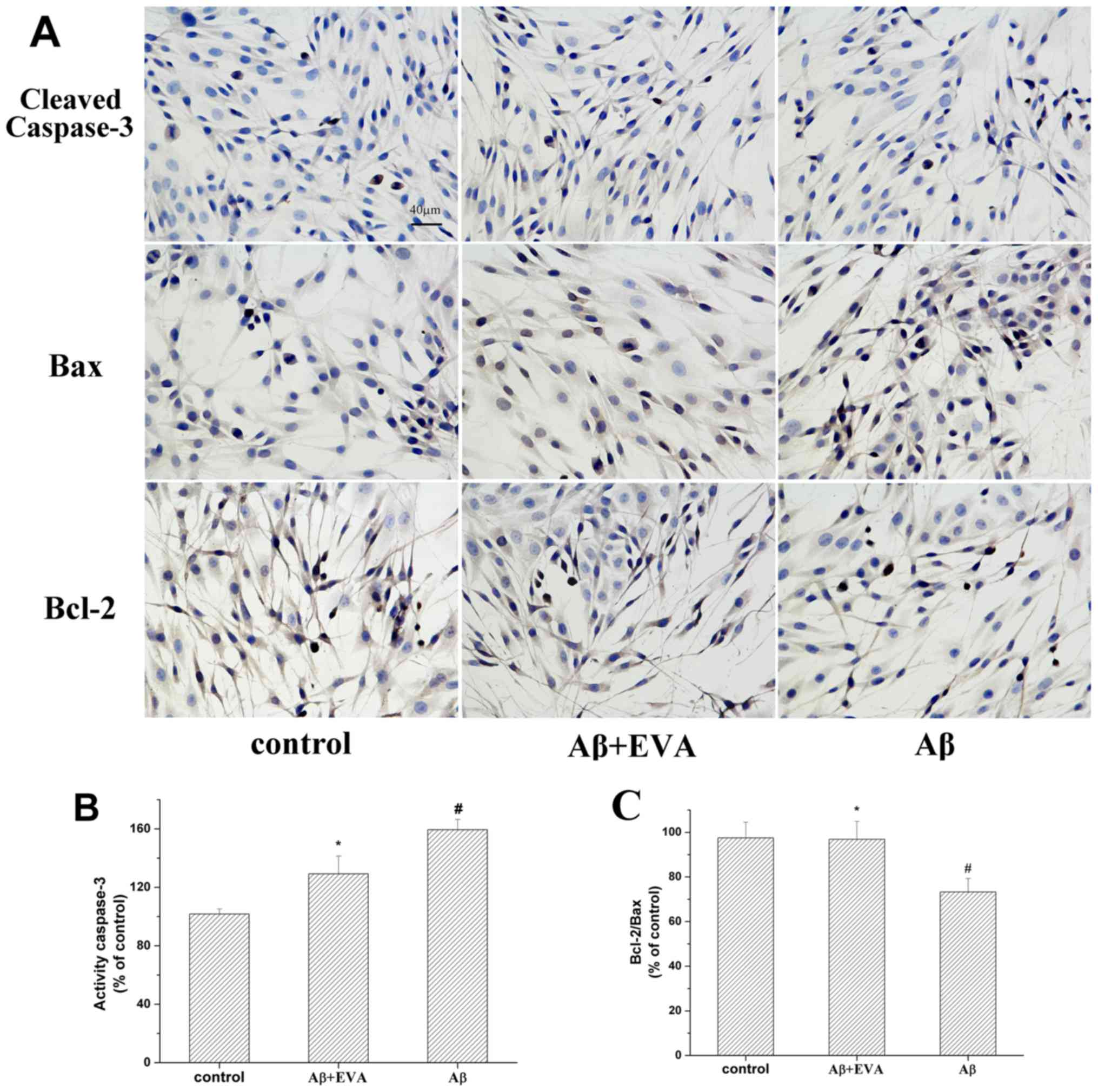
Effects of EVA on
Aβ1-42-induced apoptotic protein expression in PC12
cells
The relative expression levels of Bcl-2/Bax and
caspase-3 serve an important role in the induction of cell
apoptosis (30). The effects of EVA
on the apoptotic proteins in Aβ1-42-treated PC12 cells
were determined by the investigation of the expression levels of
Bax, Bcl-2 and cleaved-caspase-3 proteins (Fig. 7). The results revealed that treatment
with Aβ1-42 reduced the ratio of Bcl-2/Bax to 73.26%.
