Introduction
The Food and Drug Administration (FDA) approved
propylthiouracil (PTU) for the treatment of Graves' disease in 1947
(1). In nearly 70 years of clinical
application, reports of PTU-associated liver injury and failure,
and even fatality, have accumulated for adult and pediatric
patients (2–6). A warning regarding the potential risk
of severe hepatic injury associated with PTU was issued by the FDA
in 2009 (7). Therefore, it is
recommended that patients receiving PTU therapy have their liver
function closely monitored. PTU-induced liver injury primarily
manifests as differing degrees of hepatocyte necrosis (8); however, the underlying mechanisms are
largely unknown.
Gap junctions (GJs) directly connect the cytoplasm
of adjacent cells, mediating the intercellular transmission of
signaling molecules. Six transmembrane connexin (Cx) monomers are
arranged in a circle to form a hemichannel, and then two
hemichannels from neighboring plasma membranes are docked to form
the GJ (9,10). Cx expression is distinct in a variety
of tissues, and Cx32 is the major GJ protein in hepatocytes
(11,12).
GJ-mediated intercellular communication (GJIC) is
involved in a number of physiological and pathological processes
(13–15). Previous reports have suggested a role
for GJ channels in drug-induced liver injury (DILI) (16–18).
Downregulation of GJs composed of Cx32 (Cx32-GJs) could reduce the
hepatotoxicity of acetaminophen, D-galactosamine and carbon
tetrachloride (19,20). Likewise, propofol protects rat liver
cells from sevoflurane-induced cytotoxicity through inhibiting GJ
channels (21). Based on this
evidence, the inhibition of hepatic Cx32-GJs could prove to be an
effective strategy for controlling DILI. However, whether this
GJ-mediated hepatoprotection is effective against PTU toxicity, and
the potential underlying mechanism of this, remain unknown. In the
present study, the role and underlying mechanisms of GJs in
PTU-induced toxicity were explored in BRL-3A cells.
Materials and methods
Materials
PTU, carbenoxolone (CBX), anti-GAPDH and secondary
antibodies for western blotting were obtained from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). Anti-Cx32 antibody was obtained
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Cell culture
reagents, Lipofectamine 2000 and calcein acetoxymethyl ester
(Calcein-AM) were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). The Cell Counting kit-8 (CCK-8) was obtained
from Dojindo (Mashikimachi, Kumamoto, Japan). The
2′,7′-dichlorofluorescin diacetate (DCFH-DA) was from Beyotime
Institute of Biotechnology (Haimen, China). All other reagents and
chemicals were obtained from Sigma-Aldrich; Merck KGaA, unless
otherwise stated.
Cell culture
The BRL-3A rat liver cell line was purchased from
the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
Cells were cultivated in Dulbecco's modified Eagle's medium
supplemented with 10% fetal bovine serum and 100 U/ml
penicillin-streptomycin at 37°C in an atmosphere containing 5%
CO2.
CCK-8 assay
Direct toxicity was determined using a CCK-8 kit
according to the manufacturer's instructions. First, BRL-3A cells
were subjected to 0.6 and 0.8 mg/ml PTU for 24 h at 37°C, after
which they were incubated with 10% (v/v) CCK-8 reagent at 37°C for
3 h. The absorbance was read using a microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA) at a wavelength of 450 nm.
The cell viability was normalized against that of the vehicle
control.
A standard colony-formation assay
A standard colony-formation assay was used for
detecting the cytotoxicity of PTU to BRL-3A cells (22). Briefly, following exposure to PTU at
0.6 and 0.8 mg/ml for 12 h, cells were rinsed with
phosphate-buffered saline (PBS), harvested with trypsin, diluted
and seeded into 6-well plates at a density of 500 cells/well. Cells
were subsequently stained with 4% crystal violet at room
temperature 5–7 days later. Colonies consisting of ≥50 cells were
counted. The surviving fraction was evaluated by normalizing to the
colony-forming efficiency of the vehicle-treated cells.
Small interfering (si)RNA
transfection
Cx32 expression was inhibited by siRNA transfection
in BRL-3A cells. The siRNA sequences targeted against the rat Cx32
gene were as described previously (23,24):
siRNA-1, 5′-CACCAACAACACATAGAAA-3′; siRNA-2,
5′-GCATCTGCATTATCCTCAA-3′; and siRNA-3, 5′-GCCTCTCACCTGAATACAA-3′.
Cx32 siRNAs (50 nM) or the negative control siRNA (NC siRNA) were
transiently transfected into BRL-3A cells using Lipofectamine 2000,
according to the manufacturer's instructions. After 48 h
incubation, western blotting and a ‘parachute’ assay, as described
below, were performed to confirm Cx32 expression knockdown and GJIC
inhibition.
Western blotting
The procedure of western blotting was performed as
described previously (25). In
brief, cell lysates were obtained in lysis buffer (P0013; Beyotime
Institute of Biotechnology), sonicated and centrifuged at 14,167 ×
g at 4°C for 30 min. Protein concentration was determined by BCA
assay. Cell lysates (20 µg per lane) were separated using SDS-PAGE
in 10% Tris-glycine gels and transferred to nitrocellulose
membranes. Following blocking with 5% skimmed dry milk in Tris
buffered saline with Tween-20 (0.05% Tween-20) at room temperature
for 1 h, the membranes were incubated with specific antibodies
against Cx32 (sc-59948; dilution, 1:1,000) and GAPDH (G8795;
dilution, 1:2,000) overnight at 4°C. Secondary antibodies (goat
anti-mouse IgG-peroxidase conjugated; A4416; dilution, 1:4,000)
were then added at room temperature for 1 h. The immunopositive
protein bands were visualized using an Amersham Enhanced
Chemiluminescence Detection kit (GE Healthcare Life Sciences,
Little Chalfont, UK) and the intensities were detected by the
Quantity One software (Bio-Rad, version 4.6.2).
‘Parachute’ dye-coupling assay
A ‘parachute’ dye-coupling assay was performed to
evaluate GJ function as previously described (26). Cells were seeded into 12-well plates
and cultured to 80–90% confluency. Donor cells from one well were
labeled with 5 µM Calcein-AM and trypsinized, diluted and seeded
onto receiver cells at a ratio of 1:150 (donor/receiver). GJs
formed between donor cells and receiver cells during a 4-h
incubation at 37°C and were monitored using an Olympus IX71
fluorescence microscope (Olympus Corporation, Tokyo, Japan). Under
each experimental condition (treatment of CBX or (si)RNA
transfection), the average number of receiver cells containing
calcein dye per donor cell was counted and normalized to that of
the vehicle control.
