Introduction
Acute respiratory distress syndrome (ARDS) is a
severe inflammatory disease of the lungs, characterized by
hypoxemia that is caused by the severe impairment of gas exchange
and lung mechanics as a result of acute lung injury (ALI) and edema
(1). With an increasing incidence of
~200,000 cases annually in the United States, ARDS has a high rate
of mortality (30–50%) and is responsible for exorbitant health care
expenditures (1–3). While several factors are known to
induce ARDS, including hyperoxia and hemorrhage, sepsis and
pneumonia are the leading causes of the disease (2). Although a variety of interventions and
treatments have been developed for this inflammatory disorder, the
mortality rate of ARDS remains higher than acceptable (4). Therefore, there is an urgent need to
develop an effective method or drug for treating ARDS.
Lipopolysaccharides (LPSs) located in the outer
membrane of gram-negative bacteria have been reported to cause ARDS
by increasing inflammatory cytokine production in lung tissue
(5). As an important component of
the inflammatory system, neutrophils partially initiates
inflammation in patients with ALI/ARDS and contributes to the
development of ARDS-associated pulmonary edema (6). After infiltrating the lungs and
migrating into the airways, neutrophils secrete various
pro-inflammatory cytokines, including tumor necrosis factor-α
(TNF-α), interleukin (IL)-1β and IL-6, which disrupt endothelial
and epithelial barriers (6–8). It has been reported that the increased
expression of proinflammatory cytokines can be ascribed to the
NF-κB pathway (2). Activation of the
NF-κB pathway is dependent on the phosphorylation of NF-κB and its
subsequent translocation to the nucleus, resulting in gene
transcription (2,9). Proinflammatory signaling and resultant
immune responses are triggered by toll-like receptors (TLRs), which
are highly conserved pattern recognition receptors that recognize
nonendogenous pathogen-associated molecular pattern molecules
(1). Ten functional TLRs have been
identified in humans, and appear to be associated with ARDS
(10). TLR3 was revealed to
participate in hyperoxia-induced ARDS and TLR2 was demonstrated to
mediate hemorrhage-induced ARDS (11,12).
TLR4 was also identified as a pivotal regulator capable of inducing
ARDS in various murine models, including those induced by sepsis
(12,13).
Recent reports have demonstrated that certain plant
extracts help to protect against inflammation-associated diseases
(14,15). For example, phytochemicals present in
ginkgo biloba, sun ginseng and blackberries have been shown to have
therapeutic potential for endotoxin-associated diseases (16). Carnosic acid (CA) is a benzenediol
abietane diterpene found in a variety of herbal plants including
rosemary and sage (17). It is an
antioxidant that has multiple pharmacological effects, including
anti-obesity, anti-inflammatory, anti-tumor and neuroprotective
activities (18–21). CA has been revealed to protect
against myocardial injury and inhibit colitis in vivo,
potentially via anti-inflammatory activity (22). However, to the best of our knowledge,
no studies have investigated the effects of CA on ARDS.
In the present study, the effect of CA on
LPS-induced ALI in mice was investigated. The impacts on lung
injury and inflammation, as well as neutrophil infiltration and the
generation of TNF-α, IL-1β and IL-6 were determined. The ability of
CA to inhibit the NF-κB pathway, which is critically associated
with neutrophil activation and inflammatory responses, was also
assessed.
Materials and methods
Animals and treatments
Male BALB/c mice (age, ~8 weeks; weight, 21–25 g)
were purchased from the Model Animal Research Center of Nanjing
University and housed in a standard laboratory room maintained at
20–26°C and 40–70% relative humidity, under a 12 h light/dark
cycle. Mice also received free access to water and food.
Mice were randomly assigned to 5 groups (n=6/group).
A group of mice was used as control, while mice the other 4 groups
were used to establish an acute lung injury (ALI) model induced
with LPS (Sigma-Aldrich; Merck KGaA). At ~12 h prior to receiving
an intravenous (IV) injection of LPS (4 mg/kg), the mice were given
CA at the following doses: 0, 10, 20 or 40 mg/kg. A total of 6 h
later, the mice were sacrificed and samples of lung tissue were
removed for testing. CA was purchased from Dalian Meilun Biotech
Co., Ltd, dissolved in 0.5% v/v DMSO in saline and injected into
mice intraperitoneally. Bronchoalveolar lavage fluid (BALF) was
obtained for MPO analysis, and the left lungs were collected for
histopathological examination, while the, right lungs were
collected for wet-to-dry (W/D) analysis, western blotting and
reverse transcription quantitative PCR (RT-qPCR) examinations. All
animal experiments were performed at the Zhongda Hospital Southeast
University (Nanjing, China), and all experimental protocols were
approved by the Ethics Committee of the Zhongda Hospital Southeast
University.
Bronchoalveolar lavage fluid and
tissue sampling
Each mouse was anesthetized with 0.75% pentobarbital
(35 mg/kg; IV) followed by decapitation. Animals were confirmed
dead when no breathing or heart beat was detected. Following
sacrifice, neck skin was dissected and the trachea was rapidly
exposed. Next, a catheter was deeply inserted into the trachea and
the lungs were lavaged 3 times with 1 ml of cold PBS solution. The
BALF collected from each mouse was centrifuged at 420 × g for 15
min at 4°C and the supernatant was stored at −80°C until further
use. Cell pellets were re-suspended in PBS for subsequent
neutrophil purification. The lungs were cut into two parts, placed
into individual centrifuge tubes, snap frozen in liquid nitrogen
and stored at −80°C. Lung tissue (~100 mg) were placed into
centrifuge tubes and suspended in cool normal saline solution.
Tissue samples were then homogenized with homogenate rods and
centrifuged at 12,000 × g for 10 min at 4°C to collect supernatant
for myeloperoxidase (MPO), ELISA analysis and western blotting.
