Introduction
Wolcott-Rallison syndrome (WRS) is a rare autosomal
recessive disorder characterized by early-onset or permanent
neonatal diabetes mellitus, skeletal dysplasia and growth
retardation, and other variable multisystemic clinical
manifestations, wherein the majority of patients succumb in infancy
or childhood (1–5). Long-term regular insulin therapy has
been demonstrated to improve survival rates (6,7). To
date, <100 cases of WRS have been reported according to the
literature. WRS is mostly reported in individuals from countries
where consanguineous families are common (8). It has previously been demonstrated that
mutations in the gene encoding eukaryotic translation initiation
factor 2α kinase 3 (EIF2AK3) are responsible for WRS (4,5). In the
present report, a novel mutation in EIF2AK3 (c.205G>T) was
reported in a patient with WRS. Notably, both parents of this
patient, who were from unrelated families, were heterozygous
carriers for this mutation.
Case report
The patient was a Chinese boy born at 39 weeks via
natural childbirth, with a birth weight of 3.2 kg. His parents were
nonconsanguineous. In February 2016, at 3-months of age, the
patient was diagnosed with neonatal diabetes mellitus at Wuhan
Children's Hospital (Hubei, China). In June 2016, at 7-months of
age, the patient was diagnosed with WRS and admitted to the
Department of Endocrinology, Zhengzhou Children's Hospital
(Zhengzhou, China). The patient presented with vomiting and a high
blood glucose level. Experimental treatment with Glibenclamide (1
mg/kg/day; Shanxi YunPeng Pharmaceutical Co., Ltd., Taiyuan, China)
was ineffective. The patient was discharged with a regimen of
premixed insulin Novolin® 30R (1.0–1.5 U/kg; Novo
Nordisk, Bagsværd, Denmark) three times daily.
At 7 months of age, the patient experienced fever of
unknown origin, anorexia and vomiting. Anti-infective therapy was
ineffective. Physical examinations revealed the following: Height,
65 cm; weight, 7.5 kg. No abnormal physical features, lethargy, or
cardiopulmonary abnormalities were observed. The patient's liver
was enlarged, extending ~2 cm below the rib cage. Blood glucose was
detected using a glucose meter and test strips (Johnson &
Johnson, New Brunswick, NJ, USA). The patient's blood glucose
levels fluctuated between 2.7–19.0 mmol/l (normal range, 3.9–6.1
mmol/l). Blood C-peptide levels were detected using an
electrochemical luminescence method (Roche Diagnostics, Basel,
Switzerland). The patient's Blood C-peptide levels were 0.07 ng/ml,
which was markedly lower than the normal range (1.1–4.4 ng/ml).
Islet cell antibody (ICA) was detected using the indirect
immunofluorescent method, while the glutamic acid decarboxylase
antibody (GAD) was detected using immunoradiometric methods,
according to the manufacturer's protocol (Roche Diagnostics). Both
ICA and GAD tests were negative. Renal and thyroid function tests
were normal. In addition, chest X-ray, cardiac enzyme levels and
electrocardiogram were normal. A liver function test revealed
elevated alanine transaminase (ALT) levels of 1,065.9 U/l (normal
range, 0–45 U/l) and aspartate transaminase (AST) levels of 1,887.7
U/l (normal range, 0.35 U/l). An abdominal ultrasound exam revealed
an enlarged liver.
The patient received an insulin aspart injection
(NovoLog®; Medtronic, Minneapolis, MN, USA) to control
blood glucose levels, delivering a large dose (1.0–2.0 units)
before meals, with a total of 6 U/day. In addition, the patient
also received aspartate ornithine and reduced glutathione injection
(150 mg/kg; Qirui Pharmaceutical Co., Ltd., Wuhan, China) to
protect liver function, and ceftazidime (300 mg/kg; Hailing
Chemical Pharmaceutical Co., Ltd., Hainan, China) twice a day as
anti-infection treatment.
Following treatment, the patient's blood glucose
fluctuated between 4–9 mmol/l before meals, to 8–13 mmol/l after
meals. Although the patient's body temperature and blood glucose
levels became steady, the patient's liver function was evaluated,
which revealed ALT levels of 6,677.2 U/l and AST levels of 8,976.5
U/l, with systemic edema due to hypoproteinemia. Ultimately, the
patient received hemodialysis and was discharged 15 days later. The
patient had regular follow-ups for six months, during which liver
function was normal.