Observation of cell morphology
Cell morphology was observed using a FEI Quanta-400
scanning electron microscope (SEM; Thermo Fisher Scientific, Inc.).
Sterile slides were placed in 12-well plates and served as
substrates. Cells were treated with vehicle control or PTU at 0.6
and 0.8 mg/ml for 24 h, rinsed with PBS and fixed in 4%
paraformaldehyde for 1 h at room temperature. Following gradient
dehydration in a series of ethanol from 30–100%, the samples were
dried in a vacuum freeze-drying apparatus for 1 h and were treated
with gold sputtering for SEM observation.
Detection of PTU concentration in
BRL-3A cells
Reversed-phase high-performance liquid
chromatography was adopted to measure the PTU content in BRL-3A
cells (27). Briefly, following
incubation with PTU at 0.6 and 0.8 mg/ml for 24 h, cells were
washed with PBS three times, harvested by trypsinization,
resuspended in PBS and counted. Five freeze-thaw cycles were used
to lyse the cells. Methanol (HPLC-grade) was added for protein
precipitation. Following centrifugation at 14,167 × g for 10 min at
4°C, the supernatant was collected and injected into a Shimadzu
LC-20AD system (Shimadzu Corporation, Kyoto, Japan). The
chromatographic conditions used were as follows: Application of a
Luna C18 column (250×4.6 mm; 5mm; Phenomenex, Torrance,
CA, USA), methanol and water (40:60) was used as the mobile phase
at a flow rate of 1.0 ml/min, which was detected at a wavelength of
272 nm with a column temperature of 30°C. The concentrations of PTU
in the cell samples were calculated using a calibration curve
method.
Analysis of intracellular reactive
oxygen species (ROS) levels
Following exposure to PTU at 0.6 and 0.8 mg/ml for 6
h, BRL-3A cells were labeled with DCFH-DA, which is hydrolyzed by
esterase into DCFH without fluorescence. Intracellular ROS can
oxidize non-fluorescent DCFH to fluorescent DCF (28). Formation of DCF was determined using
a Perkin LS55 fluorescence spectrophotometer (PerkinElmer Inc.,
Waltham, MA, USA) with an excitation wavelength of 488 nm and an
emission wavelength of 525 nm. Fluorescence intensity was
normalized to that of the vehicle control and was regarded as a
measure of the intracellular ROS level. The DCF images were
captured via fluorescence microscopy (Olympus IX71; Olympus
Corporation).
Statistical analysis
The data were presented as the mean ± standard error
and were analyzed using IBM SPSS Statistics 19.0 (Armonk, NY, USA).
Statistical analysis was performed by Student's t-test or one-way
analysis of variance followed by Dunnett's test. In all cases,
P<0.05 was considered to indicate a statistically significant
difference.
Results
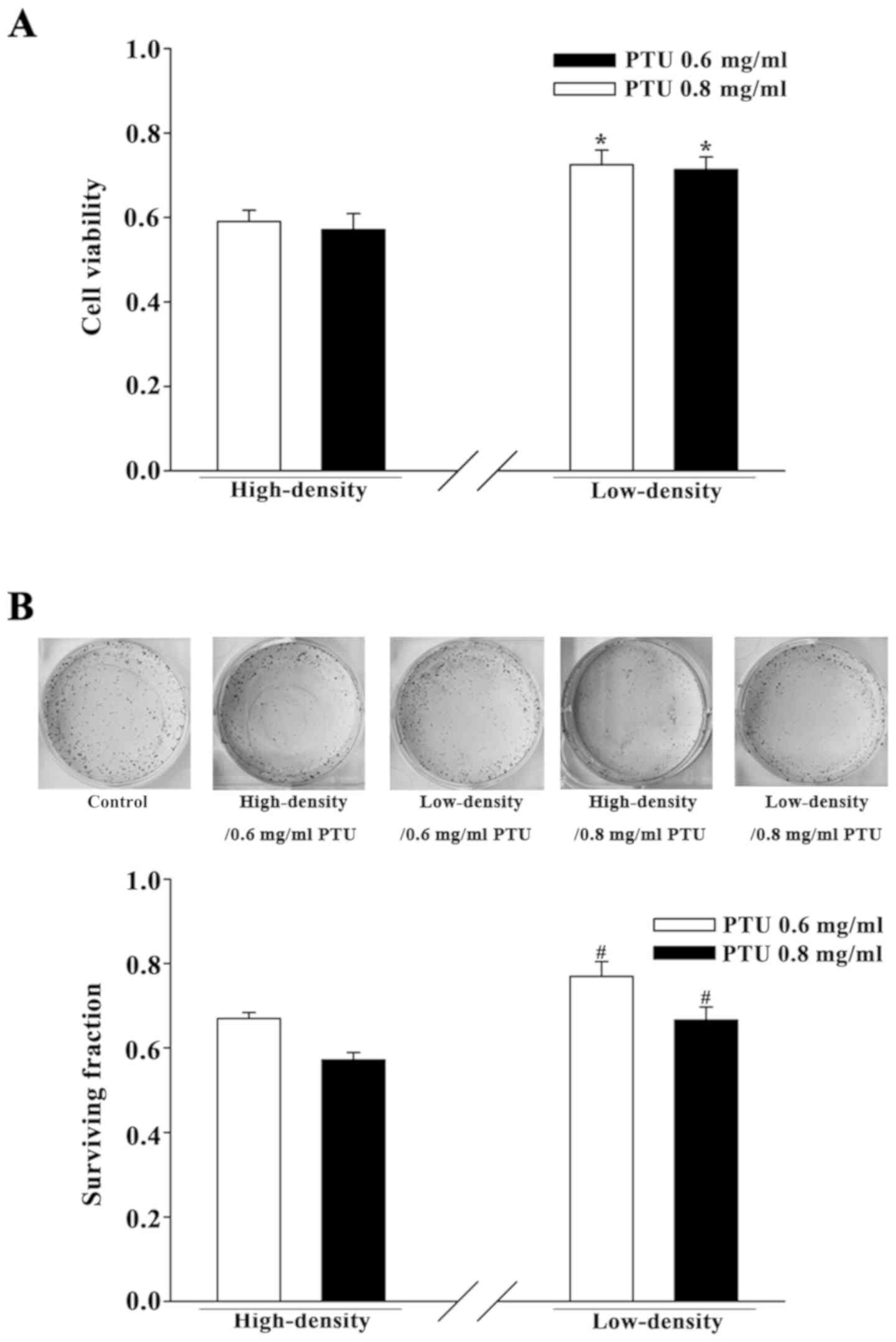
Cell density influences the cytotoxic
effect of PTU in BRL-3A cells
BRL-3A cells were cultured separately under two
cell-density conditions for initial investigation of the influence
of GJIC on PTU toxicity. At a high cell density (2×104
cells per cm2), GJs formed efficiently as the cells had
frequent contact with one another. At a low cell density
(5×103 cells per cm2), the wide dispersion of
cells infrequently permitted the formation of GJs. A preliminary
cell viability assay for concentrations screening was performed
with PTU from 0.3 to 0.8 mg/ml (data not shown). The concentrations
of 0.6 and 0.8 mg/ml PTU were chosen in the present study.