Neutrophil purification and apoptosis
analysis
Neutrophils were isolated by discontinuous Percoll
gradient centrifugation as previously described (23). In brief, 2 ml of Percoll solution
diluted with HBSS (1,090 mg/ml; Sigma-Aldrich; Merck KGaA) was
added to a tube and another 2 ml of Percoll solution (1,080 mg/ml)
was gently layered on top of the first solution. Resuspended cells
were then gently layered onto the Percoll solution, and centrifuged
at 960 × g for 15 min at 4°C. The neutrophil layer located between
the Percoll layers was collected into tubes containing PBS, which
were then centrifuged at 420 × g for 5 min at 4°C. The cells were
subsequently resuspended in pre-cooled PBS and cells were fixed
overnight at 4°C in pre-cooled 70% ethanol. After washing twice
with PBS, fixed cells were incubated with 5 µl Annexin V-FITC and
10 µl propidium iodide (PI; cat. no. AP101-30; Hangzhou Lianke
Biology Technology Co., Ltd.) at 4°C for 30 min in the dark and
then analyzed for apoptosis using a BD FACSCalibur flow cytometer
(BD Biosciences). Data were analyzed using FlowJo software (Version
7.6; FlowJo, LLC).
ELISA
ELISA kits (Biolegend, Inc.) were used to measure
the levels of IL-1β (cat. no. 432601), IL-6 (cat. no. 431304) and
TNF-α (cat. no. 430904) in BALF and lung tissue homogenates
according to the manufacturer's protocols. Samples and standards
were pipetted into a microplate pre-coated with capture antibodies
(provided as part of the aforementioned ELISA kits) and incubated
at room temperature for 2 h. After washing 3 times with 300 µl Wash
Buffer, biotinylated detection antibodies (all provided as part of
the aforementioned kits) were added and incubated for 1 hat room
temperature, followed by incubation with avidin horseradish
peroxidase (HRP) conjugate for 30 min. The immune reaction was
visualized by the addition of 3,3′,5,5′-Tetramethylbenzidine
(Beyotime Institute of Biotechnology) substrate solution followed
by incubation at room temperature in the dark for 15 min.
Subsequently, the reaction was stopped by the addition of 1 M
H2SO4 and the optical density of each well
was measured at 450 nm with a microplate reader (TECAN-GENious;
Tecan Group, Ltd.).
Lung myeloperoxidase activity
determination
MPO activity in lung homogenates was determined
using a MPO assay kit according to the manufacturer's protocol
(cat. no. A044-1-1; Nanjing Jiancheng Bioengineering Institute).
The homogenates were subsequently centrifuged at 5,000 × g for 10
min at 4°C and the supernatants were collected and used to
calculate MPO activity based on the maximal absorbance at 532 nm.
The homogenates were mixed with reagent II, followed by the
addition of reagent IV and a chromogenic agent, and left to react
for 10 min at 60°C. MPO activity was calculated based on the
maximal absorbance at 532 nm and activity was normalized to the
total protein concentration of the same sample, which was
determined using a bicinchoninic acid assay (cat. no. 23225;
Pierce; Thermo Fisher Scientific, Inc.).
Lung wet-to-dry weight (W/D)
ratio
To evaluate lung edema, mice were euthanized and the
right lungs were collected, weighed and placed in an oven at 70°C
for 48 h. Dried tissues were then weighed to calculate the W/D
ratio.
Histopathological and
immunohistochemical examinations
The left lungs of mice were fixed in 10% buffered
formalin for 24 h at room temperature. A small section of fixed
lung tissue was cut with a surgical blade, dehydrated in a series
of graded ethanol solutions (30, 60, 80, 95 and 100%), cleared with
xylene and embedded inparaffin. Embedded tissue was subsequently
cut into 5 µm sections, which were deparaffinized with xyleneand
rehydrated in a descending gradient of ethanol concentrations (100,
95, 80, 60 and 30%). Some sections were stained with hematoxylin
and eosinat room temperature for 15 min and observed under a light
microscope (magnification, ×400), while other sections were used
for immunohistochemical examinations of TLR4 and NF-κB expression.
Endogenous tissue peroxidase was inactivated by incubation with
0.5% H2O2 for 5 min at room temperature, and
antigen retrieval was performed by boilingtissue in citrate buffer
(pH 6.0) for 10 min. Tissue sections were then blocked with 1% BSA
(Beijing Solarbio Science & Technology Co., Ltd.) for 30 min at
room temperature and then incubated for 1 h at room temperature
with primary antibodies against TLR4 (cat. no. sc-293072; 1:200;
Santa CruzBiotechnology, Inc.) and NF-κB (cat. no. sc-514451;
1:200; Santa Cruz Biotechnology, Inc.). Sections were then
incubated for 20 min at room temperature with HRP-conjugated
secondary antibodies (cat. no. IH-0031; 1:5,000; Beijing Dingguo
Changsheng Biotechnology Co., Ltd.). Color was developed with a
diaminobenzidine kit (cat. no. JD091-1G; Beijing Dingguo Changsheng
Biotechnology Co., Ltd.), followed by counterstaining with
hematoxylin for 2 min at room temperature.
Reverse transcription quantitative
PCR
Total RNA was extracted from the collected mouse
lung tissues using TRIzol reagent (Takara Biotechnology Co., Ltd.)
and 3 µg of total RNA was reverse transcribed into cDNA using a
Bestar™qPCR RT kit (cat. no. DBI-2220; DBI Bioscience) according to
the manufacturer's protocol. qPCR was performed using Bestar™
SYBRGreen qPCR MasterMix (cat. no. DBI-2044; DBI Bioscience) in an
ABI 7500 RT-PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermo cycling conditions were as follows:
Denaturation at 95°C for 10 min, followed by 40 cycles of 94°C for
20 sec, 58°C for 20 sec and 72°C for 20 sec. The primers used were
as follows: TLR4 forward, 5′-CCATTGCTTGGCGAATGTTT-3′ and reverse,
5′-TGTCTCAGGCTGTTTGTTCC-3′; NF-κB forward,
5′-AGAGCAACCAAAACAGAGGG-3′ and reverse, 5′-TGCAAATTTTGACCTGTGGG-3′;
β-actin forward, 5′-CATTGCTGACAGGATGCAGA-3′ and reverse,
5′-CTGCTGGAAGGTGGACAGTGA-3′. The levels of target mRNA were
analyzed using the 2−ΔΔCq method (24) and normalized using β-actin as a
control. Each test was repeated three times.
Western blot analysis
Collected lung tissues were lysed with RIPA buffer
(Beyotime Institute of Biotechnology) supplemented with a protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA). The protein
concentrations in the lysates were determined using a BCA kit (cat.
