Introduction
Lung ischemia-reperfusion injury (LIRI) is
characterized by non-specific alveolar damage, lung edema,
pulmonary hypertension and hypoxemia (1), and has been implicated in several
clinical conditions including cardiopulmonary resuscitation, lung
transplantation and pulmonary embolism (2–4). LIRI
has a relatively complicated pathological mechanism, which serves
an important role in apoptosis. Previous studies have revealed that
cell apoptosis is associated with an increased risk of LIRI,
indicating that anti-apoptosis may be a potential target to treat
lung ischemic disease (5,6).
Heat shock protein 70 (HSP70) is highly conserved
and is activated by stress stimulation (7). The main function of HSP70 is to
participate in the regulation of intracellular protein folding and
to protect cells from stress damage (8,9). HSP70
has been demonstrated to affect anti-apoptotic functions (10). A previous study have revealed that
the overexpression of HSP70 effectively protects alveolar
epithelial cells against apoptosis and antagonizes the effects of
LIRI (11). Furthermore, the
overexpression of HSP70 also prevents proteins from misfolding and
aggregating (12,13). The overexpression of HSP70 also
attenuates ischemia-reperfusion injury (IRI) induced by lung
transplantation (14). In addition,
several studies have reported that the overexpression of HSP70
reduces heart (15) and brain
(16) IRI. However, to the best of
our knowledge, the mechanism that underlies the protective effect
of HSP70 on LIRI has not yet been elucidated.
The B-cell lymphoma (Bcl)-2 family of proteins is
comprised of pro-apoptotic proteins, including BH3 interacting
domain death agonist and Bcl-2-associated X (Bax), and
anti-apoptotic proteins including Bcl-2 and Bcl-extra large.
Jayanthi et al (17)
demonstrated that Bcl-2 and Bax regulates apoptosis and that
apoptosis is dependent on the ratio of Bcl-2/Bax; the lower the
Bcl-2/Bax ratio, the more severe the level of apoptosis.
The caspase family are enzymatic proteins that serve
key roles in the signal transduction of apoptosis (18). One of the family members, caspase-3,
is the core effector and main executor of apoptosis (19) that is positively correlated with
apoptosis. Mitogen-activated protein kinase (MAPK)/extracellular
signal-regulated kinase (ERK) and phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT) are important pathways for
maintaining cell survival (20,21). The
two pathways are activated during lung ischemia (22,23),
indicating that they may be involved in the pathological process of
pulmonary ischemia.
The present study assessed the protective effect of
HSP70 on LIRI as well as its underlying mechanism. Levels of Bcl-2,
Bax, caspase-3 and lactate dehydrogenase (LDH), and cell cycle and
apoptotic rate were analyzed to assess the protective effects of
HSP70. In addition, the expression of phosphorylated (p)-ERK and
p-AKT were determined. Finally, the involvement of the MAPK/ERK and
PI3K/AKT signaling pathways were evaluated using their respective
inhibitors.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were purchased from Hyclone (GE Healthcare Life
Sciences, Logan, UT, USA). The anti-HSP70 antibody was purchased
from Abcam (Cambridge, UK). The anti-Caspase-3, anti-Bcl-2,
anti-Bax, anti-ERK, anti-p-ERK, anti-AKT, anti-p-AKT, anti-GAPDH
and anti-β-Actin antibodies, as well as LY294002 (the PI3K/AKT
inhibitor) and U0126 (the MAPK/ERK inhibitor) were all purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture
A549 cells (Type Culture Collection of the Chinese
Academy of Sciences, Shanghai, China) were cultured and passaged in
DMEM containing 10% FBS at 37°C in a humidified atmosphere with 5%
CO2. Cells were divided into three groups: The
recombinant lentiviral infection group (lentivirus mediated
knockdown of the HSP70 gene), the lentivirus control group (empty
lentiviral vector) and the non-infection group.
Infection
A549 cells were transfected with a lentiviral
overexpression vector (1×107 TU/ml; Shanghai GeneChem
Co., Ltd., Shanghai, China) that expressed HSP70 and green
fluorescent protein (GFP). The lentiviral vector containing an
shRNA sequence (5′-GGACGAGTTTGAGCACAAG-3′), which targeted HSP70 or
the lentiviral vector backbone according to the manufacturer's
protocol. Briefly, 4×104 cells/well were seeded into
24-well plate and incubated at 37°C in 5% CO2 overnight.
According to the pretesting, 20 µl lentivirus liquid, 80 µl
Enhanced Infection Solution, 50 µg/ml polybrene (both Shanghai
GeneChem Co., Ltd.) and 800 µl DMEM were added directly onto the
cell monolayer. After incubating at 37°C for 12 h, the normal DMEM
was added. After 48 h of lentivirus transfection, GFP-transfected
cells and total cells were counted by two researchers independently
under a fluorescence microscope (each researcher randomly selected
5–7 fields of vision at a magnification of ×400 and counted more
than 700 cells). The average infection rate was calculated as
follows: Infection rate=green fluorescent cells/total cells × 100%.
Stable cells were screened by puromycin (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for culture and passaged.
Hypoxia/reoxygenation cell model
A549 cells were cultured in DMEM without glucose or
serum and placed in a hypoxic environment at 37°C for 2, 4, 6, 8
and 10 h. Hypoxic conditions were established using an Anaeropack
(Mitsubishi Gas Chemical Company, Inc., Tokyo, Japan), which
contained sodium ascorbate as the principal ingredient to absorb
oxygen and generate the same volume of carbon dioxide via oxidative
degradation in a sealed container (24). Subsequently, glucose-free DMEM
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) was replaced with normal DMEM and the cells were cultured in
a reoxygenated environment with 21% O2 and 5%
CO2 for 24 h at 37°C. U0126 or LY294002 were dissolved
in dimethyl sulfoxide (DMSO). The stock solutions were prepared
with minimal exposure to air and light, and stored at −20°C for 1 h
prior to hypoxia/reoxygenation treatment. Then 10 µM U0126 or 50 µM
LY294002 from the stock solutions were added to the uninfected A549
cells (the U0126 group) before hypoxia/reoxygenation. DMSO was
added to the non-infection, lentivirus control and lentiviral
infection groups.
Cell viability assay
A549 cells were seeded at a density of
8×103 in 96-well plates and incubated at 37°C for 24 h.
Subsequently, cells were exposed to hypoxic conditions for 2, 4, 6,
8 and 10 h and reoxygenized for a further 24 h. Finally, 10 µl of
Cell Counting kit-8 (CCK-8; Wuhan Boster Biological Technology,
Ltd., Wuhan, China) solution was added to each well according to
the manufactures protocol and incubated at 37°C for 1 h. The
absorbance of each well was then measured at 450 nm using a
microplate reader.