PTU-induced cytotoxicity was assessed using a CCK-8 assay and a
standard colony-formation assay. Fig.
1 illustrates that PTU treatment, at concentrations of 0.6 and
0.8 mg/ml, decreased the cell survival of BRL-3A cells under both
culture conditions. However, the survival was significantly greater
in the low-density condition compared with the high-density
condition. Thus, the toxicity of PTU in BRL-3A cells was reduced
under low cell-density conditions wherein there was a deficiency in
GJIC.
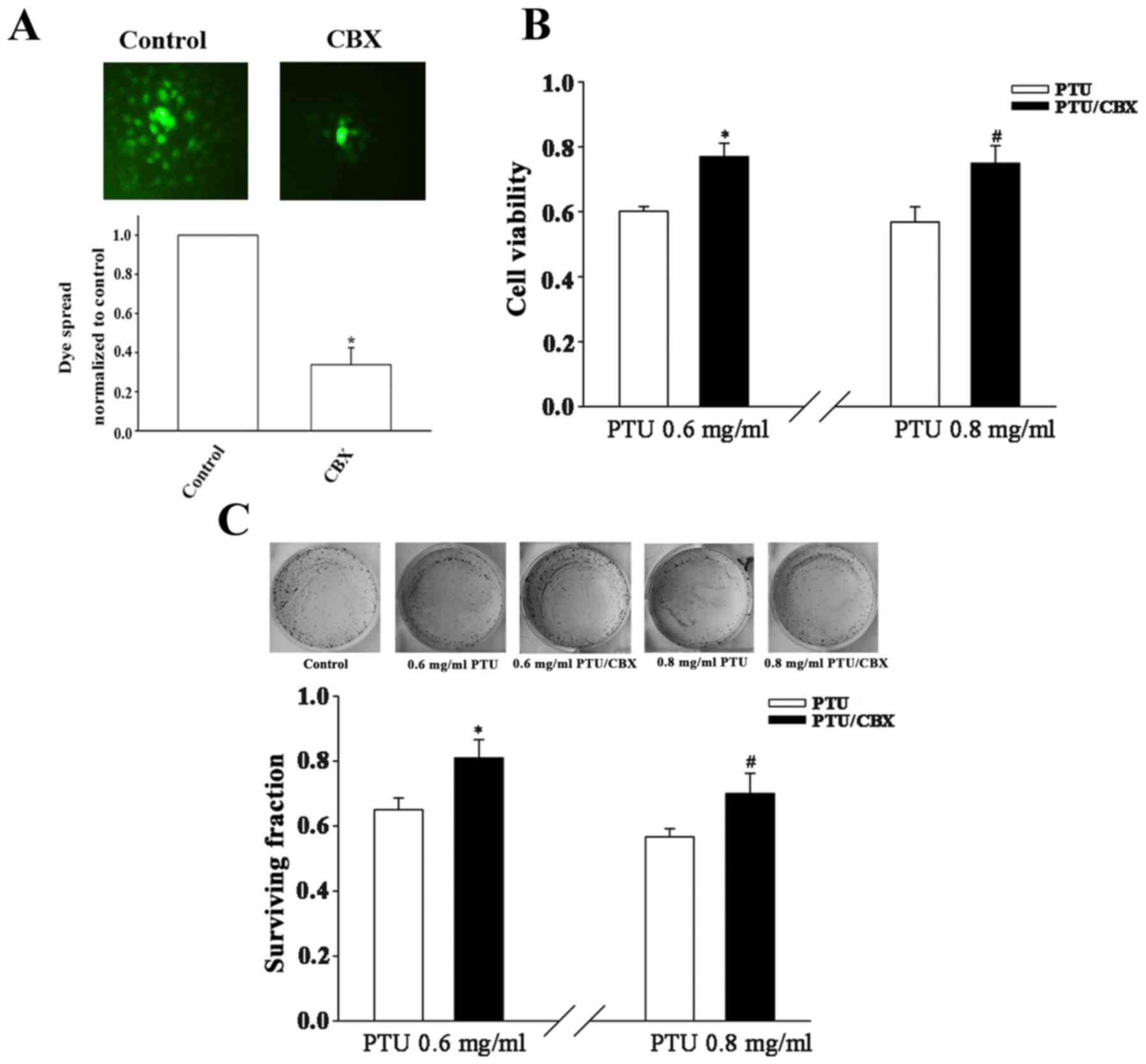
Pharmacological inhibition of GJ
function decreases PTU cytotoxicity in BRL-3A cells
To investigate whether the cell density-dependence
of PTU toxicity in BRL-3A cells is associated with GJIC, an
extensive GJ inhibitor, CBX (29),
was adopted to manipulate GJ function. BRL-3A cells were pretreated
with the CBX prior to exposure to PTU. In a subsequent ‘parachute’
assay, dye-coupling was significantly blocked by CBX pretreatment
(Fig. 2A). Under high cell-density
culture conditions, the cell survival was significantly increased
following pretreatment with 100 µM CBX for 1 h at PTU
concentrations of 0.6 and 0.8 mg/ml, as compared with PTU alone
(Fig. 2B and C). These results
indicate that the inhibition of GJs contributes to the decreased
toxicity of PTU at high BRL-3A cell densities.