During hospitalization, blood samples were collected
and DNA was extracted from the peripheral leukocytes of the patient
and his parents. Written informed consent was obtained from the
patient's parents prior to participation in the present case
report. The EF2AK3 gene was amplified by polymerase chain reaction,
followed by Sanger sequencing performed by Meiji Biomedical
Technology Co. Ltd., Shanghai, China. Briefly, blood samples (2 ml)
were collected and centrifuged at 13,400 × g for 2 min at room
temperature. Subsequently, total DNA was extracted from peripheral
leukocytes using the DNA extraction reagent (Tiangen Biotech Co.,
Ltd., Beijing, China), according to the manufacturer's protocol. To
examine the 17 exons of EIF2AK3, DNA was amplified using the
TIANamp blood DNA kit (Tiangen Biotech Co., Ltd.) using the
specific primer pairs listed in Table
I, according to the manufacturer's protocol. The following
thermocycling conditions were used for the PCR: Initial
denaturation at 94°C for 5 min; 38 cycles of 94°C for 30 sec, 62°C
for 30 sec and 72°C for 45 sec. Sequencing analysis revealed that
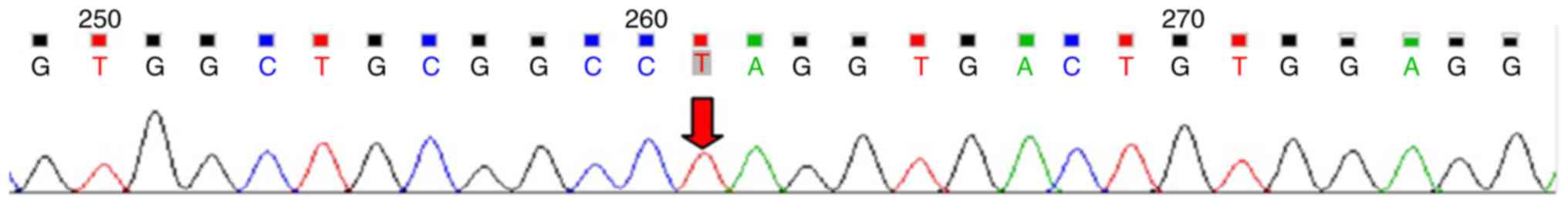
the patient was homozygous for a novel nonsense mutation,
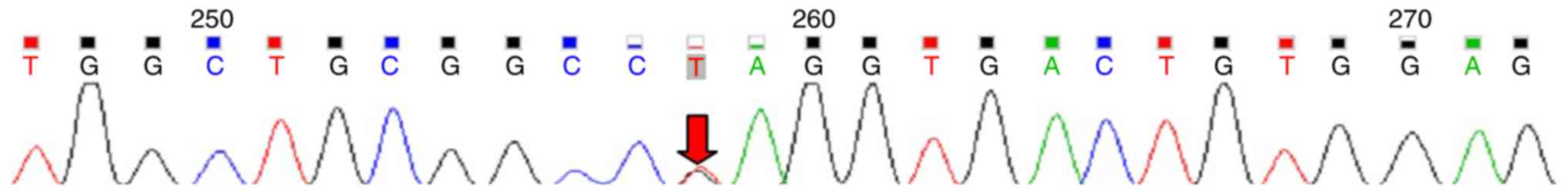
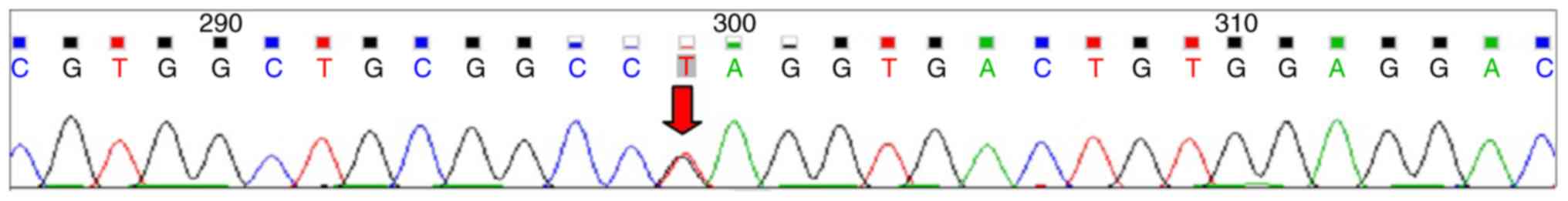
c.205G>T (P.Glu69Ter) in the EIF2AK3 gene (Fig. 1) and both parents were identified as
heterozygous carriers for the mutation (Figs. 2 and 3). To the best of our knowledge, this
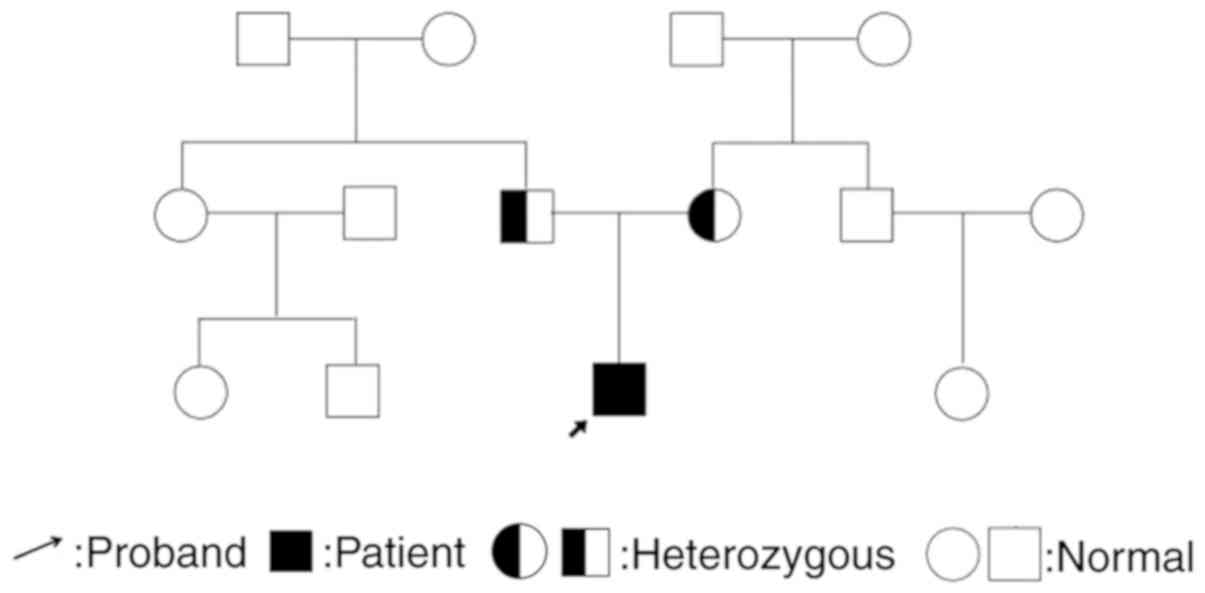
mutation has not been reported previously. Family genealogy of both
parents is presented in Fig. 4.
 | Table I.Primer pairs used for DNA
sequencing. |
Table I.
Primer pairs used for DNA
sequencing.
| Exon | Primer sequence
(5′-3′) |
|---|
|
1 | F: |
GAGAGGCAGGCGTCAGTG |
|
| R: |
CGCGCGTAAACAAGTTGC |
|
2 | F: |
TGAGCATGTGGGATAAGTCC |
|
| R: |
TGCCCTAAAGGGACACAAAC |
|
3 | F: |
TCAGGATCAAGACTCCAGCTC |
|
| R: |
TGACAACCTCAGGGGAAAAT |
|
4 | F: |
GGAGTTGGTAATCTAACTGATGC |
|
| R: |
CCAACAGCAACATTATCTGAA |
|
5 | F: |
GCCCTCTTGTGGCATAAATC |
|
| R: |
CTGGGAGAGGAAGAACCGTA |
|
6 | F: |
GCCCTCTTGTGGCATAAATC |
|
| R: |
GGCACTCCTGAAGTAGGAAGG |
|
7 | F: |
CCCTCCCTGTTTTTGTTGAA |
|
| R: |
GGGCAAAGACAGTCAGGATT |
|
8 | F: |
CTGGGCCATTTGTTTAACTT |
|
| R: |
TGAAATTGTCTCCCAAGATG |
|
9 | F: |
TAGTTAAAGACGGGCCTATT |
|
| R: |
CAAGAGTAGCTTTGGTGGAG |
| 10 | F: |