23225; Pierce; Thermo Fisher Scientific, Inc.). Equal quantities of
protein (20 µg) from the lysates were separated using SDS-PAGE (10%
gel) and the separated protein bands were transferred onto
polyvinylidene difluoride membranes (Bio-Rad Laboratories, Inc.),
which were subsequently blocked with 5% non-fat dry milk in TBST
(Tris-buffered saline with 0.1% Tween-20) at room temperature for 1
h. The membranes were then washed and incubated overnight at 4°C
with primary antibodies against TLR4 (cat. no. sc-52962; 1:1,000;
Santa Cruz Biotechnology, Inc.), NF-κB (cat. no. sc-514451; 1:200;
Santa Cruz Biotechnology, Inc.), p-NF-κB (cat. no. sc-135768;
1:1,000; Santa Cruz Biotechnology, Inc.) and GAPDH (cat. no.
ab8245; 1:2,000; Abcam). The antibody against NF-κB or p-NF-κB
recognized the p65 or p-p65 subunit, respectively. Membranes were
subsequently washed with TBST and then incubated with
HRP-conjugated secondary antibodies at 37°C for 1 h (cat. no.
BA1050; 1:5,000; Boster Biological Technology). Immunostained
protein bands were visualized with a chemiluminescent substrate
(ECL-Plus; GE Healthcare). The assay was repeated three times.
Semi-quantitative analysis was performed using Image-Pro Plus 6.0
software (Media Cybernetics, Inc.).
Statistical analysis
Data are expressed as the mean ± standard deviation.
One-way ANOVA and a two-tailed Student's t-test were used to
compare the differences in parameters between experimental groups.
All statistical analyses were performed using IBM SPSS Statistics
for Windows, version 21.0 (IBM Corp.) and P<0.05 was considered
to indicate a statistically significant difference.
Results
Effect of CA on LPS-induced ALI
To observe the effect of CA on lung injuries caused
by LPS, the lungs were collected 6 h after LPS treatment and
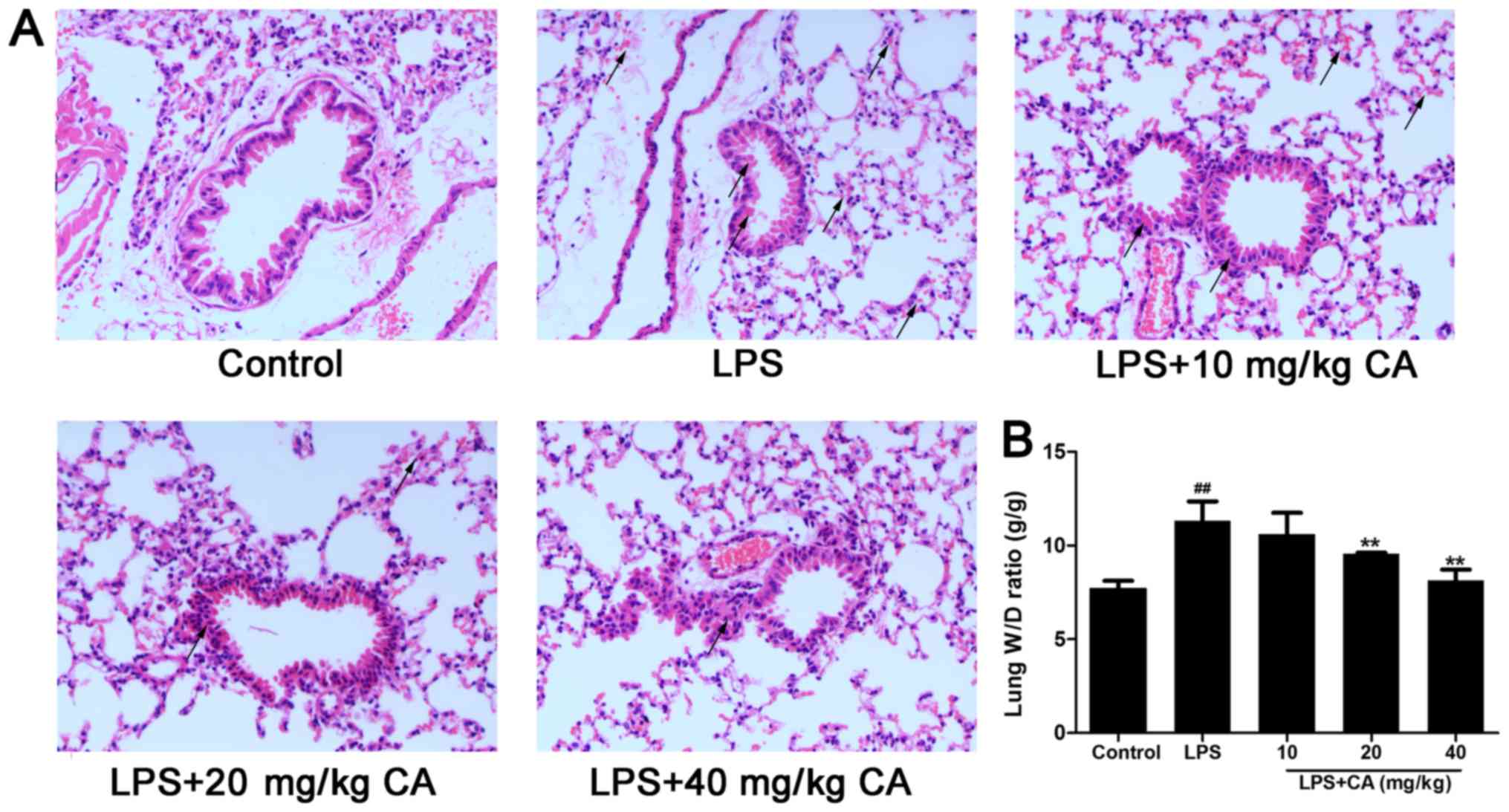
subjected to a histologic examination. As presented in Fig. 1A, lungs in the control group
exhibited no signs of hemorrhage or edema and little evidence of
inflammatory cell infiltration. In contrast, LPS treatment alone
induced hemorrhage and pathologic alterations typically associated
with ALI, including hemorrhage, accumulation of inflammatory cells
in the alveolar space, alveolar wall thickening and edema of the
lung interstitium and alveoli, as indicated with black arrows
(Fig. 1A). Consistent with the afore
mentioned histopathological observations, the W/D ratio was
significantly increased after LPS treatment (Fig. 1B). However, the increase induced by
LPS was significantly inhibited by CA treatment at 20 and 40 mg/kg,
indicating that CA protected the lungs from injuries induced by
LPS.