Apoptosis analysis
Following 6 h of hypoxia and 24 h of reoxygenation,
the three groups of cells were collected with 0.25% trypsin. An
Annexin V-allophycocyanin/propidium iodide (PI) staining kit
(eBioscience; Thermo Fisher Scientific, Inc.) was utilized for the
detection of apoptosis according to the manufacturer's protocol.
Following washing with PBS and binding buffer, the cells were
resuspended in Annexin V binding buffer (1×106
cells/ml). Subsequently, 5 µl of fluorochrome-conjugated Annexin V
was added to 100 µl of cell suspension and incubated for 10–15 min
at room temperature. Cells were then further washed with binding
buffer and resuspended in 200 µl of binding buffer.
A total of 5 µl of the PI staining solution was
added to the cell suspension, which was then analyzed using a
FACStar Plus flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA). Data analysis was performed with BD Accuri C6 software (BD
Biosciences).
Cell cycle analysis
A PI Flow Cytometry kit (Abcam) was used to
determine cell cycle status with a flow cytometer. The three groups
of cells were cultured under hypoxic conditions for 6 h and then
reoxygenated for 24 h prior to being harvested into a single cell
suspension. According to the manufacturer's protocol, the cells
were fixed in 66% ethanol at 4°C for 2 h, rehydration in PBS and
stained with PI + RNase from the PI Flow Cytometry kit at 4°C for
30 min. Finally, the fluorescence intensity of PI was measured at
488 nM on FL2 using a flow cytometer. Data analysis was performed
with FlowJo VX10 software (FlowJo LLC, Ashland, OR, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) detection
Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) and reverse transcribed into cDNA
with PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan).
Briefly, 1 µg total RNA of each sample was used for each reaction
in a 20 µl reaction mixture. After removing genomic DNA using gDNA
Eraser (Takara Bio, Inc.), cDNA was synthesized at 37°C for 15 min.
qPCR was performed using cDNA as a template with the FastStart
Essential DNA Green Master kit (Roche Diagnostics, Basel,
Switzerland) and β-actin as a reference. The reaction conditions
included an initial pre-denaturation at 95°C for 10 min, followed
by 45 cycles of 95°C for 15 sec, 60°C for 20 sec and 72°C for 15
sec. After the termination of the PCR, the production was analyzed
by the LightCycler96 System (Roche Diagnostics, GmbH, Mannheim,
Germany) automatically. The primers used were as follows: β-actin
forward, 5′-GATGAGATTGGCATGGCTTT-3′ and reverse,
5′-CACCTTCACCGTTCCAGTTT-3′; HSP70 forward,
5′-CAAGAAGAAGGTGCTGGACA-3′ and reverse, 5′-GTACAGTCCGCTGATGATGG-3′.
The relative level of HSP70 mRNA was calculated using the
2−ΔΔCq method (25).
Western blot analysis
Following hypoxia-reoxygenation treatment, the cells
in each group were collected. Cells were lysed with RIPA lysis
buffer containing phenylmethylsulfonyl fluoride (both Beyotime
Institute of Biotechnology, Shanghai, China) within 20 min. The
concentration was determined with BCA Protein Assay kit (Beijing
Transgen Biotech Co., Ltd., Beijing, China) and adjusted to a
concentration of 40 µg per well. The protein sample and loading
buffer were mixed in proportion and denatured at 95°C for 5 min,
and then subjected to SDS-PAGE (10% stacking gel and 5% separating
gel) with the conditions, 80 V for 40 min and 110 V for 80 min.
Following electrophoresis, the proteins were transferred to a
polyvinylidene difluroide membrane. The membrane was then blocked
for 2 h with 5% skim milk solution at room temperature, washed
three times with TBST solution and incubated with the following
primary antibodies overnight at 4°C: Anti-HSP70 (cat. no. 47455;
1:1,000), anti-AKT (cat. no. 4691; 1:1,000), anti-p-AKT (cat. no.
4060; 1:2,000), anti-ERK (cat. no. 4695; 1:1,000), anti-p-ERK (cat.
no. 4370; 1:2,000), anti-caspase-3 (cat. no. 9662; 1:1,000),
anti-Bcl-2 (cat. no. 4223; 1:1,000), anti-Bax (cat. no. 5023;
1:1,000), anti-GADPH (cat. no. 5174; 1:1,000) and anti-β-actin
(cat. no. 4970; 1:1,000). Samples were then incubated with
horseradish peroxidase-conjugated rabbit anti-mouse (cat. no. 6728;
1:2,000; Abcam) and goat anti-rabbit (cat. no. 7074; 1:2,000; Cell
Signaling Technology, Inc.) immunoglobulin G secondary antibodies
for 2 h at room temperature. Developing in darkroom with West Pico
ECL Kit (Millipore, Billerica, MA, USA). Quantity One 4.4 software
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used to analyze
the integrated optical density of each band.
LDH assay
The LDH assay kit (cat. no C0017) was purchased from
Beyotime Institute of Biotechnology. All measurements were
collected in accordance with the manufacturer's protocol.