AAGACTGGAGGGATAGCAGT |
|
| R: |
AGATCTTAGGTCATTTCTTCTTTG |
| 11 | F: |
TGAACTGATTTTCACATTACCAC |
|
| R: |
AATTGGCAGCACTTAGAACC |
| 12 | F: |
GCCTTCAGGGTTGTCTTACT |
|
| R: |
CATTGTAATCACACAAGCAAA |
| 13 (1) | F: |
ACAGAGGGTGCAGTTCAGGT |
|
| R: |
CACAATGGTTGCCAATATCC |
| 13 (2) | F: |
AAGGTCAAGGGAGAGAACCT |
|
| R: |
ACCCTCTGCTCTCAGATGCTT |
| 14 | F: |
CATGCACACCCACTGTACTT |
|
| R: |
CTGGAACACTACTGCCAGTTT |
| 15 | F: |
CTTTGGGATTCAATAATGCT |
|
| R: |
CCAATCTGCTGGTATTAAGAA |
| 16 | F: |
TGTGGAATCTGTGGGATGTC |
|
| R: |
TGCTAAGGACCGCTTACGTT |
| 17 | F: |
TTTTGCCAGCACTGATTTTA |
|
| R: |
TTTCAAGTCTGCAATTTTGG |
Discussion
WRS is a rare autosomal recessive disease, which was
initially described by Wolcott and Rallison in 1972 (1). It is characterized by early-onset
diabetes mellitus, skeletal dysplasia and growth retardation. Other
symptoms include severe liver and renal dysfunction, and central
hypothyroidism (2–5). The majority of cases have been reported
in countries with a high incidence of marriage between first
cousins (9). Mutations in EIF2AK3,
which is located at chromosome 2p12, are responsible for this
disorder (4,5), and encodes for pancreatic endoplasmic
reticulum kinase (PERK) (7). PERK is
a major endoplasmic reticulum stress transducer in cells. Mutations
in EIF2AK3 disrupt the expression and/or function of PERK, which
blocks β cell development and impairs gluconeogenesis (6). This gene is highly expressed in the
nucleus of pancreatic β cells and bone tissues (10), which comprise the major sites used
for diagnosis of this disease. EIF2AK3 is expressed in the liver
and kidney at a lower level, which leads to liver and kidney
dysfunction, or even liver failure for patients with WRS (10). The prognosis of WRS is poor. Patients
typically succumb to hepatic failure and renal failure at an early
age (7). However, one patient was
previously reported to live until 35 years of age and died without
any symptoms (7).
According to previous studies, mutations of EIF2AK3
include nonsense, missense and framework drift mutations, among
other types (7,11). In the present case report, the
patient was homozygous for the c.205G>T (P.Glu69Ter) mutation of
the EIF2AK3 gene which was identified to be a novel nonsense
mutation. To the best of our knowledge, this mutation site has not
yet been reported in China (6,12). In