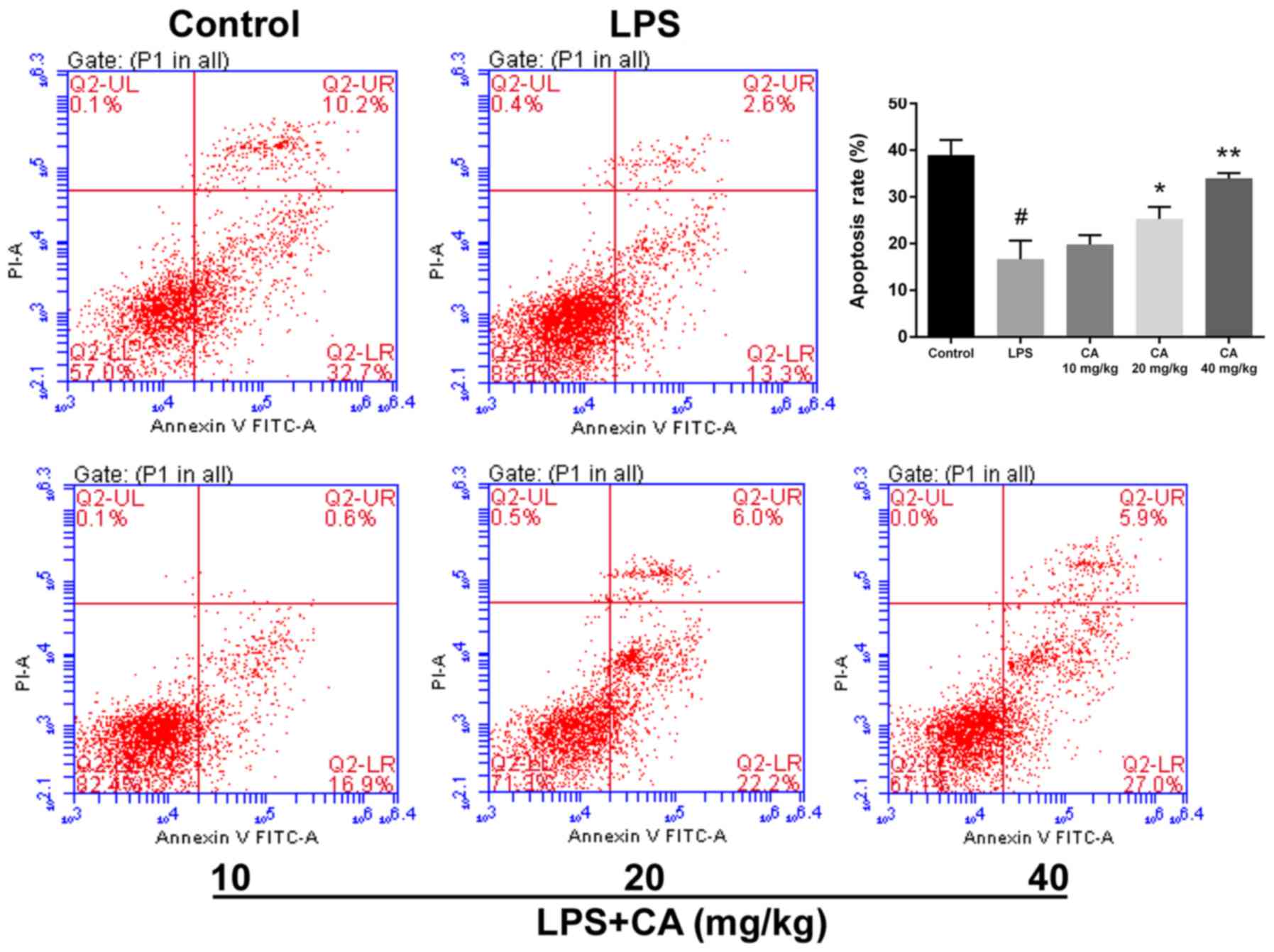
Effect of CA on neutrophil apoptosis
in LPS-induced ALI
Neutrophils are an important contributor to ARDS
(25). To compare the viability of
neutrophils, cell apoptosis was determined via flow cytometry. As
presented in Fig. 2, LPS treatment
significantly decreased cell apoptosis compared with the control.
In mice that received CA at doses of 20 and 40 mg/kg, apoptosis was
significantly increased compared with that of the LPS group.
Additionally, the rate of apoptosis in the CA-treated groups
increased with higher doses and gradually approached the rate of
apoptosis observed in the control group. At 40 mg/kg, there was no
statistical difference from the control group. These results
indicated that CA treatment significantly inhibited the suppression
of neutrophil apoptosis by LPS.