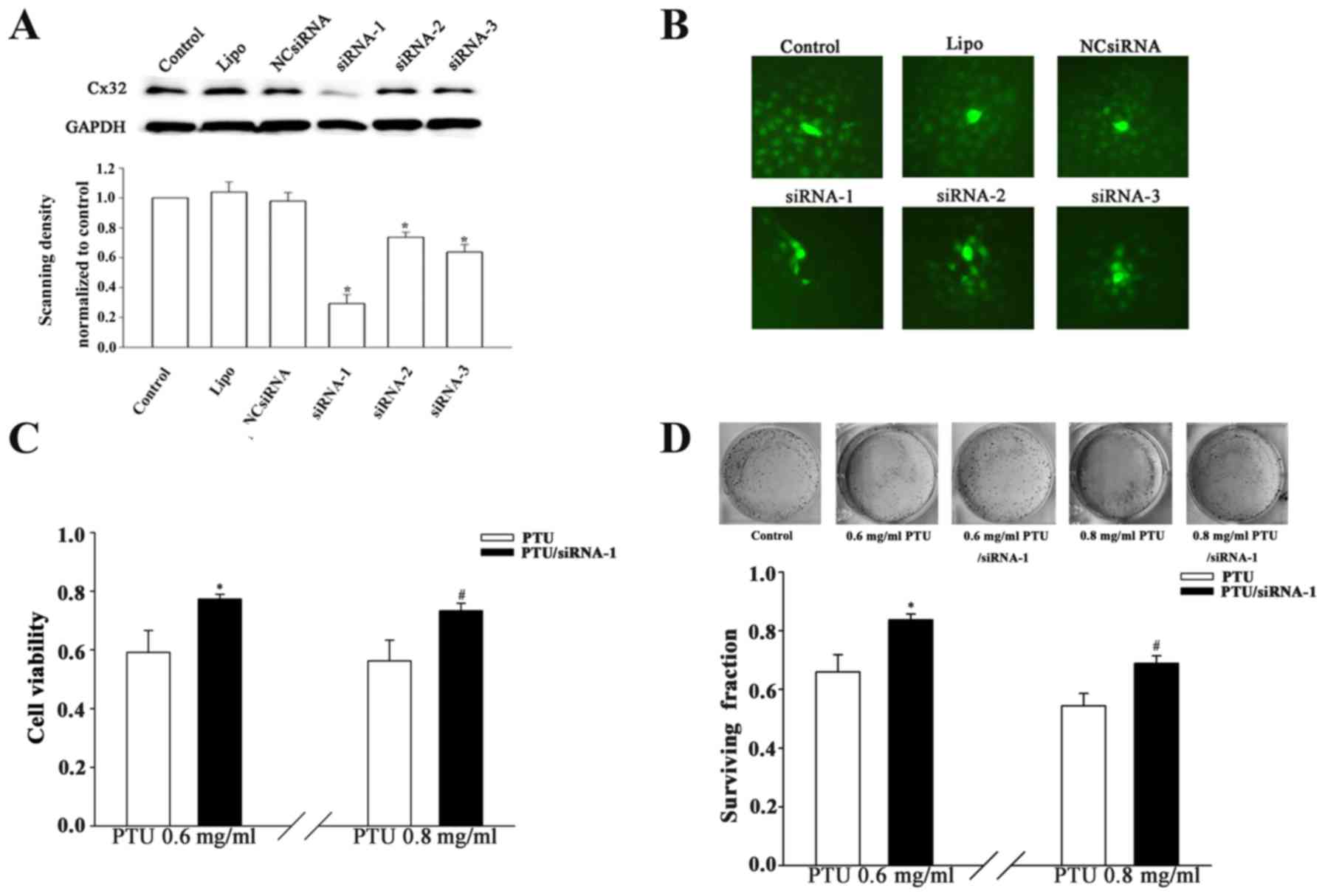
Suppression of Cx32-GJ function by
Cx32 knockdown attenuates PTU cytotoxicity
Cx32 is a key GJ constituent protein in liver cells
(12). Specific knockdown of the
Cx32 gene was conducted to confirm the effect of Cx32-GJ on PTU
cytotoxicity. As indicated in Fig. 3A
and B, the downregulation of Cx32 expression significantly
suppressed the spread of calcein dye through GJs in
siRNA-transfected cells. Furthermore, Cx32-knockdown (siRNA-1
transfection) increased the cell viability by factors of 1.31 and
1.29 in the presence of 0.6 and 0.8 mg/ml PTU, respectively, at
high cell densities (Fig. 3C).
Likewise, clonogenic capacity was improved at these two
concentrations when Cx32-GJs were inhibited (Fig. 3D). The results indicate that Cx32-GJ
serves an important role in the PTU-induced cytotoxicity of BRL-3A
cells.
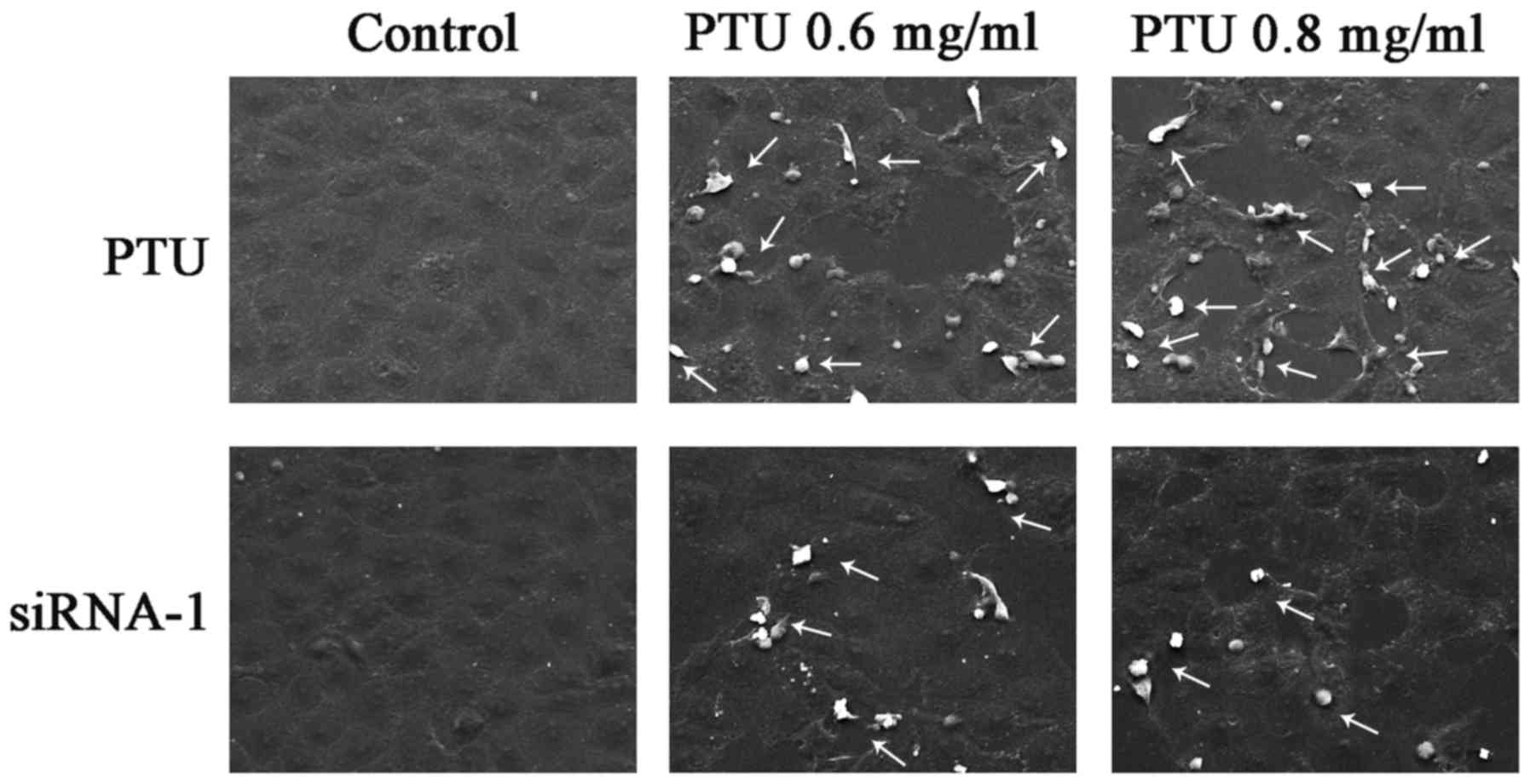
Cx32-GJs influence cell morphology and
necrosis
PTU has been demonstrated to induce necrosis in
liver injury (8). To assess the
necrosis of BRL-3A cells subjected to PTU in the absence of
Cx32-GJ, SEM images were captured. Fig.