Statistical analysis
Data were analyzed using SPSS 23.0 statistical
software (IBM Corp., Armonk, NY, USA). All results were presented
as the mean ± standard deviation. One-way analysis of variance
followed by a Least Significant Difference test was performed to
compare differences between the means in more than two groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Lentiviral transfection inhibits the
expression of HSP70
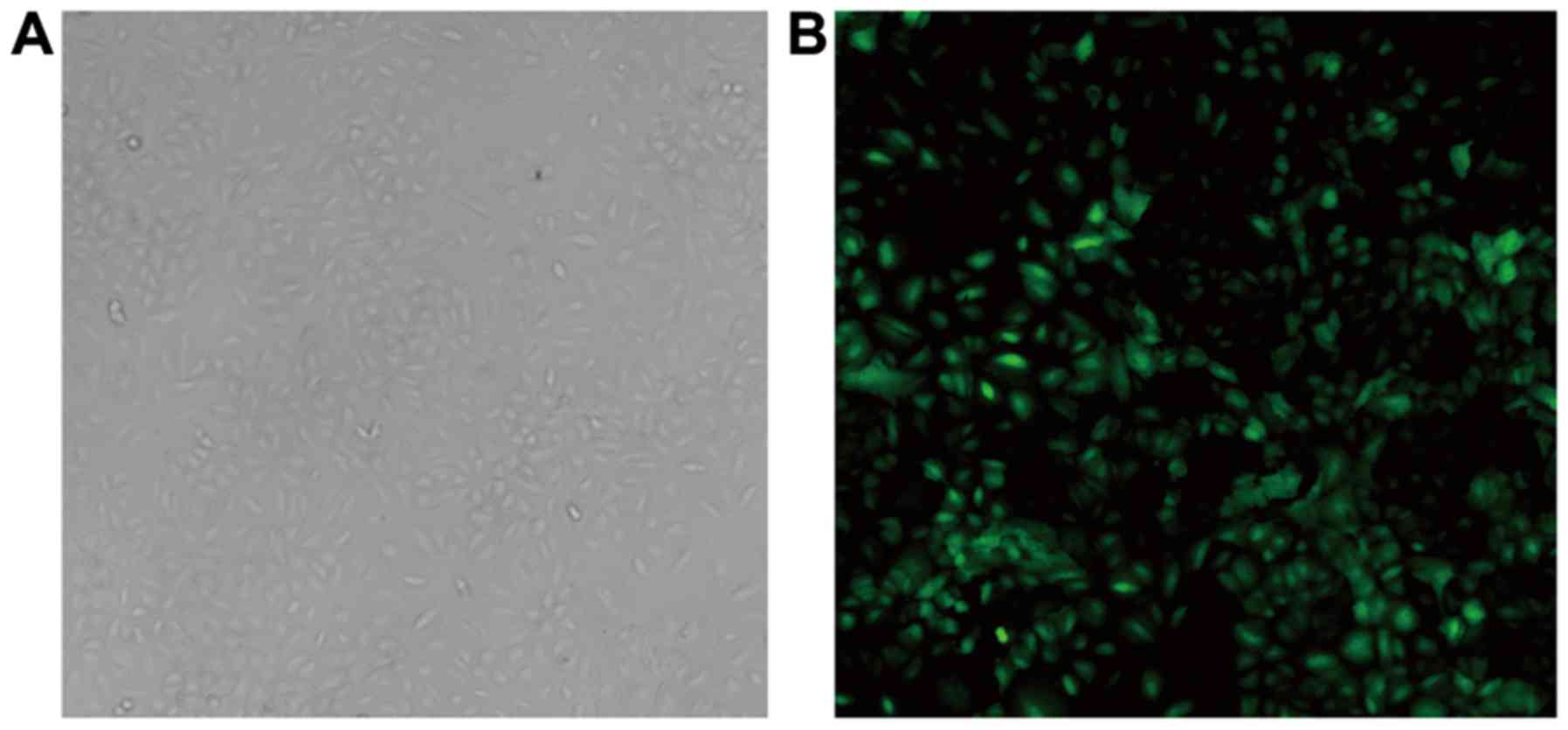
At 48 h post-transfection, A549 cells appeared to be
polygon-shaped, plump and interconnected under the fluorescence
microscope (Fig. 1). The infection
rate of A549 cells, was ~80%. Compared with the lentivirus control
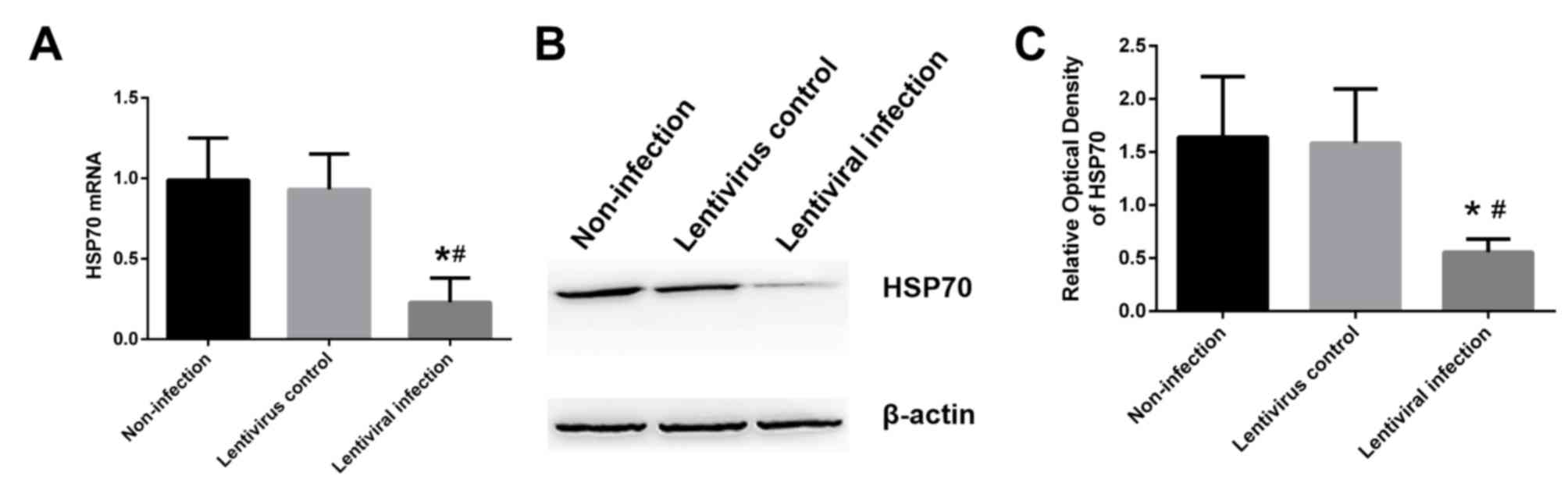
group and the non-infection group, a significant decrease was
observed in the mRNA (Fig. 2A) and
protein (Fig. 2B and C) levels of
HSP70 in the lentivirus infection group (P<0.05).
HSP70 silencing decreases A549 cell
viability
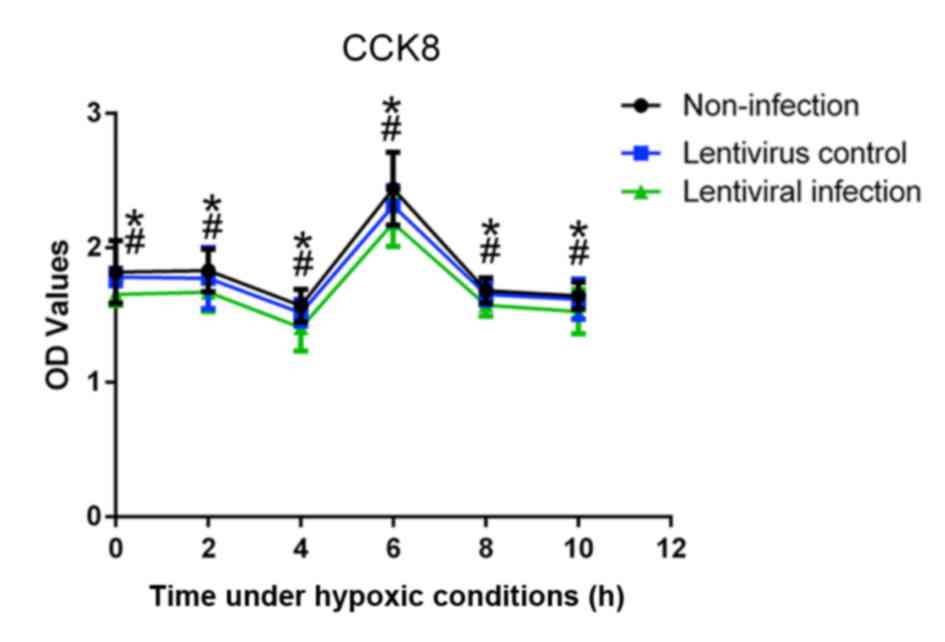
In order to select the effective duration of
hypoxia, the cell viability under hypoxic conditions was measured.