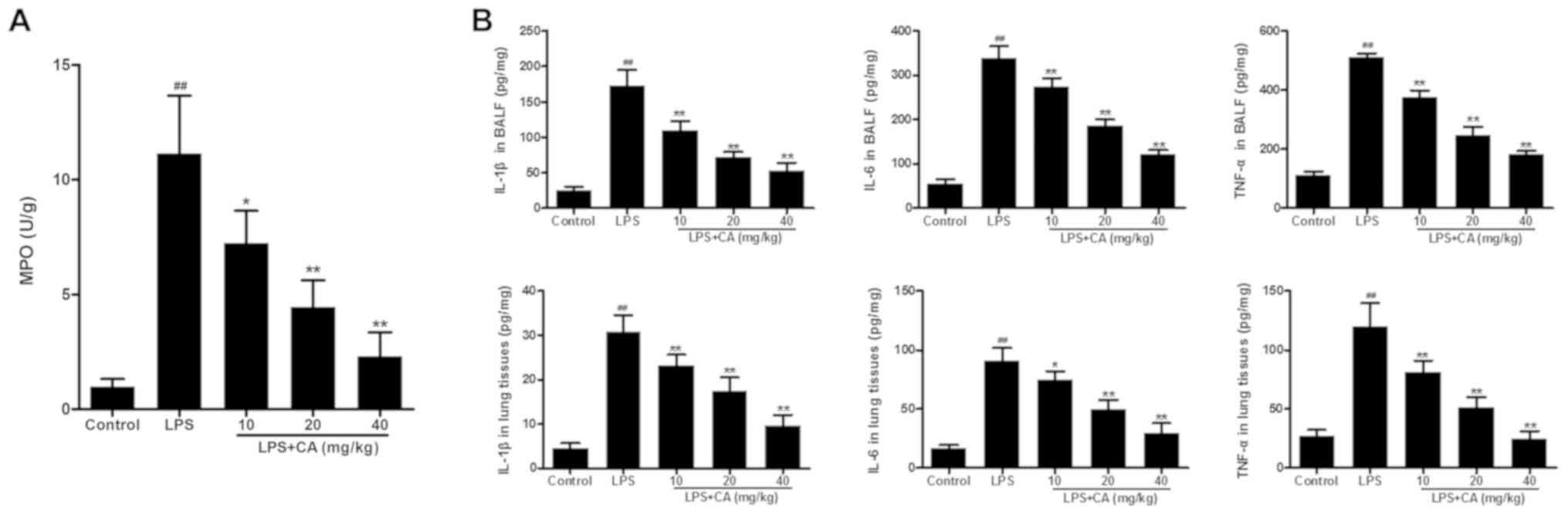
Effect of CA on MPO activity and
IL-1β, IL-6 and TNF-α levels
In the present study, MPO activity was used as an
indicator of neutrophil infiltration. As presented in Fig. 3A, significantly increased MPO
activity was observed in the LPS group, when compared with MPO
activity in the control group. Pretreatment with CA significantly
reduced MPO activity in LPS-challenged lungs in a dose-dependent
manner. These results indicated that LPS induced the infiltration
of neutrophils into lung tissue and that the effect was
significantly inhibited by CA treatment.
After confirming neutrophil infiltration, the levels
of IL-1β, IL-6 and TNF-α were determined to investigate the effect
of CA on LPS-induced inflammation. As presentedin Fig. 3B, the levels of IL-1β, IL-6 and TNF-α
in the BALF and lung tissues of the LPS group were significantly
increased compared with those in the control group. Pretreatment
with CA significantly reduced IL-1β, IL-6 and TNF-α levels in
LPS-challenged lungs in a dose-dependent manner. These results
indicated that LPS induced inflammation in the lungs and that this
inflammation could be significantly inhibited by CA treatment.
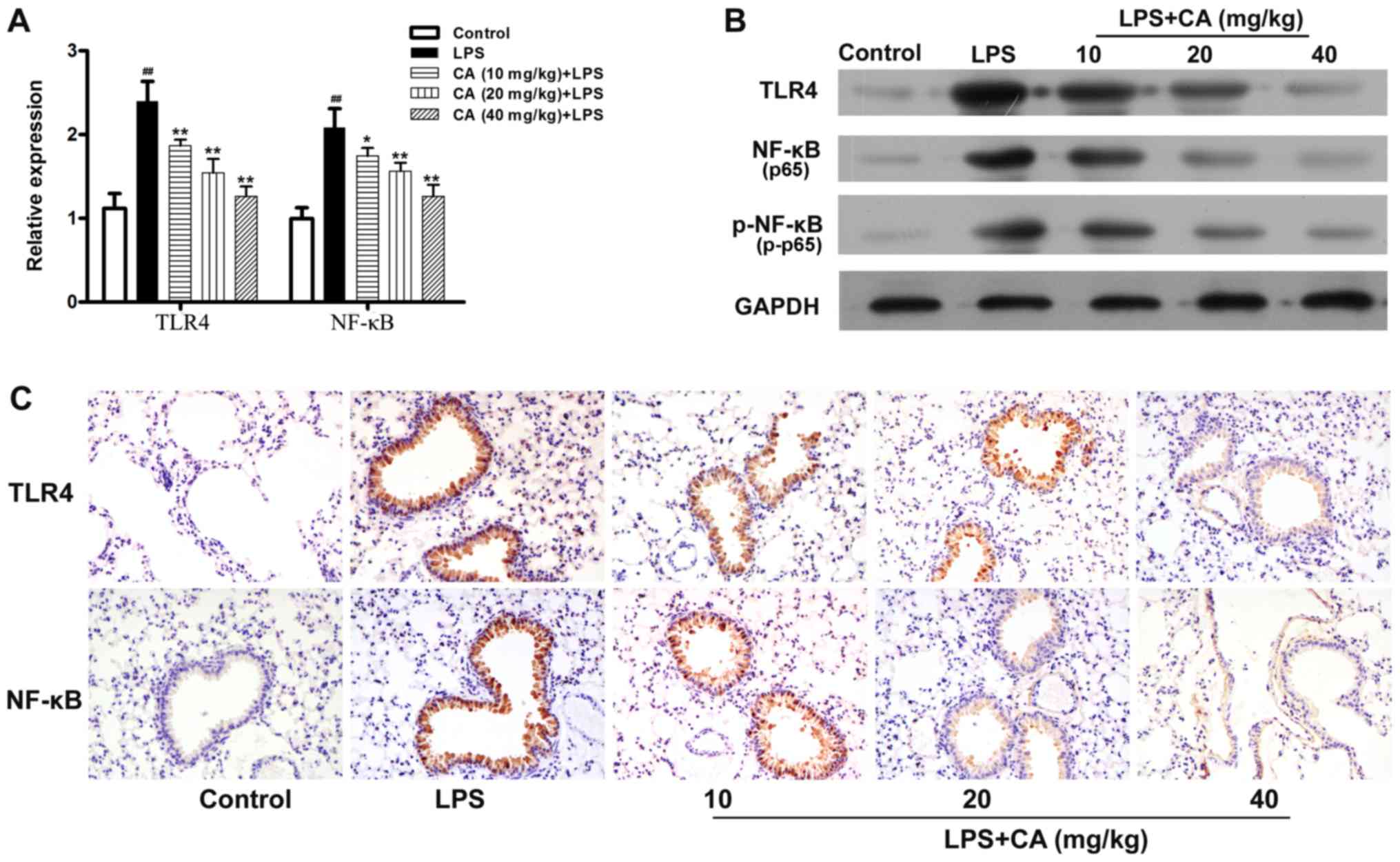
Effects of CA on TLR4 and NF-κB
expression
TLR4 and NF-κB serve critical roles in LPS-induced
inflammation (26). In the present
study, the expression of these inflammatory mediators were examined
to investigate the mechanism by which CA inhibited LPS-induced
inflammation. As presented in Fig. 4A
and B, the levels of TLR4 and NF-κB mRNA and protein
expressions in the LPS-treated group were significantly increased
compared with the levels in the control group. However, the ability
of LPS to promote the expression of TLR4 and NF-κB mRNA, and
protein was significantly inhibited by pretreatment with CA.