Pretreatment with 100 µM of EVA partially reversed this change in
Aβ1-42-treated PC12 cells. Furthermore, treatment with
Aβ1-42 increased the levels of cleaved-caspase-3 to
159.44% in PC12 cells compared with those of the normal control
cells. Pretreatment with 100 µM of EVA inhibited the increase of
the expression levels of cleaved-caspase-3 induced by
Aβ1-42. These results suggested that pretreatment with
EVA may be effective for the prevention of
Aβ1-42-induced cell apoptosis in PC12 cells.
Discussion
EVA has been used as an antioxidant agent for
oxidative injury. This compound has been examined for antioxidant
properties via in vivo and in vitro studies (21,28,29). The
present study investigated the effect of EVA on
Aβ1-42-induced PC12 cells, a common in vitro
model of AD (31). Although the
precise molecular mechanism underlying the Aβ-mediated neuronal
apoptosis remains unclear, the majority of the studies indicated
that oxidative stress is a hallmark of Aβ-induced neuronal toxicity
in AD (11–13,32). The
PC12 cell line is a useful model system to study
Aβ1-42-induced cytotoxicity due to its particular
sensitivity to Aβ peptides (33).
Furthermore, PC12 cells are relatively easy to culture and survive
longer than primary cultured neurons (33). Therefore, PC12 cells were selected to
investigate the neuroprotective effects of EVA against
Aβ1-42-induced cell damage. Furthermore, several animal
models have been used to determine whether compounds such as
Aβ1-42 may protect against AD, including a transgenic
animal model and a rat model induced by intrahippocampal injection
of Aβ1-42 in the brain (34,35). The
main aim addressed by the present study was whether EVA could
protect against Aβ1-42-induced injury in PC12 cells and
the investigation of the associated mechanism underlying this
process.
Aβ is considered an inducer of neuronal damage
(5). In the present study, treatment
with Aβ1-42resulted in a decrease in the percentage of
viable cells. Furthermore, the increase in LDH activity of
Aβ1-42-treated PC12 cells implied an increase in the
number of damaged plasma membranes and an increase in the overall
cell death (36). However, treatment
with EVA increased the viability of cells treated
withAβ1-42 in a dose-dependent manner. In addition, the
morphological characteristics of Aβ1-42-treated PC12
cells were investigated by the TUNEL assay.
Aβ1-42-treated PC12 cells that were pretreated with EVA
exhibited a decrease in the level of apoptosis. These results
indicated that pretreatment with different concentrations of EVA
could efficiently prevent cytotoxicity and apoptosis induction by
Aβ1-42 in PC12 cells. Additionally, no cellular toxicity
was induced by treatment with 100 µM of EVA, which demonstrated
good biocompatibility of this compound. Additionally, it was
previously reported that EVA alone did not alter the ROS levels and
metalloproteinase-9 expression in the LPS-stimulated RAW264.7
macrophage cells (22). Therefore,
the group treated with EVA alone was not included in the other
experiments in the current study.
Oxidative stress is one of the most important
factors that induce cellular injury and apoptosis in several
neurodegenerative disorders (17).
Excessive levels ROS and lipid peroxidation are markers of
oxidative stress (17). Aβ induced
cytotoxicity through the overproduction of intracellular ROS
following superoxide accumulation in the mitochondria of PC12 cells
(37). The levels of MDA, a
decomposition product of lipid hydroperoxides, are representative
of oxidative damage to the cells and tissues (38). In the present study, EVA reduced the
production of ROS and MDA in a dose-dependent manner. This result
demonstrated that EVA may induce protective effects on
Aβ1-42-treated PC12 cells by reducing the overproduction
of ROS.