The results of the CCK-8 assay revealed that, at 6 h, A549 cells
had the highest marked viability compared with other time points
(Fig. 3). In addition, the viability
of the lentivirus infection group was significantly lower than that
of the lentivirus control and non-infection groups at all time
points (P<0.05).
HSP70 silencing increases the number
of apoptotic A549 cells following hypoxia and reoxygenation
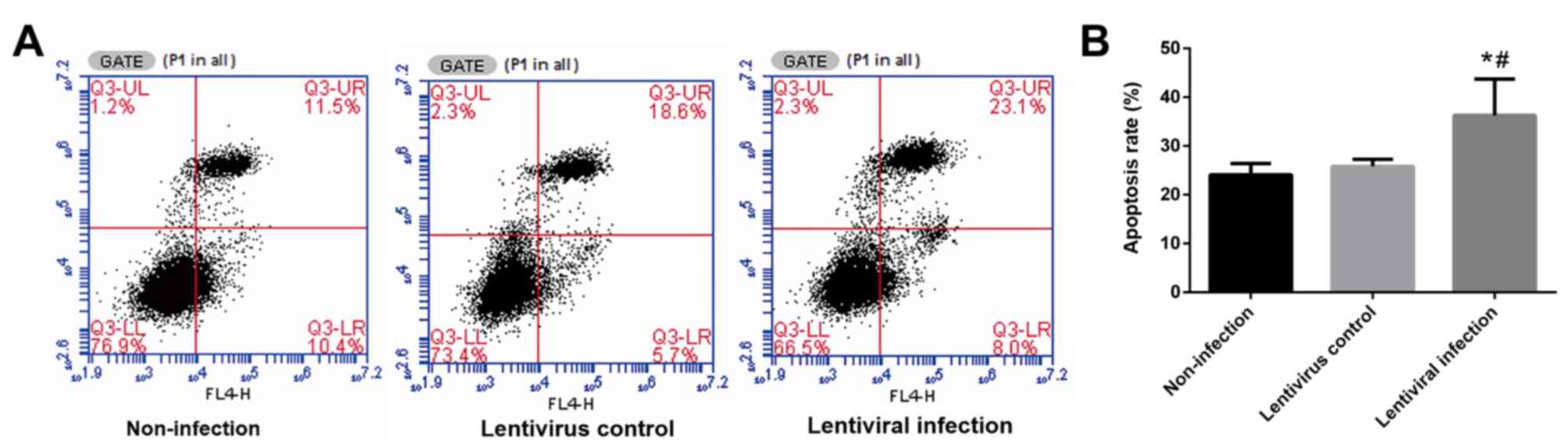
Following 6 h of hypoxia and 24 h of reoxygenation,
the rate of apoptosis in the lentiviral infection group was
significantly higher when compared with the other two groups
(P<0.05; Fig. 4).
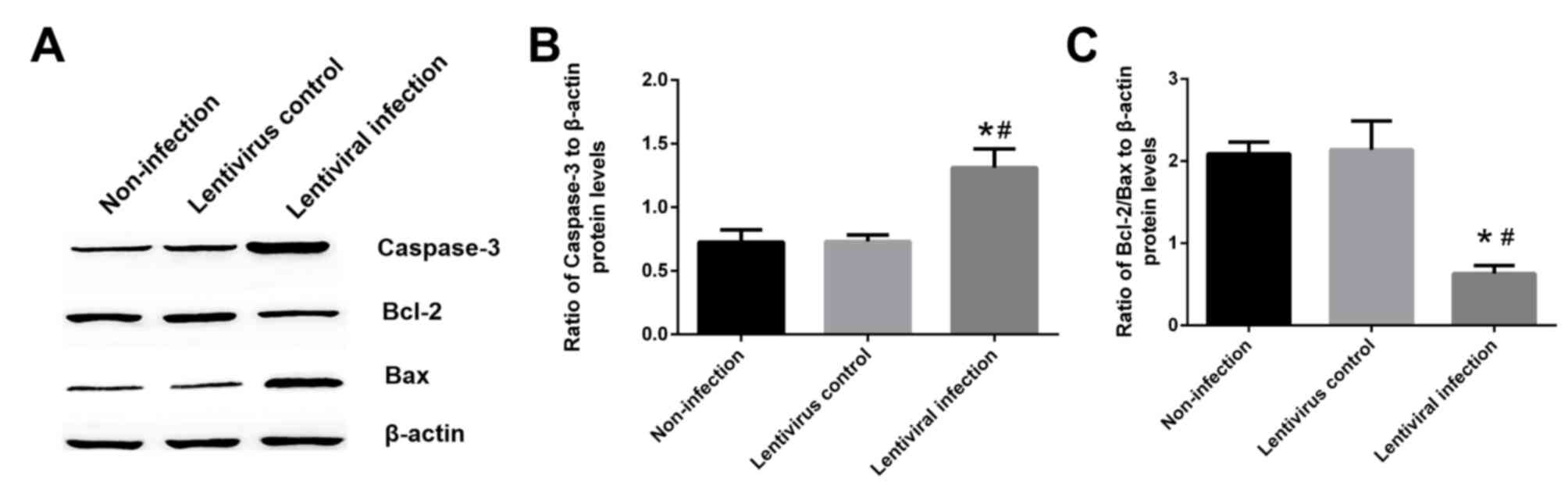
Hsp70 silencing increases caspase-3
expression and decreases the ratio of Bcl-2/Bax
The results of western blotting revealed that the
expression of caspase-3 in the lentiviral infection group was
increased by ~80% when compared with the non-infection and
lentivirus control groups (P<0.05; Fig. 5A and B). Furthermore, the ratio of
Bcl-2/Bax was decreased by 70.1% when compared with the lentivirus
control group (P<0.05; Fig. 5A and
C).