Additionally, it was observed that the phosphorylation level of
NF-κB was significantly increased by LPS treatment and that this
increase was inhibited by CA pretreatment in a dose-dependent
manner (Fig. 4B).
Subsequently, immunohistochemistry was used to
examine the expression of TLR4 and NF-κB in lung tissue. As
presented in Fig. 4C, lung tissue
from the control mice displayed very little positive staining for
TLR4 and p65. After LPS treatment, the levels of TLR4 and p65
expression were significantly increased in the lung tissue
epithelial cells. The LPS-induced increases in TLR4 and p65 were
not significantly inhibited by pretreatment with CA.
Discussion
Inflammation has been traditionally considered to be
an important defense response induced by an infection or injury
(27). However, inflammation can
induce severe damage to various organs, including kidney (28), liver (29) and lungs (30). The use of drugs derived from plants,
such as CA from Salvia officinalis, has been revealed to be
beneficial in reversing inflammation-induced tissue injuries
(17). In the present study, the
effect of CA on LPS-induced inflammation and ALI was investigated
in a mouse model of lung injury induced by LPS. This model is
commonly used to determine the effectiveness of anti-inflammatory
agents (31). The results of the
present study demonstrated that CA significantly attenuated
LPS-induced inflammation and injuries in lungs.
CA is a major bioactive component of rosemary
extracts. The extensive use of rosemary extracts in medicines has
verified the strong pharmaceutical activity of CA (23,32,33).
Recent studies identified the tissue-protective role of CA in
several types of organ injury, including Parkinson's disease,
colitis and acute myocardial injury (22,32,33). The
protective effect of CA is mainly ascribed to its anti-inflammatory
and anti-oxidation properties (34).
It has been revealed that CA treatment can inhibit the infiltration
of inflammatory cells, including macrophages and neutrophils
(33,35). Consistent with that report, the
decreased infiltration of inflammatory cells was observed in the
present study and this was further supported by reduced levels of
MPO activity, a neutrophil marker. The initiation and development
of ARDS can be attributed to inflammatory cells, such as
macrophages and neutrophils (36).
In response to LPS exposure, alveolar macrophages become activated
and subsequently induce neutrophil infiltration (36). Infiltrated inflammatory cells produce
large quantities of reactive oxygen species (ROS) that exacerbate
inflammatory responses and significantly contribute to ALI
(37). CA exerts an antioxidant
effect by scavenging ROS and promoting the generation of secondary
anti-oxidants (34). Neutrophils
participate in ALI largely due to their production of
proinflammatory cytokines, including TNF-α, that delay apoptosis
(38). Neutrophils normally serve an
important role in body defense; however, the long-term survival of
neutrophils at inflammatory sites can aggravate tissue damage
(39). During an inflammatory
response, neutrophil clearance, achieved via efficient apoptosis,
is the primary mechanism that limits lung injuries and promotes the
dissipation of inflammation (40). A
previous study has shown that delayed neutrophil apoptosis,
prolonged neutrophil survival times and the continuous release of
cytotoxic factors (including oxygen free radicals) by neutrophils
into lung tissue result in lung injuries (41). The results of the present study
demonstrated that injection of LPS caused significant lung injuries
in rats and this significantly reduced the apoptosis of neutrophils
in BALF, indicating that the delayed apoptosis of neutrophils
contributed to lung injury in the rats and could potentially be
alleviated by increasing the apoptosis rate of neutrophils. In
addition to inhibiting inflammatory cell infiltration, the
anti-inflammatory activity of CA may also be due to an increase in
apoptosis in LPS-treated neutrophils, which may contribute to the
CA protection of LPS-induced ALI.
The anti-inflammatory activity of CA can be
attributed to its ability to inhibit the production of
proinflammatory cytokines, which are increased during the early
stages of ALI and are crucial for its occurrence and progression
(42). A previous study demonstrated
that CA inhibited the production of TNF-α in a macrophage cell
line, RAW 264.7, stimulated with LPS (43). In addition, increasing evidence
indicates that CA markedly inhibits the LPS-induced production of
TNF-a, IL-1β and IL-6 in vitro (43,44). In
a previous study, CA was determined to inhibit the increased
production of TNF-α and IL-6 in rats with LPS-induced liver
injuries and inhibit IL-1β, IL-17A and IFN-γ production in a rat
model of colitis induced by dextran sulfate sodium (17). In the present study, it was
determined that CA treatment significantly inhibited the
LPS-induced increases of TNF-a, IL-6 and IL-1β levels. These
cytokinesare secreted by the endothelium, epithelium and alveolar
macrophages in response to an initial inflammatory insult (36). The current results indicated that CA
inhibited the further exacerbation of endothelial and epithelial
injuries induced by LPS, and thereby dampened the inflammatory
process in ALI.
Several recent studies have demonstrated that the
NF-κB pathway serves a critical role in ALI (45–47). In
the present study, after LPS treatment, NF-κB is activated by TLR4,
leading to increases in the production of TNF-α, IL-1β and IL-6.
These cytokines activate NF-κB to initiate a cycle that broadens
the original immune response (42).
It has been revealed that CA inhibits NF-κB activation and the
downstream signaling process, leading to reduced levels of TNF-α,
IL-1β and IL-6 (48). Furthermore,
CA has also been demonstrated to block signaling pathways upstream
of NF-κB, including the Syk/Src, PI3K and Akt pathways (45). Consistent with a previous study
(49), the results of the current
study indicated that NF-κB activation was markedly increased in
LPS-induced lung injury. Although upstream signaling was not
investigated in the present study, the results indicated that
Syk/Src pathway inhibition may be involved in mediating the
protective effect of CA, as Syk has been demonstrated to be a
direct enzymatic target of CA (45).