Excessive production of ROS is associated with
oxidative stress, resulting in cellular injury and apoptosis in a
wide variety of cell types (13).
Under physiological conditions, intracellular ROS levels are
accurately regulated by free radical scavengers and by a precise
antioxidant defense system (39).
The clearance of ROS is achieved with various antioxidants
(39,40). The most widely investigated cellular
antioxidant system comprises the enzymes SOD, CAT and GSH-Px, which
are associated with a direct removal of ROS (41–43). It
has been demonstrated that SOD can transform superoxide anions to
hydrogen peroxide, which is subsequently scavenged by CAT (41,42).
GSH-Px converts lipid hydroperoxides to their corresponding
alcohols and free hydrogen peroxide to water (42). Following administration of
Aβ1-42, overproduction of ROS led to disturbances in the
endogenous antioxidant balance of PC12 cells. According to the
current results, enzyme activities of SOD, CAT and GSH-Px were
decreased in Aβ1-42-induced PC12 cells. However, EVA
could attenuate the activity levels of SOD, CAT and GSH-Px in a
dose-dependent manner, which suggested that the protective effect
of EVA may be partially involved in its antioxidative effect.
It is generally accepted that ROS is required for
the activation of the mitochondria-dependent apoptotic pathways in
neurodegenerative diseases (44).
Mitochondrial membrane potential is an indicator of mitochondrial
dysfunction to cells and tissues (45). Mitochondrial dysfunction is caused by
an irreversible depolarization of the mitochondria (30). This process is usually encountered in
various neurodegenerative diseases in association with
Aβ1-42-induced neuronal toxicity (39). Concomitantly, mitochondrial
dysfunction further initiates ROS production in the mitochondria
and their subsequent release into the cytoplasm, leading to
oxidative stress via activation of apoptotic signaling (46). In the present study, the
mitochondrial membrane potential of Aβ1-42-treated PC12
cells was decreased. However, EVA was found to be effective in
improving Aβ1-42-induced mitochondrial membrane
depolarization in PC12 cells. This result provides evidence that
EVA may exert neuroprotective effects by stabilizing the
mitochondrial membrane potential.
The decrease in the mitochondrial membrane potential
influences the regulation of apoptotic proteins, including the
Bcl-2 family proteins (45).
Caspase-3, Bax and Bcl-2 proteins regulate the mitochondrial
apoptotic pathway (47,48). Bcl-2 is an anti-apoptotic protein,
while Bax is a pro-apoptotic protein which can enhance programmed
cell death (13). Furthermore, under
oxidative stress and mitochondrial membrane depolarization
conditions, the decreased Bcl-2/Bax ratio can directly lead to the
activation of caspase-3 (19,30).
Several studies have suggested that caspase-3 is activated in the
hippocampus of AD rats as well as in Aβ1-42-induced PC12
cells (13,14,34). It
has been previously demonstrated that activation of caspase-3 is
involved in the cytotoxicity and apoptosis of PC12 cells (49). The present study indicated that the
ratio of Bcl-2/Bax was significantly decreased in
Aβ1-42-induced cells, and that the expression levels of
the cleaved form of caspase-3 were significantly increased
following Aβ1-42 administration. However, pretreatment
with EVA reversed the alterations induced by Aβ1-42.
These results suggested that EVA may prevent
Aβ1-42-induced cytotoxicity and apoptosis in PC12 cells
via regulation of the protein Bcl-2, Bax and caspase-3.
In conclusion, the present study investigated the
protective effect of EVA on Aβ1-42-induced neuronal
toxicity. EVA protected Aβ1-42-induced PC12 cell damage
and this protective effect was mediated by the inhibition of
oxidative stress and prevention of cell apoptosis. The present
study provides novel data with regard to the effect of EVA and may
provide a novel strategy for the treatment of AD. Furthermore, a
future study will involve an intrahippocampal injection of
Aβ1-42 in the brain of a rat model to verify the
appropriate concentration and administration route of EVA in
vivo.
Acknowledgements
The authors would like to thank Dr Manda Williams
(Department of Bioengineering, University of Washington, Seattle,
WA, USA) for language editing.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81603050), the
Foundation of Sichuan Provincial People's Hospital (grant no.