4 illustrates the typical morphological characteristics of cell
necrosis, including cell swelling and spillovers of cellular
content (as indicated by the arrows), which were clearly noted
following PTU treatment, but were less obvious following
siRNA-mediated Cx32-GJ inhibition. These results suggest that the
suppression Cx32-GJ attenuates PTU hepatotoxicity, which is partly
associated with a decrease in PTU-induced necrosis.
Inhibition of Cx32-GJ decreases
intracellular PTU accumulation
GJs facilitate direct intercellular communication
between neighboring cells. PTU, as a small molecule, could be
transmitted through GJs. To assess whether the PTU concentration in
BRL-3A cells could be reduced by blocking Cx32-GJ function, the
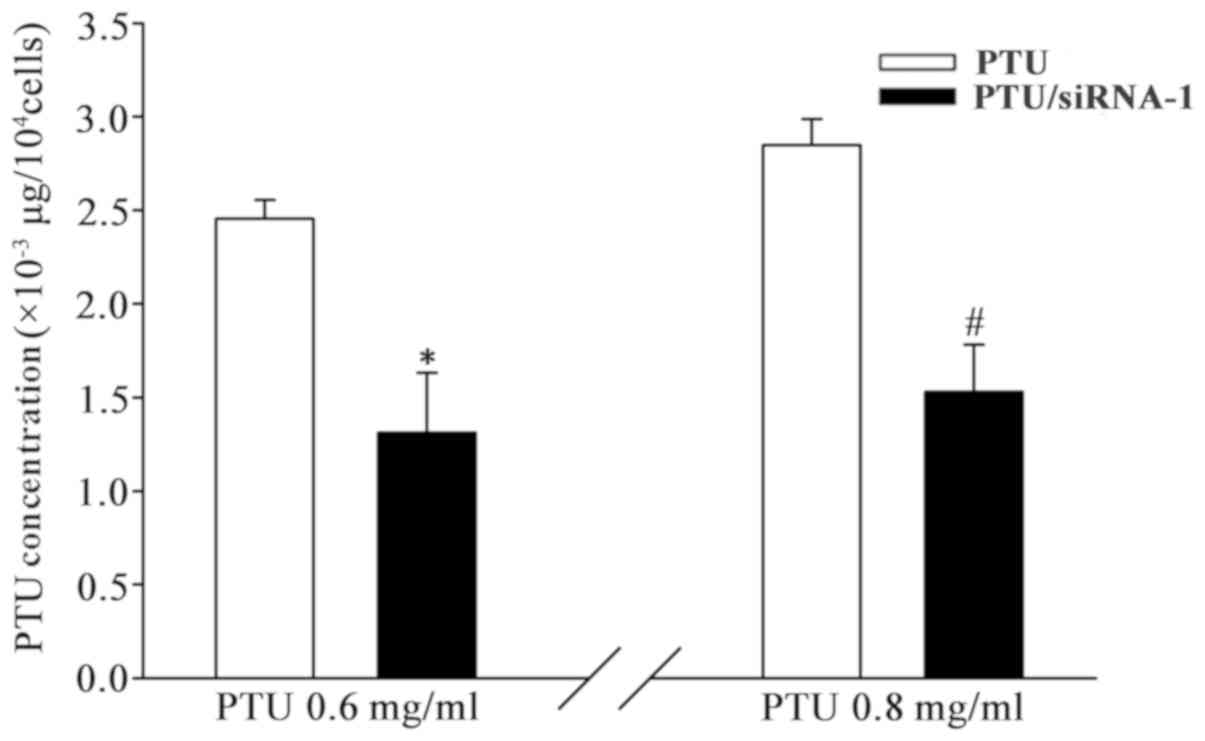
intracellular PTU content was determined. As indicated in Fig. 5, the level of cellular PTU [PTU (µg)
per 104 cells] significantly decreased when cells were
transfected with Cx32 siRNA-1. The reduction of PTU uptake in
BRL-3A cells between those with and without Cx32-GJ were
significant at 0.6 and 0.8 mg/ml PTU. These results demonstrate
that PTU accumulation is reduced when Cx32-GJ is downregulated.