The present study also revealed that TLR4 expression was
significantly increased in LPS-injured lung tissue, which was
significantly inhibited after treatment with CA, indicating that
the inflammatory response is limited by inhibiting the upstream
targets of TLR4. Consistent with the current results, a previous
study reported that CA exerted an inhibitory effect on the
expression and phosphorylation of NF-κB, as well as on the
expression of TLR4 in various inflammatory disorders, including
diabetes (50). Another study
reported that CA induced cytotoxicity in breast cancer cells
(51). In the current study, no
toxic effects of CA were detected and no mortality occurred in all
CA-treatment groups were equal. However, further studies are
required to determine whether repeated long-term administration of
CA produces toxic effects.
In summary, the current results indicated that CA
alleviated the injury of ALI by regulating immune responses and
this regulation may result from the ability of CA to inhibit the
expression of TLR4 and the phosphorylation of NF-κB. These results
strongly indicated that CA may be a promising agent for the
treatment of ARDS.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Science and
Technology Support Program of Suqian (grant no. S201628).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QL conceived and designed the present study. LL
performed the experiments. HS and KC completed data analysis and
interpretation. QL and KC drafted and revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols involving animals were
approved by the Ethics Committee of the Zhongda Hospital Southeast
University (Nanjing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Han S and Mallampalli RK: The acute
respiratory distress syndrome: From mechanism to translation. J
Immunol. 194:855–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gonzales JN, Lucas R and Verin AD: The
acute respiratory distress syndrome: Mechanisms and perspective
therapeutic approaches. Austin J Vasc Med. 2(pii):
10092015.PubMed/NCBI
|
|
3
|
Koh Y: Update in acute respiratory
distress syndrome. J Intensive Care. 2:22014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geboers DG, de Beer FM, Tuip-de Boer AM,
van der Poll T, Horn J, Cremer OL, Bonten MJ, Ong DS, Schultz MJ
and Bos LD: Plasma suPAR as a prognostic biological marker for ICU
mortality in ARDS patients. Intensive Care Med. 41:1281–1290. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peng J, Wei D, Fu Z, Li D, Tan Y, Xu T,
Zhou J and Zhang T: Punicalagin ameliorates
lipopolysaccharide-induced acute respiratory distress syndrome in
mice. Inflammation. 38:493–499. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bdeir K, Higazi AA, Kulikovskaya I,
Christofidou-Solomidou M, Vinogradov SA, Allen TC, Idell S,
Linzmeier R, Ganz T and Cines DB: Neutrophil alpha-defensins cause
lung injury by disrupting the capillary-epithelial barrier. Am J
Respir Crit Care Med. 181:935–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cribbs SK and Martin GS: Stem cells in
sepsis and acute lung injury. Am J Med Sci. 341:325–332. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou X, Dai Q and Huang X: Neutrophils in
acute lung injury. Front Biosci (Landmark Ed). 17:2278–2283. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu T, Wang DX, Zhang W, Liao XQ, Guan X,
Bo H, Sun JY, Huang NW, He J, Zhang YK, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-κB. PLoS One. 8:e564072013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murray LA, Knight DA, McAlonan L,
Argentieri R, Joshi A, Shaheen F, Cunningham M, Alexopolou L,
Flavell RA, Sarisky RT and Hogaboam CM: Deleterious role of TLR3
during hyperoxia-induced acute lung injury. Am J Respir Crit Care
Med. 178:1227–1237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barsness KA, Arcaroli J, Harken AH,
Abraham E, Banerjee A, Reznikov L and McIntyre RC:
Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J
Physiol Regul Integr Comp Physiol. 287:R592–R599. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeyaseelan S, Chu HW, Young SK, Freeman MW
and Worthen GS: Distinct roles of pattern recognition receptors
CD14 and Toll-like receptor 4 in acute lung injury. Infect Immun.
73:1754–1763. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kazemi S, Shirzad H and Rafieian-Kopaei M:
Recent findings in molecular basis of inflammation and
anti-inflammatory plants. Curr Pharm Des. 24:1551–1562. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nardi GM, Farias Januario AG, Freire CG,
Megiolaro F, Schneider K, Perazzoli MR, Do Nascimento SR, Gon AC,
Mariano LN, Wagner G, et al: Anti-inflammatory activity of berry
fruits in mice model of inflammation is based on oxidative stress
modulation. Pharmacognosy Res. 8 (Suppl 1):S42–S49. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiang Q, Liu Z, Wang Y, Xiao H, Wu W, Xiao
C and Liu X: Carnosic acid attenuates lipopolysaccharide-induced
liver injury in rats via fortifying cellular antioxidant defense
system. Food Chem Toxicol. 53:1–9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Sun JJ, Chen GY, Wang WW, Xie ZT,
Tang GF and Wei SD: Carnosic acid nanoparticles suppress liver
ischemia/reperfusion injury by inhibition of ROS, Caspases and
NF-κB signaling pathway in mice. Biomed Pharmacother. 82:237–246.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang T, Takikawa Y, Satoh T, Yoshioka Y,
Kosaka K, Tatemichi Y and Suzuki K: Carnosic acid prevents obesity
and hepatic steatosis in ob/ob mice. Hepatol Res. 41:87–92. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng CH, Su JD, Chyau CC, Sung TY, Ho SS,
Peng CC and Peng RY: Supercritical fluid extracts of rosemary
leaves exhibit potent anti-inflammation and anti-tumor effects.
Biosci Biotechnol Biochem. 71:2223–2232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin HB, Choi MS, Ryu B, Lee NR, Kim HI,
Choi HE, Chang J, Lee KT, Jang DS and Inn KS: Antiviral activity of
carnosic acid against respiratory syncytial virus. Virol J.
10:3032013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Oliveira MR: The dietary components
carnosic acid and carnosol as neuroprotective agents: A mechanistic
view. Mol Neurobiol. 53:6155–6168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kocak C, Kocak FE, Akcilar R, Isiklar OO,
Kocak H, Bayat Z, Simsek H, Taser F and Altuntas I: Molecular and
biochemical evidence on the protective effects of embelin and
carnosic acid in isoproterenol-induced acute myocardial injury in
rats. Life Sci. 147:15–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma HJ, Huang XL, Liu Y and Fan YM: Sulfur
dioxide attenuates LPS-induced acute lung injury via enhancing
polymorphonuclear neutrophil apoptosis. Acta Pharmacol Sin.