2016LY01) and the National Key Specialty Construction Project of
Clinical Pharmacy (grant no. 30305030698).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ and YZ conceived and designed the current study.
YT, LZ and LB collected samples and performed the experiments. YT
and JC analyzed the data. LZ and JS drafted the manuscript. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brookmeyer R, Johnson E, Ziegler-Graham K
and Arrighi HM: Forecasting the global burden of Alzheimer's
disease. Alzheimers Dement. 3:186–191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Selkoe DJ: Alzheimer's disease: Genes,
proteins and therapy. Physiol Rev. 81:741–766. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Epperly T, Dunay MA and Boice JL:
Alzheimer disease: Pharmacologic and nonpharmacologic therapies for
cognitive and functional symptoms. Am Fam Physician. 95:771–778.
2017.PubMed/NCBI
|
|
4
|
Gao LB, Yu XF, Chen Q and Zhou D:
Alzheimer's disease therapeutics: Current and future therapies.
Minerva Med. 107:108–113. 2016.PubMed/NCBI
|
|
5
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zheng WH, Bastianetto S, Mennicken F, Ma W
and Kar S: Amyloid beta peptide induces tau phosphorylation and
loss of cholinergic neurons in rat primary septal cultures.
Neuroscience. 115:201–211. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi SJ, Kim JK, Kim HK, Harris K, Kim CJ,
Park GG, Park CS and Shin DH: 2,4-Di-tert-butylphenol from sweet
potato protects against oxidative stress in PC12 cells and in mice.
J Med Food. 16:977–983. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Butterfield DA, Castegna A, Lauderback CM
and Drake J: Evidence that amyloid beta-peptide-induced lipid
peroxidation and its sequelae in Alzheimer's disease brain
contribute to neuronal death. Neurobiol Aging. 23:655–664. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crack PJ, Cimdins K, Ali U, Hertzog PJ and
Iannello RC: Lack of glutathione peroxidase-1 exacerbates
Abeta-mediated neurotoxicity in cortical neurons. J Neural Transm
(Vienna). 113:645–657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Selkoe DJ: Soluble oligomers of the
amyloid beta-protein impair synaptic plasticity and behavior. Behav
Brain Res. 192:106–113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu JF, Chu SF, Ning N, Yuan YH, Xue W,
Chen NH and Zhang JT: Protective effect of (−)clausenamide against
Abeta-induced neurotoxicity in differentiated PC12 cells. Neurosci
Lett. 483:78–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li G, Ma R, Huang C, Tang Q, Fu Q, Liu H,
Hu B and Xiang J: Protective effect of erythropoietin on
beta-amyloid-induced PC12 cell death through antioxidant
mechanisms. Neurosci Lett. 442:143–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xian YF, Lin ZX, Mao QQ, Ip SP, Su ZR and
Lai XP: Protective effect of isorhynchophylline against
beta-amyloid-induced neurotoxicity in PC12 cells. Cell Mol
Neurobiol. 32:353–360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Turunc Bayrakdar E, Uyanikgil Y, Kanit L,
Koylu E and Yalcin A: Nicotinamide treatment reduces the levels of
oxidative stress, apoptosis, and PARP-1 activity in
Aβ(1–42)-induced rat model of Alzheimer's disease. Free Radical
Res. 48:146–158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu Y, Dong Y, Tucker D, Wang R, Ahmed ME,
Brann D and Zhang Q: Treadmill exercise exerts neuroprotection and
regulates microglial polarization and oxidative stress in a
streptozotocin-induced rat model of sporadic Alzheimer's disease. J
Alzheimers Dis. 56:1469–1484. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deguchi K, Hayashi T, Nagotani S, Sehara
Y, Zhang H, Tsuchiya A, Ohta Y, Tomiyama K, Morimoto N, Miyazaki M,
et al: Reduction of cerebral infarction in rats by biliverdin
associated with amelioration of oxidative stress. Brain Res.