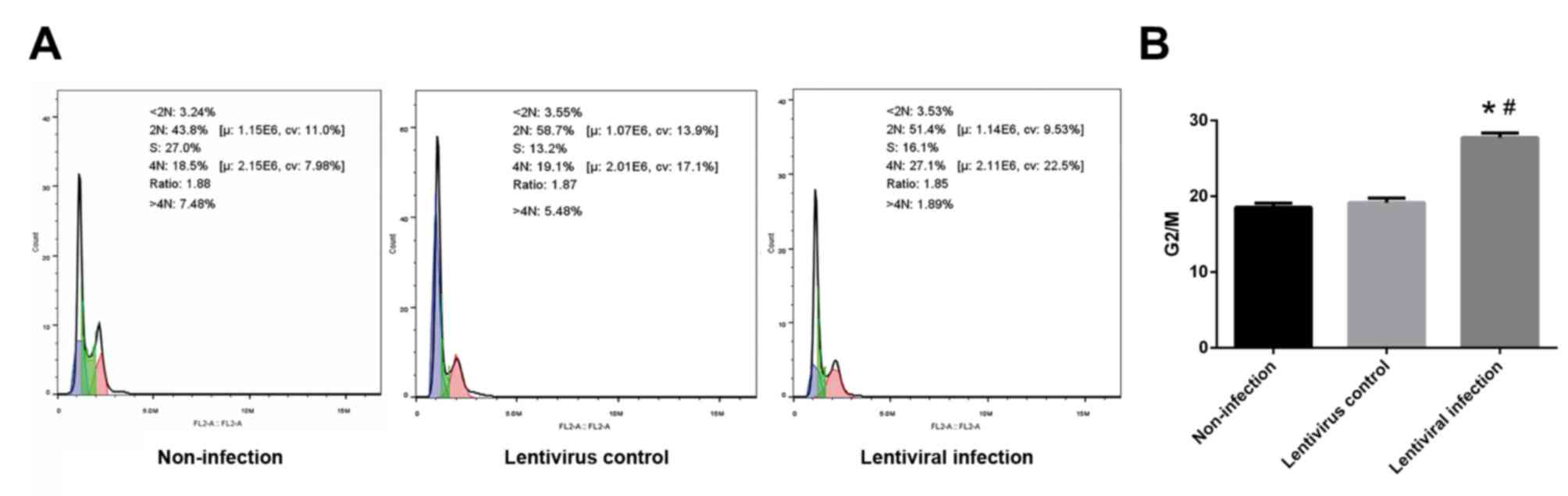
HSP70 silencing causes G2/M
arrest
The ratio of G2 and M phase cells in the lentiviral
infection group was significantly higher than that of the
non-infection and lentivirus control groups (P<0.05; Fig. 6).
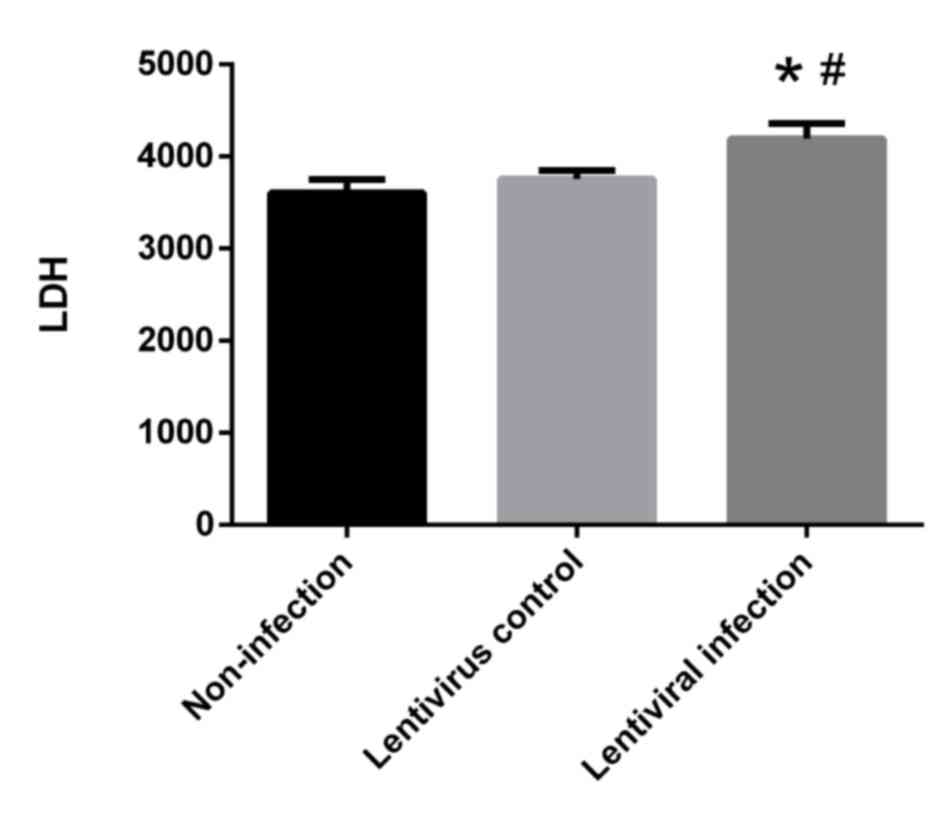
HSP70 silencing promotes cellular LDH
release
LDH levels in the lentiviral infection group were
significantly higher when compared with the non-infection and
lentivirus control groups (P<0.05; Fig. 7). These results indicate that HSP70
silencing promotes the release of LDH.
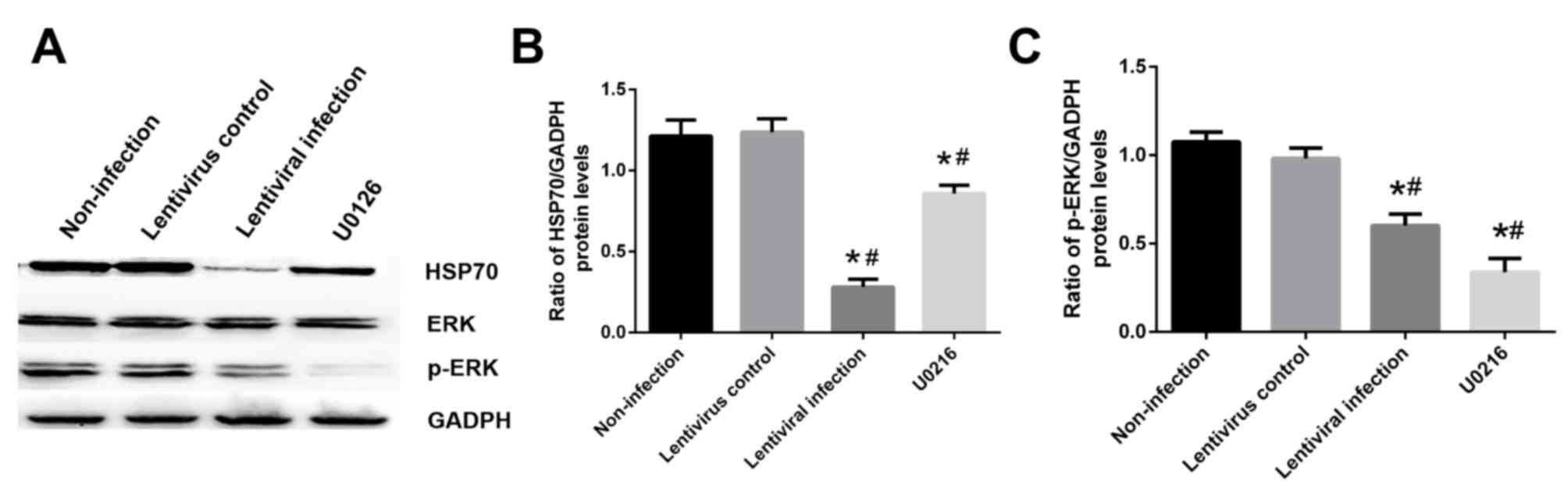
Influence of HSP70 on the MAPK/ERK
signaling pathway
Cells were exposed to hypoxia for 6 h and
reoxygenation for 24 h with or without 10 µM U0126 pretreatment.