recent years, other novel mutations in EIF2AK3 have also been
reported (13–16). In the present case report, the
patient's parents were both heterozygous carriers for the same
mutation. Although WRS typically occurs in families with
consanguineous marriage (7,10), the present case report details a rare
case where the patient's parents are nonconsanguineous, but carried
the same mutation site.
The present patient presented permanent neonatal
diabetes mellitus and transient liver failure, but no central
hypothyroidism or kidney dysfunction, which demonstrates the
diversity of clinical manifestations of the disease. It may
therefore be hypothesized that this is due to variable expression
of the EIF2AK3 gene, other modified genes, environmental
factors and differences in disease management. The patient did not
undergo skeletal X-ray, so it could not be determined whether he
exhibited epiphyseal dysplasia.
In conclusion, the incidence of WRS is very low. In
patients with neonatal diabetes mellitus, particularly those with
multiple system manifestations, a diagnosis of WRS should be
considered. A detailed history and physical examination may also be
beneficial for diagnosis and treatment of WRS. Genetic testing, as
the gold standard in diagnosing this disease, may provide guidance
for prenatal diagnosis.
Acknowledgements
The authors would like to acknowledge and thank the
patient's family members for their voluntary participation, as well
as Professor Yiping Shen (Division of Genetics and Genomics, Boston
Children's Hospital, Boston, MA, USA) for guiding this case
report.
Funding
The present case report was supported by a grant
from the Henan Province Science and Technology Department,
Zhengzhou, China (grant no. 142102310139).
Availability of data and materials
Not applicable.
Authors' contributions
HW contributed to the conception and design. AH and
HYW performed the data analyses and prepared the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from the
patient's parents prior to participation in the present case
report.
Patient consent for publication
Written informed consent was obtained from the
patient's parents prior to participation in the present case
report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wolcott CD and Rallison ML: Infancy-onset
diabetes mellitus and multiple epiphyesal dysplasia. J Pediatr.