33:983–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grommes J and Soehnlein O: Contribution of
neutrophils to acute lung injury. Mol Med. 17:293–307. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li YY, Zhang GY, He JP, Zhang DD, Kong XX,
Yuan HM and Chen FL: Ufm1 inhibits LPS-induced endothelial cell
inflammatory responses through the NF-κB signaling pathway. Int J
Mol Med. 39:1119–1126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chovatiya R and Medzhitov R: Stress,
inflammation, and defense of homeostasis. Mol Cell. 54:281–288.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ueda N and Takasawa K: Impact of
inflammation on ferritin, hepcidin and the management of iron
deficiency anemia in chronic kidney disease. Nutrients. 10(pii):
E11732018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li S, Hong M, Tan HY, Wang N and Feng Y:
Insights into the role and interdependence of oxidative stress and
inflammation in liver diseases. Oxid Med Cell Longev.
2016:42340612016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bhargava R, Janssen W, Altmann C,
Andrés-Hernando A, Okamura K, Vandivier RW, Ahuja N and Faubel S:
Intratracheal IL-6 protects against lung inflammation in direct,
but not indirect, causes of acute lung injury in mice. PLoS One.
8:e614052013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen H, Bai C and Wang X: The value of the
lipopolysaccharide-induced acute lung injury model in respiratory
medicine. Expert Rev Respir Med. 4:773–783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin CY, Chen JH, Fu RH and Tsai CW:
Induction of Pi form of glutathione S-transferase by carnosic acid
is mediated through PI3K/Akt/NF-κB pathway and protects against
neurotoxicity. Chem Res Toxicol. 27:1958–1966. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang N, Xia Z, Shao N, Li B, Xue L, Peng
Y, Zhi F and Yang Y: Carnosic acid prevents dextran sulfate
sodium-induced acute colitis associated with the regulation of the
Keap1/Nrf2 pathway. Sci Rep. 7:110362017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xia G, Wang X, Sun H, Qin Y and Fu M:
Carnosic acid (CA) attenuates collagen-induced arthritis in db/db
mice via inflammation suppression by regulating ROS-dependent p38
pathway. Free Radic Biol Med. 108:418–432. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Osakabe N, Yasuda A, Natsume M and
Yoshikawa T: Rosmarinic acid inhibits epidermal inflammatory
responses: Anticarcinogenic effect of Perilla frutescens extract in
the murine two-stage skin model. Carcinogenesis. 25:549–557. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Williams AE and Chambers RC: The mercurial
nature of neutrophils: Still an enigma in ARDS? Am J Physiol Lung
Cell Mol Physiol. 306:L217–L230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dong Z and Yuan Y: Accelerated
inflammation and oxidative stress induced by LPS in acute lung
injury: Inhibition by ST1926. Int J Mol Med. 41:3405–3421.
2018.PubMed/NCBI
|
|
38
|
Lin WC, Lin CF, Chen CL, Chen CW and Lin
YS: Inhibition of neutrophil apoptosis via sphingolipid signaling
in acute lung injury. J Pharmacol Exp Ther. 339:45–53. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Selders GS, Fetz AE, Radic MZ and Bowlin
GL: An overview of the role of neutrophils in innate immunity,
inflammation and host-biomaterial integration. Regen Biomater.
4:55–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee WL and Downey GP: Neutrophil
activation and acute lung injury. Curr Opin Crit Care. 7:1–7. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhenglong Y, Qiaolian X and Haibo Q: Role
of neutrophil apoptosis in the lung of rats with acute lung injury.
Chin J Crit Care Med. 25:503–505. 2005.
|
|
42
|
Zhang X, Shang F, Hui L, Zang K and Sun G:
The alleviative effects of metformin for lipopolysaccharide-induced
acute lung injury rat model and its underlying mechanism. Saudi
Pharm J. 25:666–670. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kuo CF, Su JD, Chiu CH, Peng CC, Chang CH,
Sung TY, Huang SH, Lee WC and Chyau CC: Anti-inflammatory effects
of supercritical carbon dioxide extract and its isolated carnosic
acid from Rosmarinus officinalis leaves. J Agric Food Chem.
59:3674–3685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang T, Zhang J, Sun L, Zhu X, Li J, Wang
J, Chen H, Bao R, Deng X, Hou J and Liu Y: Combined effects of a
neutrophil elastase inhibitor (sivelestat sodium) and a free
radical scavenger (edaravone) on lipopolysaccharide-induced acute
lung injury in rats. Inflamm Res. 61:563–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu SF and Malik AB: NF-kappa B activation
as a pathological mechanism of septic shock and inflammation. Am J
Physiol Lung Cell Mol Physiol. 290:L622–L645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Everhart MB, Han W, Sherrill TP, Arutiunov
M, Polosukhin VV, Burke JR, Sadikot RT, Christman JW, Yull FE and
Blackwell TS: Duration and intensity of NF-kappaB activity
determine the severity of endotoxin-induced acute lung injury. J
Immunol. 176:4995–5005. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu S, Feng G, Wang GL and Liu GJ: p38MAPK
inhibition attenuates LPS-induced acute lung injury involvement of
NF-kappaB pathway. Eur J Pharmacol. 584:159–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tang B, Tang F, Wang Z, Qi G, Liang X, Li
B, Yuan S, Liu J, Yu S and He S: Upregulation of
Akt/NF-κB-regulated inflammation and Akt/Bad-related apoptosis
signaling pathway involved in hepatic carcinoma process:
Suppression by carnosic acid nanoparticle. Int J Nanomedicine.
11:6401–6420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Oh J, Yu T, Choi SJ, Yang Y, Baek HS, An
SA, Kwon LK, Kim J, Rho HS, Shin SS, et al: Syk/Src
pathway-targeted inhibition of skin inflammatory responses by
carnosic acid. Mediators Inflamm. 2012:7813752012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Park MY and Mun ST: Carnosic acid inhibits
TLR4-MyD88 signaling pathway in LPS-stimulated 3T3-L1 adipocytes.
Nutr Res Pract. 8:516–520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yildiz-Ozturk E, Gulce-Iz S, Anil M and
Yesil-Celiktas O: Cytotoxic responses of carnosic acid and
doxorubicin on breast cancer cells in butterfly-shaped microchips
in comparison to 2Dand 3D culture. Cytotechnology. 69:337–347.
2017. View Article : Google Scholar : PubMed/NCBI
|