1188:1–8. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dumont M and Beal MF: Neuroprotective
strategies involving ROS in Alzheimer disease. Free Radical Biol
Med. 51:1014–1026. 2011. View Article : Google Scholar
|
|
18
|
Calabrese V, Guagliano E, Sapienza M,
Panebianco M, Calafato S, Puleo E, Pennisi G, Mancuso C,
Butterfield DA and Stella AG: Redox regulation of cellular stress
response in aging and neurodegenerative disorders: Role of
vitagenes. Neurochem Res. 32:757–773. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Ji X, Zhang J, Shi G, Zhu X and Wang
K: Paeoniflorin attenuates Aβ25-35-induced neurotoxicity in PC12
cells by preventing mitochondrial dysfunction. Folia Neuropathol.
52:285–290. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu SL, Chen JC, Li CC, Lo HY, Ho TY and
Hsiang CY: Vanillin improves and prevents trinitrobenzene sulfonic
acid-induced colitis in mice. J Pharmacol Exp Ther. 330:370–376.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JH, Lee HO, Cho YJ, Kim J, Chun J,
Choi J, Lee Y and Jung WH: A vanillin derivative causes
mitochondrial dysfunction and triggers oxidative stress in
Cryptococcus neoformans. PLoS One. 9:e891222014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jung HJ, Song YS, Kim K, Lim CJ and Park
EH: Assessment of the anti-angiogenic, anti-inflammatory and
antinociceptive properties of ethyl vanillin. Arch Pharmacal Res.
33:309–316. 2010. View Article : Google Scholar
|
|
23
|
Lirdprapamongkol K, Sakurai H, Kawasaki N,
Choo MK, Saitoh Y, Aozuka Y, Singhirunnusorn P, Ruchirawat S,
Svasti J and Saiki I: Vanillin suppresses in vitro invasion and in
vivo metastasis of mouse breast cancer cells. Eur J Pharm Sci.
25:57–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Makni M, Chtourou Y, Fetoui H, Garoui el
EM, Boudawara T and Zeghal N: Evaluation of the antioxidant,
anti-inflammatory and hepatoprotective properties of vanillin in
carbon tetrachloride-treated rats. Eur J Pharmacol. 668:133–139.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tai A, Sawano T, Yazama F and Ito H:
Evaluation of antioxidant activity of vanillin by using multiple
antioxidant assays. Biochim Biophys Acta. 1810:170–177. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kwon J, Kim J, Park S, Khang G, Kang PM
and Lee D: Inflammation-responsive antioxidant nanoparticles based
on a polymeric prodrug of vanillin. Biomacromolecules.
14:1618–1626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dhanalakshmi C, Janakiraman U, Manivasagam
T, Justin Thenmozhi A, Essa MM, Kalandar A, Khan MA and Guillemin
GJ: Vanillin attenuated behavioural impairments, neurochemical
deficts, oxidative stress and apoptosis against rotenone induced
rat model of Parkinson's disease. Neurochem Res. 41:1899–1910.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tai A, Sawano T and Yazama F: Antioxidant
properties of ethyl vanillin in vitro and in vivo. Biosci
Biotechnol Biochem. 75:2346–2350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen XM, Wei M, Zhang HM, Luo CH, Chen YK
and Chen Y: Effect of vanillin and ethyl vanillin on cytochrome
P450 activity in vitro and in vivo. Food Chem Toxicol.
50:1897–1901. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu Y, Sun X, Gong T, He Q and Zhang Z:
antioxidant and antiapoptotic effects of 1,1,1anillin on cytochrome
P450 activity in vitro and in1-one on protecting PC12 cells from
Aβ-induced injury. Mol Pharmaceutics. 11:428–435. 2013. View Article : Google Scholar
|
|
31
|
Kinarivala N, Shah K, Abbruscato TJ and
Trippier PC: Passage variation of PC12 cells results in
inconsistent susceptibility to externally induced apoptosis. ACS
Chem Neurosci. 8:82–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Goldsbury C, Whiteman IT, Jeong EV and Lim
YA: Oxidative stress increases levels of endogenous amyloid-β
peptides secreted from primary chick brain neurons. Aging Cell.