Inhibition of Cx32-GJ prevents ROS
transmission
To investigate the possibility of ROS transmission
through GJs, intracellular ROS levels in the presence and absence
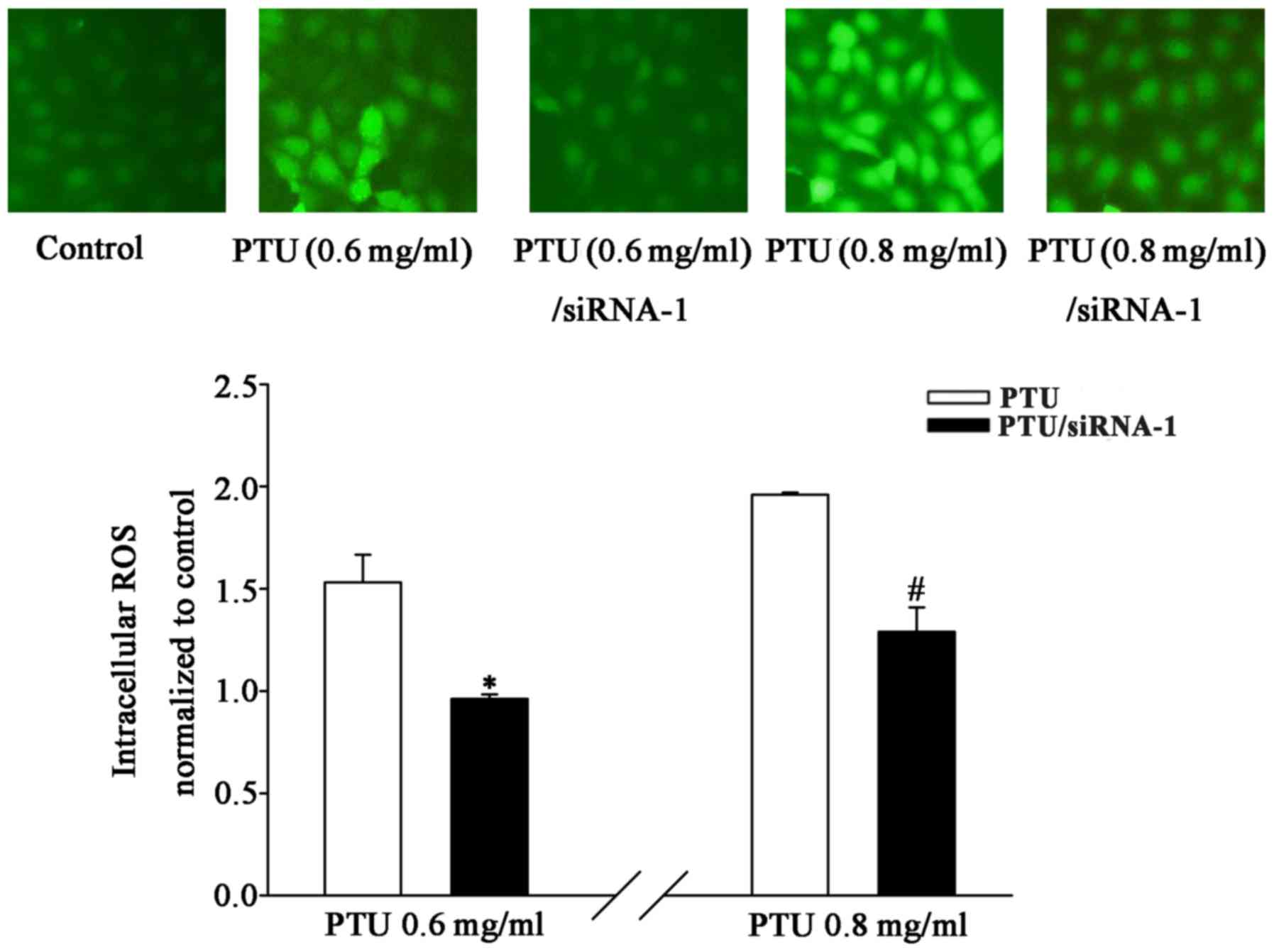
of Cx32-GJ were detected. The results revealed that PTU-induced
toxicity was accompanied by an increase in ROS production at
concentrations ≤0.8 mg/ml. Fig. 6
illustrates that Cx32 siRNA-1 transfection markedly suppressed the
PTU-induced increase in DCF fluorescence, which was quantified by
fluorescence spectrophotometry. The levels of intracellular ROS
were significantly reduced in the Cx32-knockdown group at PTU
concentrations of 0.6 and 0.8 mg/ml. These findings suggest that
the attenuation of PTU-induced toxicity by Cx32-GJ downregulation
may be associated with the inhibition of ROS transmission.
Discussion
In the present study, it was demonstrated that
PTU-induced toxicity depends on GJs in BRL-3A cells. The
investigation indicated that the inhibition of GJs could protect
against PTU-induced liver cell damage in vitro. Furthermore,
PTU-induced necrosis was decreased following specific blocking of
Cx32-GJs. Results from the current study illustrated that
inhibition of GJ has a protective effect in PTU-induced liver
toxicity and that GJ-attenuated cytotoxicity is associated, in
part, with the reduction of necrosis.
Recent studies have demonstrated that the inhibition
of hepatic Cx32-GJs can attenuate the liver toxicity associated
with various types of drugs, suggesting a promising new therapeutic
target for DILI (19,20). An ‘injury signal’ responsible for
inducing apoptosis or necrosis diffuses through GJ channels to
cause damage or death in the adjacent cells, which is known as the
‘bystander effect’. This effect has been observed in herpes simplex
virus thymidine kinase and in the radiotherapy of cancer (30–32). In
DILI, blocking the propagation of ‘injury signals’ among cells
through Cx32-GJ may be key to this hepatoprotective effect. The
‘injury signals’ mediated by GJs have been widely investigated,
particularly in carcinoma cells (25,33).
However, their intrinsic properties have not been identified. The
relevant signals are considered to be the drugs or their toxic
metabolites, calcium, inositol 1,4,5-triphosphate (IP3),
or other signaling molecules triggering cell damage (34–37).
There are ~21 Cx isoforms in mammals, which assemble
into junction channels with selective permeability to molecules
(9). For example, Cx32-GJs exhibit
higher permeability to adenosine, cAMP and IP3 than do
Cx43-GJs, while ATP is conducted through Cx43-GJs more effectively
than through Cx32-GJs (38–40). In the present work, inhibition of GJ
function using extensive GJ blocker (CBX) and Cx32 siRNAs decreases
PTU cytotoxicity, in which Cx32-GJ have an essential role on this
hepatoprotective effect. Therefore, in the present study, the
possible ‘injury signals’ transmitted through Cx32-GJs should have
the following characteristics: i) The ability to permeate through
Cx32 channels; and ii) the ability to influence the cytotoxicity of
PTU. It was considered that PTU and ROS are the potential ‘injury
signals’ involved in Cx32-GJ-mediated PTU toxicity.
The molecular weight of PTU is 170.2 Da, which is
less than the limit for passage through GJ channels (1–2 kDa);
therefore, it could be transferred via GJs by passive diffusion.