The results revealed that treatment with 10 µM U0126 effectively
inhibited ERK phosphorylation and decreased HSP70 expression
compared with the non-infection group (Fig. 8A and B). In addition, HSP70 silencing
decreased the level of p-ERK compared with the non-infection group
(P<0.05; Fig. 8A and C).
Influence of HSP70 on the PI3K/AKT
signaling pathway
Cells were exposed to hypoxia for 6 h and
reoxygenation for 24 h with or without 50 µM LY294002 pretreatment.
The results revealed that treatment with 50 µM LY294002 effectively
inhibited AKT phosphorylation and decreased HSP70 expression
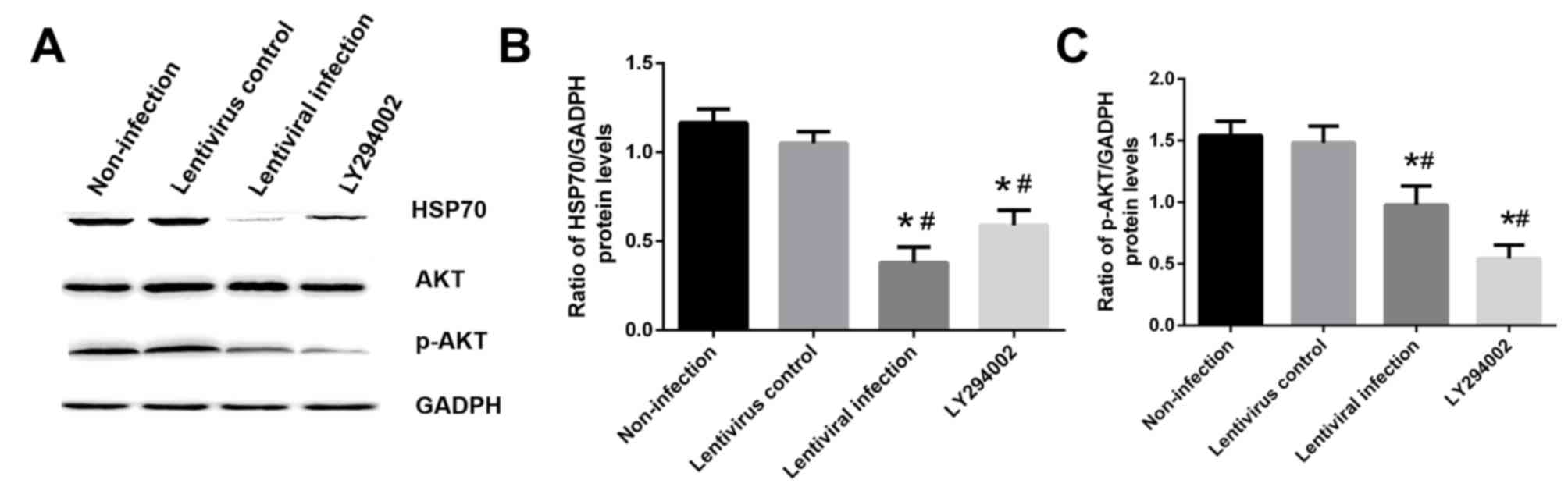
compared with the non-infection group (P<0.05; Fig. 9A and B). In addition, HSP70 silencing
decreased the level of p-AKT compared with the non-infection group
(P<0.05; Fig. 9A and C).
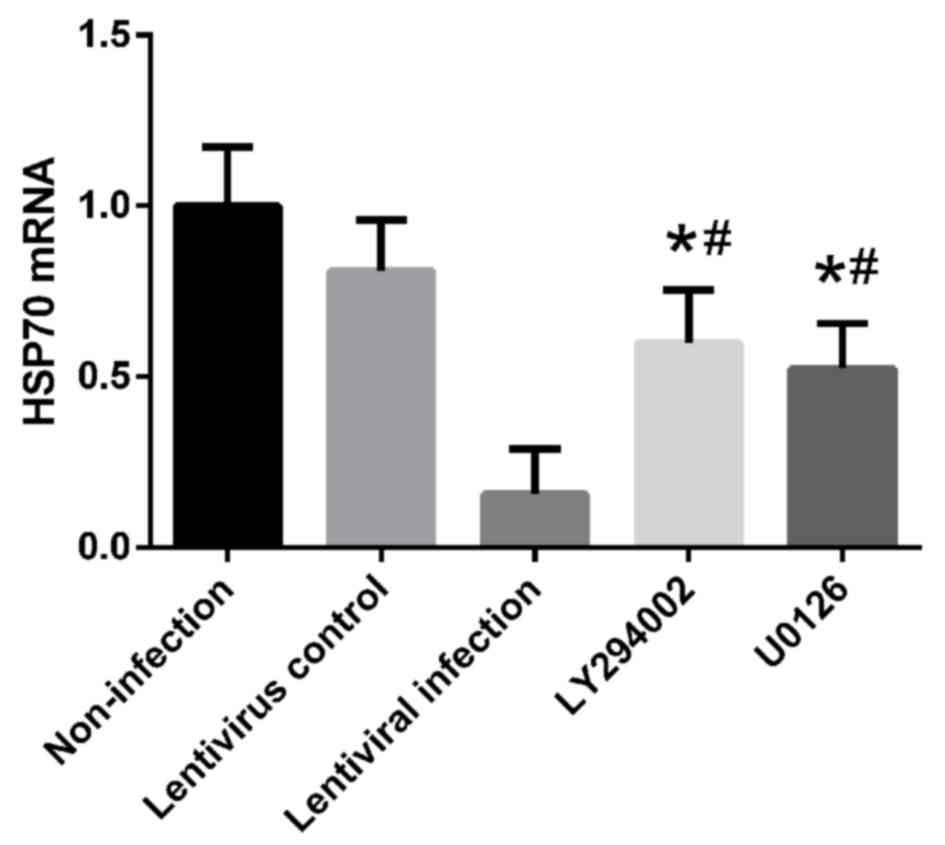
Treatment with 10 µM U0126 and 50 µM LY294002 also reduced the mRNA
level of HSP70 compared with the non-infection group (Fig. 10).