80:292–297. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bin-Abbas B, Shabid S, Hainu B and
Ai-Ashwel A: Wolcott-Rallison syndrome: Clinical, radiological and
histological findings in a Saudi child. Ann Saudi Med. 21:73–74.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brickwood S, Bonthron DT, AI-Gazali LI,
Piper K, Heam T, Wilson DI and Hanley NA: Wolcott-Rallison
syndrome: Pathogenic insights into neonatal diabetes from new
mutation and expression studies of EIF2AK3. J Med Genet.
40:685–689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iyer S, Korada M, Rainbow L, Kirk J, Brown
RM, Shaw N and Barrett TG: Wolcott-Rallison syndrome: A clinical
and genetic study of three children, novel mutation in EIF2AK3 and
review of the literature. Acta Pardiatr. 3:1195–1201. 2004.
|
|
5
|
Engelmann G, Meyburg J, Shahbek N, Al-Ali
M, Hairetis MH, Baker AJ, Rodenburg RJ, Wenning D, Flechtenmacher
C, Ellard S, et al: Recurrent acute liver failure and
mitochondriopathy in a case of Wolcott-Rallison syndrome. J Inherit
Metab Dis. 31:540–546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feng DR, Meng Y, Zhao SM, Shi HP, Wang WC
and Huang SZ: Two novel EIF2AK3 mutations in a Chinese boy with
Wolcott-Rallison syndrome. Zhonghua Er Ke Za Zhi. 49:301–305.
2011.(In Chinese). PubMed/NCBI
|
|
7
|
Senée V, Vattem KM, Delépine M, Rainbow
LA, Haton C, Lecoq A, Shaw NJ, Robert JJ, Rooman R, Diatloff-Zito
C, et al: Wolcott-Rallison Syndrome: Clinical, genetic, and
functional study of EIF2AK3 mutations and suggestion of genetic
heterogeneity. Diabetes. 53:1876–1883. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rubio-Cabezas O, Patch AM, Minton JA,
Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan
CR, Jefferson IG, et al: Wolcott-Rallison syndrome is the most
common genetic cause of permanent neonatal diabetes in
consanguineous families. J Clin Endocrinol Metab. 94:4162–4170.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ozbek MN, Senée V, Aydemir S, Kotan LD,
Mungan NO, Yuksel B, Julier C and Topaloglu AK: Wolcott-Rallison
syndrome due to the same mutation (W522X) in EIF2AK3 in two
unrelated families and review of the literature. Pediatr Diabetes.
11:279–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tzakis AG, Nunnelley MJ, Tekin A, Buccini
LD, Garcia J, Uchida K, Neville HL, Nares MA, Ruiz P and Bodamer O:
Liver, pancreas and kidney transplantation for the treatment of
Wolcott-Rallison syndrome. Am J Transplant. 15:565–567. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delépine M, Nicolino M, Barrett T,
Golamaully M, Lathrop GM and Julier C: EIF2AK3, encoding
translation initiation factor 2-alpha kinase 3, is mutated in
patients with Wolcott-Rallison syndrome. Nat Genet. 25:406–409.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sang Y, Liu M, Yang W, Yan J, Chengzhu and
Ni G: A novel EIF2AK3 mutation leading to Wolcott-Rallison syndrome
in a Chinese child. J Pediatr Endocrinol Metab. 24:181–184. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gürbüz F, Yüksel B and Topaloğlu AK:
Wolcott-rallison syndrome with novel EIF2AK3 gene mutation. J Clin
Res Pediatr Endocrinol. 8:496–497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Al-Sinani S, Al-Yaarubi S, Sharef SW,
Al-Murshedi F and Al-Maamari W: Novel mutation in wolcott-rallison
syndrome with variable expression in two omani siblings. Oman Med
J. 30:138–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Franco E, Flanagan SE, Houghton JA,
Lango Allen H, Mackay DJ, Temple IK, Ellard S and Hattersley AT:
The effect of early, comprehensive genomic testing on clinical care
in neonatal diabetes: An international cohort study. Lancet.
386:957–963. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Al-Aama JY, Al-Zahrani HS, Jelani M, Sabir
HS, Al-Saeedi SA and Ahmed S: Novel splice site mutation in EIF2AK3
gene causes Wolcott-Rallison syndrome in a consanguineous family
from Saudi Arabia. Congenit Anom (Kyoto). 58:39–40. 2018.
View Article : Google Scholar : PubMed/NCBI
|