Furthermore, PTU could markedly suppress glutathione
S-transferase (GST) activity to interfere with the
antioxidation of glutathione (41).
Therefore, the present study was performed to investigate the
possibility that PTU is a GJ-permeable ‘injury signal’. The results
indicated that PTU uptake was decreased in cells deficient in
functional Cx32 channels. This decline may not only be conducive to
the restoration of GST activity; it may also weaken allergic
responses caused by PTU acting as a hapten.
Oxidative stress was observed in hypothyroid rats
induced by administration of drinking water containing 0.05% PTU
for 8 weeks (42,43). Oxidative stress is an important
pathophysiological basis of liver injury, in which ROS is the
essential mediator; it induces lipid peroxidation of the cellular
and mitochondrial membranes, causing changes in permeability and
affecting signal transduction pathways (e.g., ROS/Protein kinase C,
ROS/c-Jun N-terminal kinase (44–46).
Previous studies identified that ROS can spread through Cx32
channels, acting as toxic molecules (19,47). The
experimental results in the present study suggested that ROS
induced by PTU-exposure were regulated by Cx32-GJs. Rat hepatocytes
deficient in Cx32 could prevent the passage of ROS between cells.
Thus, ROS could be PTU-induced ‘injury signals’ with selective Cx32
channel permeability.
Immortalized BRL-3A cells endogenously expressing
Cx32 and Cx43 is a simple model for investigating the role of GJ in
PTU-induced hepatotoxicity in vitro. However, the
limitations of using only one rat liver cell line are the
differences in species and primary hepatocytes (such as confounding
factors arising from the complicated hemodynamic system), the
effects of GJs on the PTU-induced cytotoxicity in human
hepatocytes, and in vivo, remain to be explored.
In conclusion, the role of GJs in PTU-induced
cytotoxicity was investigated in BRL-3A cells. The results
indicated that the suppression of Cx32-GJ channels protected
against PTU-cytotoxicity through reducing necrosis. Furthermore,
PTU and ROS appeared to affect the Cx32-GJ-mediated
hepatoprotective effect, acting as ‘injury signals’.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81400619), the
Scientific Research Project of Education Department of Guangdong
Province (grant no. 2017KTSCX079), the Funding for College
Students' Science and Technology Innovation and Cultivation of
Guangdong Province (grant no. pdjhb0220).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
NT designed the current study. NT, ZC, HC, LC and BC
performed the experiments. NT, ZC and BL analyzed the data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Landau RL: Landmark perspective: Treatment
of hyperthyroidism. JAMA. 251:1747–1748. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Glinoer D and Cooper DS: The
propylthiouracil dilemma. Curr Opin Endocrinol Diabetes Obes.
19:402–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malozowski S and Chiesa A:
Propylthiouracil-induced hepatotoxicity and death. Hopefully, never
more. J Clin Endocrinol Metab. 95:3161–3163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang J, Li LF, Xu Q, Zhang J, Weng WW, Zhu
YJ and Dong MJ: Analysis of 90 cases of antithyroid drug-induced
severe hepatotoxicity over 13 years in China. Thyroid. 25:278–283.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang MT, Lee WJ, Huang TY, Chu CL and
Hsieh CH: Antithyroid drug-related hepatotoxicity in
hyperthyroidism patients: A population-based cohort study. Br J
Clin Pharmacol. 78:619–629. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu DB, Chen EQ, Bai L and Tang H:
Propylthiouracil-induced liver failure and artificial liver support
systems: A case report and review of the literature. Ther Clin Risk
Manag. 13:65–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bahn RS, Burch HS, Cooper DS, Garber JR,
Greenlee CM, Klein IL, Laurberg P, McDougall IR, Rivkees SA, Ross
D, et al: The role of propylthiouracil in the management of graves'
disease in adults: Report of a meeting jointly sponsored by the
american thyroid association and the food and drug administration.
Thyroid. 19:673–674. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Williams KV, Nayak S, Becker D, Reyes J
and Burmeister LA: Fifty years of experience with
propylthiouracil-associated hepatotoxicity: What have we learned? J
Clin Endocrinol Metab. 82:1727–1733. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maeda S and Tsukihara T: Structure of the
gap junction channel and its implications for its biological
functions. Cell Mol Life Sci. 68:1115–1129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sosinsky GE and Nicholson BJ: Structural
organization of gap junction channels. Biochim Biophys Acta.
1711:99–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harris AL: Emerging issues of connexin
channels: Biophysics fills the gap. Q Rev Biophys. 34:325–472.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koffler LD, Fernstrom MJ, Akiyama TE,
Gonzalez FJ and Ruch RJ: Positive regulation of connexin32
transcription by hepatocyte nuclear factor-1alpha. Arch Biochem
Biophys. 407:160–167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zoidl G and Dermietzel R: Gap junctions in
inherited human disease. Pflugers Arch. 460:451–466. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scott DA, Kraft ML, Stone EM, Sheffield VC
and Smith RJ: Connexin mutations and hearing loss. Nature.
391:321998. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Belousov AB, Fontes JD, Freitas-Andrade M
and Naus CC: Gap junctions and hemichannels: Communicating cell
death in neurodevelopment and disease. BMC Cell Biol. 18 (Suppl
1):42017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park WJ, Park JW, Erez-Roman R,
Kogot-Levin A, Bame JR, Tirosh B, Saada A, Merrill AH Jr,
Pewzner-Jung Y and Futerman AH: Protection of a ceramide synthase 2
null mouse from drug-induced liver injury: Role of gap junction
dysfunction and connexin 32 mislocalization. J Biol Chem.
288:30904–30916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maurel M and Rosenbaum J: Closing the gap
on drug-induced liver injury. Hepatology. 56:781–783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naiki-Ito A, Asamoto M, Naiki T, Ogawa K,
Takahashi S, Sato S and Shirai T: Gap junction dysfunction reduces
acetaminophen hepatotoxicity with impact on apoptotic signaling and
connexin 43 protein induction in rat. Toxicol Pathol. 38:280–286.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patel SJ, Milwid JM, King KR, Bohr S,
Iracheta-Vellve A, Li M, Vitalo A, Parekkadan B, Jindal R and
Yarmush ML: Gap junction inhibition prevents drug-induced liver
toxicity and fulminant hepatic failure. Nat Biotechnol. 30:179–183.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Asamoto M, Hokaiwado N, Murasaki T and
Shirai T: Connexin 32 dominant-negative mutant transgenic rats are
resistant to hepatic damage by chemicals. Hepatology. 40:205–210.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang F, Li S, Gan X, Wang R and Chen Z:
Propofol inhibits gap junctions by attenuating sevoflurane-induced
cytotoxicity against rat liver cells in vitro. Eur J Anaesthesiol.