7:771–775. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harper JD and Lansbury PT Jr: Models of
amyloid seeding in Alzheimer's disease and scrapie: Mechanistic
truths and physiological consequences of the time-dependent
solubility of amyloid proteins. Annu Rev Biochem. 66:385–407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jantaratnotai N, Ryu JK, Schwab C, McGeer
PL and McLarnon JG: Comparison of vascular perturbations in an
Aβ-injected animal model and in AD brain. Int J Alzheimers Dis
2011. 2011.
|
|
35
|
Ryu JK and McLarnon JG: A leaky
blood-brain barrier, fibrinogen infiltration and microglial
reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med.
13:2911–2925. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beal MF: Oxidatively modified proteins in
aging and disease. Free Radical Biol Med. 32:797–803. 2002.
View Article : Google Scholar
|
|
37
|
Naoi M, Maruyama W, Yi H, Inaba K, Akao Y
and Shamoto-Nagai M: Mitochondria in neurodegenerative disorders:
Regulation of the redox state and death signaling leading to
neuronal death and survival. J Neural Transm (Vienna).
116:1371–1381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Benedı́ J, Arroyo R, Romero C,
Martı́n-Aragón S and Villar AM: Antioxidant properties and
protective effects of a standardized extract of Hypericum
perforatum on hydrogen peroxide-induced oxidative damage in PC12
cells. Life Sci. 75:1263–1276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen H, Yoshioka H, Kim GS, Jung JE, Okami
N, Sakata H, Maier CM, Narasimhan P, Goeders CE and Chan PH:
Oxidative stress in ischemic brain damage: Mechanisms of cell death
and potential molecular targets for neuroprotection. Antioxid Redox
Signaling. 14:1505–1517. 2011. View Article : Google Scholar
|
|
40
|
Tong Y, Chuan J, Bai L, Shi J, Zhong L,
Duan X and Zhu Y: The protective effect of shikonin on renal
tubular epithelial cell injury induced by high glucose. Biomed
Pharmacother. 98:701–708. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiao X, Liu J, Hu J, Zhu X, Yang H, Wang C
and Zhang Y: Protective effects of protopine on hydrogen
peroxide-induced oxidative injury of PC12 cells via Ca(2+)
antagonism and antioxidant mechanisms. Eur J Pharmacol. 591:21–27.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mueller SG, Trabesinger AH, Boesiger P and
Wieser HG: Brain glutathione levels in patients with epilepsy
measured by in vivo (1)H-MRS. Neurology. 57:1422–1427. 2001.
|
|
43
|
Ghanta S, Banerjee A, Poddar A and
Chattopadhyay S: Oxidative DNA damage preventive activity and
antioxidant potential of Stevia rebaudiana (Bertoni) Bertoni, a
natural sweetener. J Agric Food Chem. 55:10962–10967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee IK, Kang KA, Zhang R, Kim BJ, Kang SS
and Hyun JW: Mitochondria protection of baicalein against oxidative
damage via induction of manganese superoxide dismutase. Environ
Toxicol Pharmacol. 31:233–241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen JX and Yan SD: Pathogenic role of
[correction of mitochondral] amyloid-beta peptide. Expert Rev
Neurother. 7:1517–1525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Iijima T, Mishima T, Tohyama M, Akagawa K
and Iwao Y: Mitochondrial membrane potential and intracellular ATP
content after transient experimental ischemia in the cultured
hippocampal neuron. Neurochem Int. 43:263–269. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Peng HY, Du JR, Zhang GY, Kuang X, Liu YX,
Qian ZM and Wang CY: Neuroprotective effect of Z-ligustilide
against permanent focal ischemic damage in rats. Biol Pharm Bull.
30:309–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tsujimoto Y: Role of Bcl-2 family proteins
in apoptosis: Apoptosomes or mitochondria? Genes Cells. 3:697–707.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tong Y, Bai L, Gong R, Chuan J, Duan X and
Zhu Y: Shikonin protects PC12 cells against β-amyloid
peptide-induced cell injury through antioxidant and antiapoptotic
activities. Sci Rep. 8:262018. View Article : Google Scholar : PubMed/NCBI
|