Discussion
LIRI causes severe lung injury and involves a series
of pathological processes including oxidative stress, inflammation
and apoptosis (26,27). Apoptosis is an important method of
cell death following LIRI (28).
HSP70, a member of the most conserved family of HSPs, usually
serves as a regulator and signaling molecule in certain cellular
processes, such as cell damage resistance and repair, and protects
cells from various stress-induced injuries, including apoptosis,
oxidative stress and ischemic-reperfusion injury (29). The high expression of HSP72 in
alveolar epithelial cells induced by dimethylarsinic acid has been
demonstrated to inhibit cell apoptosis (30). Lipopolysaccharide-induced acute lung
injury can also be protected against via an increased HSP70
expression (31). In addition, the
overexpression of HSP70 exhibits a protective effect on LIRI
(11). As aforementioned, HSP70 is
an anti-apoptotic protein that provides protection against
hypoxic/reoxygenation injuries (32). In the current study, HSP70 silenced
A549 cells exhibited a marked reduction in Bcl-2 and a marked
increase of Bax and caspase-3. Furthermore, apoptotic rates were
increased, indicating an increase in cellular apoptosis. LDH is an
important indicator of cell injury and as such was assessed in the
present study. The results of LDH analysis revealed that HSP70
silencing increased the release of LDH in cells, indicating an
increase in cell injury. The results of the current study therefore
revealed that HSP70 silencing aggravated cell injury and apoptosis
in a model of hypoxia-reoxygenation. Thus, it was inferred that the
cytoprotective effect of HSP70 on LIRI may be associated with
anti-apoptosis and the mitigation of cell injury. Interestingly,
the authors of the current study found that the proliferation of
A549 cells increased following 6 h of hypoxia. This may be because
a short period of hypoxia stimulates cell proliferation, but
long-term hypoxia reduces cell activity (33,34).
All phases, including G2/M, have detection points
for cell cycle regulation. DNA damage caused by various factors can
lead to G2/M phase arrest, which is closely associated with
apoptosis (35). The results of the
present study revealed that HSP70 silencing resulted in G2/M arrest
in A549 cells following hypoxia/reoxygenation, reducing cell
proliferation. It is possible that apoptosis induced by HSP70
silencing may therefore be associated with cell cycle arrest.
ERK is a serine/threonine protein kinase and belongs
to the MAPK family (36). It is an
important transduction protein that transmits mitogen signals
within the body (37). AKT is a
direct target gene of PI3K. Activating PI3K can phosphorylate Akt,
which causes downstream phosphorylation cascades and interactions
between target proteins, thus regulating cell survival and
apoptosis (38). It has been
demonstrated that ERK (39) and AKT
(40) enhance cell survival by
stimulating phosphorylation in downstream targets, thereby serving
a protective role in the lung. Furthermore, the anti-apoptotic
effect of HSP70 is closely associated with the ERK (41) and AKT (42) pathways. To gain further insight into
the mechanism by which HSP70 protects against
hypoxia/reoxygenation-induced apoptosis, the present study assessed
the role of the MAPK/ERK and PI3K/AKT signaling pathways. The
results revealed that decreased levels of HSP70 were accompanied
with a decrease in p-ERK and p-AKT, indicating that HSP70 is
required for ERK and AKT phosphorylation. In addition, the
inhibition of ERK and AKT phosphorylation by their respective
inhibitors also resulted in a decrease in HSP70 expression.
Therefore, the present results indicate that HSP70 may function in
an anti-apoptotic manner via the MAPK/ERK and PI3K/AKT signaling
pathways.
In conclusion, HSP70 provided protection against
hypoxia/reoxygenation. Silencing HSP70 was revealed to decrease
cell viability and increase apoptosis in LIRI, which was
accompanied by the inhibition of the ERK and AKT signaling
pathways. The present study also highlighted the potential role of
HSP70 in lung protection. Therefore, it is hypothesized that HSP70
may be a potential target for the treatment of LIRI. Although A549
cells have many features of normal type II alveolar epithelial
cells, there are also differences between them (43). Therefore, the in-vivo
protective effects of HSP70 require further study.
Acknowledgements
The authors would like to thank Dr. Bing Luo and Dr.
Wen Liu from Qingdao University (Qingdao, China) for their help in
the preparation and modification of this manuscript.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81571938
and 81501706).
Authors' contributions
CZ performed the experiments, collected and analyzed
the data, and prepared the manuscript. JL collected the data. KW
analyzed the data. QL conceived and designed the study, and
performed the literature search. YQ conceived and designed the
study, and reviewed manuscript.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
de Perrot M, Liu M, Waddell TK and
Keshavjee S: Ischemia-reperfusion-induced lung injury. Am J Respir
Crit Care Med. 167:490–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cottini SR, Lerch N, de Perrot M,
Treggiari MM, Spiliopoulos A, Nicod L and Ricou B: Risk factors for
reperfusion injury after lung transplantation. Intensive Care Med.
32:557–563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimamoto A, Pohlman TH, Shomura S,
Tarukawa T, Takao M and Shimpo H: Toll-like receptor 4 mediates
lung ischemia-reperfusion injury. Ann Thorac Surg. 82:2017–2023.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ambrosio G and Tritto I: Reperfusion
injury: Experimental evidence and clinical implications. Am Heart
J. 138:S69–S75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quadri SM, Segall L, de Perrot M, Han B,
Edwards V, Jones N, Waddell TK, Liu M and Keshavjee S: Caspase
inhibition improves ischemia-reperfusion injury after lung
transplantation. Am J Transplant. 5:292–299. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fischer S, Maclean AA, Liu M, Cardella JA,
Slutsky AS, Suga M, Moreira JF and Keshavjee S: Dynamic changes in
apoptotic and necrotic cell death correlate with severity of
ischemia-reperfusion injury in lung transplantation. Am J Respir
Crit Care Med. 162:1932–1939. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Radons J: The human HSP70 family of
chaperones: Where do we stand? Cell Stress Chaperones. 21:379–404.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Eden W, van der Zee R and Prakken B:
Heat-shock proteins induce T-cell regulation of chronic
inflammation. Nat Rev Immunol. 5:318–330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mayer MP and Bukau B: Hsp70 chaperones:
Cellular functions and molecular mechanism. Cell Mol Life Sci.
62:670–684. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao Y, Han C, Huang H, Xin Y, Xu Y, Luo L
and Yin Z: Heat shock protein 70 together with its co-chaperone
CHIP inhibits TNF-alpha induced apoptosis by promoting proteasomal
degradation of apoptosis signal-regulating kinase1. Apoptosis.