31:219–224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Q, You T, Yuan D, Han X, Hong X, He
B, Wang L, Tong X, Tao L and Harris AL: Cisplatin and oxaliplatin
inhibit gap junctional communication by direct action and by
reduction of connexin expression, thereby counteracting cytotoxic
efficacy. J Pharmacol Exp Ther. 333:903–911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang N, Liu J, Chen B, Zhang Y, Yu M, Cai
Z and Chen H: Effects of gap junction intercellular communication
on the docetaxel-induced cytotoxicity in rat hepatocytes. Mol Med
Rep. 15:2689–2694. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang R, Huang F, Chen Z and Li S:
Downregulation of connexin 32 attenuates hypoxia/reoxygenation
injury in liver cells. J Biochem Mol Toxicol. 29:189–197. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong X, Wang Q, Yang Y, Zheng S, Tong X,
Zhang S, Tao L and Harris AL: Gap junctions propagate opposite
effects in normal and tumor testicular cells in response to
cisplatin. Cancer Lett. 317:165–171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Tao L, Fan L, Peng Y, Yang K,
Zhao Y, Song Q and Wang Q: Different gap junction-propagated
effects on cisplatin transfer result in opposite responses to
cisplatin in normal cells versus tumor cells. Sci Rep. 5:125632015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang N, Chen H, Cai Z, Zhan P, Pan T,
Zhang Y and Wu X: Development and validation of RP-HPLC method for
determination of propylthiouracil in hepatic cells. Curr Pharm
Anal. 14:219–222. 2018. View Article : Google Scholar
|
|
28
|
Tian YY, An LJ, Jiang L, Duan YL, Chen J
and Jiang B: Catalpol protects dopaminergic neurons from
LPS-induced neurotoxicity in mesencephalic neuron-glia cultures.
Life Sci. 80:193–199. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Davidson JS and Baumgarten IM:
Glycyrrhetinic acid derivatives: A novel class of inhibitors of
gap-junctional intercellular communication. Structure-activity
relationships. J Pharmacol Exp Ther. 246:1104–1107. 1988.PubMed/NCBI
|
|
30
|
Vrionis FD, Wu JK, Qi P, Waltzman M,
Cherington V and Spray DC: The bystander effect exerted by tumor
cells expressing the herpes simplex virus thymidine kinase (HSVtk)
gene is dependent on connexin expression and cell communication via
gap junctions. Gene Ther. 4:577–585. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hamada N, Matsumoto H, Hara T and
Kobayashi Y: Intercellular and intracellular signaling pathways
mediating ionizing radiation-induced bystander effects. J Radiat
Res. 48:87–95. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prise KM and O'Sullivan JM:
Radiation-induced bystander signalling in cancer therapy. Nat Rev
Cancer. 9:351–360. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krutovskikh VA, Piccoli C and Yamasaki H:
Gap junction intercellular communication propagates cell death in
cancerous cells. Oncogene. 21:1989–1999. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krysko DV, Leybaert L, Vandenabeele P and
D'Herde K: Gap junctions and the propagation of cell survival and
cell death signals. Apoptosis. 10:459–469. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin JH, Weigel H, Cotrina ML, Liu S, Bueno
E, Hansen AJ, Hansen TW, Goldman S and Nedergaard M:
Gap-junction-mediated propagation and amplification of cell injury.
Nat Neurosci. 1:7431998. View
Article : Google Scholar
|
|
36
|
Decrock E, Krysko DV, Vinken M, Kaczmarek
A, Crispino G, Bol M, Wang N, De Bock M, De Vuyst E, Naus CC, et
al: Transfer of IP3 through gap junctions is critical, but not
sufficient, for the spread of apoptosis. Cell Death Differ.
19:947–957. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Decrock E, Vinken M, Bol M, D'Herde K,
Rogiers V, Vandenabeele P, Krysko DV, Bultynck G and Leybaert L:
Calcium and connexin-based intercellular communication, a deadly
catch? Cell Calcium. 50:310–321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Niessen H, Harz H, Bedner P, Krämer K and
Willecke K: Selective permeability of different connexin channels
to the second messenger inositol 1,4,5-trisphosphate. J Cell Sci.
113:1365–1372. 2000.PubMed/NCBI
|
|
39
|
Goldberg GS, Moreno AP and Lampe PD: Gap
junctions between cells expressing connexin 43 or 32 show inverse
permselectivity to adenosine and ATP. J Biol Chem. 277:36725–36730.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bevans CG, Kordel M, Rhee SK and Harris
AL: Isoform composition of connexin channels determines selectivity
among second messengers and uncharged molecules. J Biol Chem.
273:2808–2816. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kariya K, Sawahata T, Okuno S and Lee E:
Inhibition of hepatic glutathione transferases by propylthiouracil
and its metabolites. Biochem Pharmacol. 35:1475–1479. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ye J, Zhong X, Du Y, Cai C and Pan T: Role
of levothyroxine and vitamin E supplementation in the treatment of
oxidative stress-induced injury and apoptosis of myocardial cells
in hypothyroid rats. J Endocrinol Invest. 40:713–719. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tas S, Dirican M, Sarandöl E and Serdar Z:
The effect of taurine supplementation on oxidative stress in
experimental hypothyroidism. Cell Biochem Funct. 24:153–158. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schattenberg JM, Wang Y, Rigoli RM, Koop
DR and Czaja MJ: CYP2E1 overexpression alters hepatocyte death from
menadione and fatty acids by activation of ERK1/2 signaling.
Hepatology. 39:444–455. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zheng L, Wang C, Luo T, Lu B, Ma H, Zhou
Z, Zhu D, Chi G, Ge P and Luo Y: JNK activation contributes to
oxidative stress-induced parthanatos in glioma cells via increase
of intracellular ROS production. Mol Neurobiol. 54:3492–3505. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Muriel P: Role of free radicals in liver
diseases. Hepatol Int. 3:526–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Luo C, Yuan D, Li X, Yao W, Luo G, Chi X,
Li H, Irwin MG, Xia Z and Hei Z: Propofol attenuated acute kidney
injury after orthotopic liver transplantation via inhibiting gap
junction composed of connexin 32. Anesthesiology. 122:72–86. 2015.
View Article : Google Scholar : PubMed/NCBI
|