15:822–833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu S, Xu J, Fang C, Shi C, Zhang X, Yu B
and Yin Y: Over-expression of heat shock protein 70 protects mice
against lung ischemia/reperfusion injury through SIRT1/AMPK/eNOS
pathway. Am J Transl Res. 8:4394–4404. 2016.PubMed/NCBI
|
|
12
|
Paul S and Mahanta S: Association of
heat-shock proteins in various neurodegenerative disorders: Is it a
master key to open the therapeutic door? Mol Cell Biochem.
386:45–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamashima T: Hsp70.1 and related lysosomal
factors for necrotic neuronal death. J Neurochem. 120:477–494.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hiratsuka M, Mora BN, Yano M, Mohanakumar
T and Patterson GA: Gene transfer of heat shock protein 70 protects
lung grafts from ischemia-reperfusion injury. Ann Thorac Surg.
67:1421–1427. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Zhang Y, Li C, Xie J, Liu Y, Zhu W,
Zhang X, Jiang S, Liu L and Ding Z: HSPA12B attenuates cardiac
dysfunction and remodelling after myocardial infarction through an
eNOS-dependent mechanism. Cardiovasc Res. 99:674–684. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chi W, Meng F, Li Y, Wang Q, Wang G, Han
S, Wang P and Li J: Downregulation of miRNA-134 protects neural
cells against ischemic injury in N2A cells and mouse brain with
ischemic stroke by targeting HSPA12B. Neuroscience. 277:111–122.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jayanthi S, Deng X, Bordelon M, McCoy MT
and Cadet JL: Methamphetamine causes differential regulation of
pro-death and anti-death Bcl-2 genes in the mouse neocortex. FASEB
J. 15:1745–1752. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nuñez G, Benedict MA, Hu Y and Inohara N:
Caspases: The proteases of the apoptotic pathway. Oncogene.
17:3237–3245. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A,
Wu Q, Zhang J and Hong Y: Caspases: A molecular switch node in the
crosstalk between autophagy and apoptosis. Int J Biol Sci.
10:1072–1083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi Z, Qi S, Gui L, Shen L and Feng Z:
Daphnetin protects oxidative stress-induced neuronal apoptosis via
regulation of MAPK signaling and HSP70 expression. Oncol Lett.
12:1959–1964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Ding W, Sun H, Zhou Q, Huang J,
Li X, Xie Y and Chen J: Salidroside protects PC12 cells from
MPP+-induced apoptosis via activation of the PI3K/Akt
pathway. Food Chem Toxicol. 50:2591–2597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shoji T, Yoshida S, Mitsunari M, Miyake N,
Tsukihara S, Iwabe T, Harada T and Terakawa N: Involvement of p38
MAP kinase in lipopolysaccharide-induced production of pro- and
anti-inflammatory cytokines and prostaglandin E(2) in human
choriodecidua. J Reprod Immunol. 75:82–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
López-Neblina F and Toledo-Pereyra LH:
Phosphoregulation of signal transduction pathways in ischemia and
reperfusion. J Surg Res. 134:292–299. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kamiya T, Kwon AH, Kanemaki T, Matsui Y,
Uetsuji S, Okumura T and Kamiyama Y: A simplified model of hypoxic
injury in primary cultured rat hepatocytes. In Vitro Cell Dev Biol
Anim. 34:131–137. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ng CS, Wan S, Arifi AA and Yim AP:
Inflammatory response to pulmonary ischemia-reperfusion injury.
Surg Today. 36:205–214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ng CS, Wan S and Yim AP: Pulmonary
ischaemia-reperfusion injury: Role of apoptosis. Eur Respir J.
25:356–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Antoniou KM, Pataka A, Bouros D and
Siafakas NM: Pathogenetic pathways and novel pharmacotherapeutic
targets in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther.
20:453–461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giffard RG and Yenari MA: Many mechanisms
for hsp70 protection from cerebral ischemia. J Neurosurg
Anesthesiol. 16:53–61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kato K, Yamanaka K, Hasegawa A and Okada
S: Dimethylarsinic acid exposure causes accumulation of Hsp72 in
cell nuclei and suppresses apoptosis in human alveolar cultured
(L-132) cells. Biol Pharm Bull. 22:1185–1188. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Dong Y and Cai Y: Alanyl-glutamine
prophylactically protects against lipopolysaccharide-induced acute
lung injury by enhancing the expression of HSP70. Mol Med Rep.
16:2807–2813. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Higashi T, Takechi H, Uemura Y, Kikuchi H
and Nagata K: Differential induction of mRNA species encoding
several classes of stress proteins following focal cerebral
ischemia in rats. Brain Res. 650:239–248. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang H, Okamoto M, Panzhinskiy E, Zawada
WM and Das M: PKCδ/midkine pathway drives hypoxia-induced
proliferation and differentiation of human lung epithelial cells.
Am J Physiol Cell Physiol. 306:C648–C658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu Z, Huang J, Li Q, Yang J, Zhong L and
Zeng Q: Effect of hypoxia on infiltration and migration of lung
cancer cells and expression of MMP-2 and TIMP-2. Zhongguo Fei Ai Za
Zhi. 8:270–273. 2005.(In Chinese). PubMed/NCBI
|
|
35
|
Pietenpol JA and Stewart ZA: Cell cycle
checkpoint signaling: Cell cycle arrest versus apoptosis.
Toxicology 181–182. 475–481. 2002. View Article : Google Scholar
|
|
36
|
Tanoue T and Nishida E: Molecular
recognitions in the MAP kinase cascades. Cell Signal. 15:455–462.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chao X, Zao J, Xiao-Yi G, Li-Jun M and Tao
S: Blocking of PI3K/AKT induces apoptosis by its effect on NF-κB
activity in gastric carcinoma cell line SGC7901. Biomed
Pharmacother. 64:600–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Plotkin LI and Bellido T:
Bisphosphonate-induced, hemichannel-mediated, anti-apoptosis
through the Src/ERK pathway: A gap junction-independent action of
connexin43. Cell Commun Adhes. 8:377–382. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bezler M, Hengstler JG and Ullrich A:
Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances
apoptosis of ovarian cancer cells. Mol Oncol. 6:516–529. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qi Z, Qi S, Gui L, Shen L and Feng Z:
Daphnetin protects oxidative stress-induced neuronal apoptosis via
regulation of MAPK signaling and HSP70 expression. Oncol Lett.
12:1959–1964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ji K, Xue L, Cheng J and Bai Y:
Preconditioning of H2S inhalation protects against cerebral
ischemia/reperfusion injury by induction of HSP70 through
PI3K/Akt/Nrf2 pathway. Brain Res Bull. 121:68–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oraldi M, Saracino S, Maggiora M,
Chiaravalloti A, Buemi C, Martinasso G, Paiuzzi E, Thompson D,
Vasiliou V and Canuto RA: Importance of inverse correlation between
ALDH3A1 and PPARγ in tumor cells and tissue regeneration. Chem Biol
Interact. 191:171–176. 2011. View Article : Google Scholar : PubMed/NCBI
|