Introduction
The 46,XY sex reversal syndrome (SRS) is
characterized by a 46,XY karyotype, normal female external
genitalia, completely undeveloped (‘streak’) gonads, no sperm
production and the presence of normal Müllerian structures. The
syndrome occurs with an estimated incidence of 1:5,000 (1). Individuals with an underlying SRS,
particularly those with specific Y chromosomal material in their
karyotype, have an increased risk of developing a type II germ cell
tumor/cancer (GCC) (1). GCCs arise
from primordial germ cells or gonocytes and may be subdivided into
seminomas/dysgerminomas and non-seminomas with carcinoma in
situ or gonadoblastomas as precursor lesions (2). This study presents a unique case with
bilateral gonadoblastoma and dysgerminoma in a girl presenting with
primary amenorrhea at the age of 17 years, who was initially
diagnosed with 46,XY SRS.
Case report
A 17-year-old girl was admitted to the General
Hospital of Tianjin Medical University (Tianjin, China) with no
menarche and continuous height growth. The patient was treated with
artificial estrogen and progesterone to induce an artificial
menstrual cycle for two months, but only a small amount of pink
secretion was observed in the vagina. The patient was born
following a full-term normal delivery to non-consanguineous
parents, and her mother denied the use of any sex hormone drugs or
exposure to radioactive substances during pregnancy. The younger
brother of the patient exhibited a normal phenotype. Physical
examination showed that the patient had relatively long upper
extremities, and the arm span was greater than the height of the
patient. The patient exhibited a female appearance and voice, with
little subcutaneous fat, no beard or laryngeal prominence, and
hypoplastic breasts with a light areola. No palpable mass was
identified in the groin or labia majora. The patient exhibited
female external genitalia, with normal labia majora and minora,
sparse pubic hair and a visible vaginal orifice. The anal
examination showed a small uterus, a mass of ~4.0×3.0 cm in the
right adnexa, and another mass of ~3.0×3.0 cm in the left adnexa,
both of which were hard and mobile. The B-mode ultrasound showed an
infantile uterus, the endometrium appeared as a thin echogenic
line, and the masses in the adnexa were non-homogeneous measuring
~45×15 mm (right) and ~33×14 mm (left), respectively. Serum sex
hormone analysis revealed that the follicle-stimulating hormone
level was 104.5 IU/l, the luteinizing hormone level was 43.1 IU/l,
the estradiol level was <10 pg/ml and the testosterone level was
55 ng/dl. The patient exhibited a slightly higher level of serum
testosterone than normal and a low level of serum estrogen.
Peripheral blood chromosome analysis showed the 46,XY karyotype. No
gene deletions were detected at sY84, sY86, sY127, sY134, sY254 and
sY255 of the sex determining region Y (SRY) gene. The
patient was diagnosed with 46,XY female SRS, simple gonadal
dysgenesis and unclear bilateral gonadal mass.
During the laparotomy, the small uterus was apparent
with a size of 5.0×5.0 cm. The right gonad measured 4.0×3.0×3.0 cm,
had an ovary-like appearance with a complete capsule, and was
white, smooth and hard in texture; while the left gonad measured
3.0×3.0×3.0 cm and had a similar appearance to the right gonad. The
uterine tubes were narrow. No testes or associated tissues were
identified in the region between the superficial and deep inguinal
rings and the urinary bladder, and the bifurcation of the common
iliac artery. Bilateral gonadectomy was performed for simple
gonadal dysgenesis. The patient was then pathologically diagnosed
with bilateral ovarian gonadoblastoma and right dysgerminoma. The
patient was genetically male due to her 46,XY karyotype, but
socially and psychologically female in every respect; thus, an
important aspect of postoperative treatment was to maintain the
female characteristics. Estrogen (Premarin) and medroxyprogesterone
acetate replacement therapy was administered. A four year follow-up
showed that the patient treated with a sequential therapy of
estrogen and progesterone had a regular menstrual cycle and no
tumor recurrence.
This study was approved by the Ethics committee of
Tianjin Medical University (Tianjin, China) and patient informed
consent was obtained.
Discussion
Although the genetic sex of an individual is
determined at fertilization, it is at the embryonic stage that the
sexual differentiation of the reproductive system begins. The
primordial gonad is bipotential and can differentiate into a testis
or an ovary, depending on the SRY gene located in the short
arm of the Y-chromosome. The absence of SRY permits the
bipotential gonad to differentiate into an ovary at the eighth week
of the embryo, leading to the female phenotype. The mutation,
deletion or translocation of SRY can affect the binding of
the SRY proteins with DNA, and consequently contribute to sex
reversal (3). In the present case,
the patient with a positive SRY gene on the Y-chromosome had
no testes, but had an ovarian cortex and hypoplastic female
external genitalia. This finding indicates that the SRY gene
is not the only gene responsible for testis development, and there
may be other genes besides the SRY gene involved as well
(4). Duplication of the DAX1 (also
known as NR0B1) and WNT4 genes, as well as
haploinsufficiency of the SOX9, SF1, WT1 and
DMRT1-DMRT2 genes, have been considered responsible
for the development of 46,XY sex reversal (5,6) and
therefore the SRY gene does not play a significant role in
the etiology of the disease in this case. No gene deletions
including sY84, sY86, sY127, sY134, sY254 and sY255 loci of the
SRY gene were detected; future studies are required to focus
on other loci of the SRY gene. Although no genetic
abnormality was detected in this patient, there may be
abnormalities in the content and function of SRY protein, which may
result in the lack of testis development and the lack of secretion
of testosterone and Müllerian-inhibiting factor. As a result, the
Wolffian duct degenerates and does not differentiate into the male
reproductive tract. By contrast, in the absence of
Müllerian-inhibiting factor, the Müllerian duct differentiates into
the oviducts, uterus, cervix and upper vagina. However, such female
patients without the presence of XX chromosomes do not have a
properly developed uterus or ovary, which is characterized by
primary amenorrhea, high height, poorly developed secondary sex
characteristics and external genitalia. The discordance between
chromosomal and gonadal sex is determined as SRS (4), including 46,XX male and 46,XY female
SRS.
The undeveloped primordial gonad does not have a
normal physiological function, and thus has a potential risk of
malignancy. Patients with chromosomal disorders of sex development
(DSD) that have specific Y-chromosomal material in their karyotype,
the gonadoblastoma on the Y-chromosome (GBY) region, have an
increased risk of developing GCC (7). GCC risk varies, but is estimated to
be >30% in patients with simple gonadal dysgenesis and is often
bilateral (8).
GCC may be induced by the interaction of abnormal
streak gonads with the intra-abdominal environment, or by gene
mutation. As a result, gonadectomy should be performed on the
diseased side or bilaterally for prophylactic purposes. In the
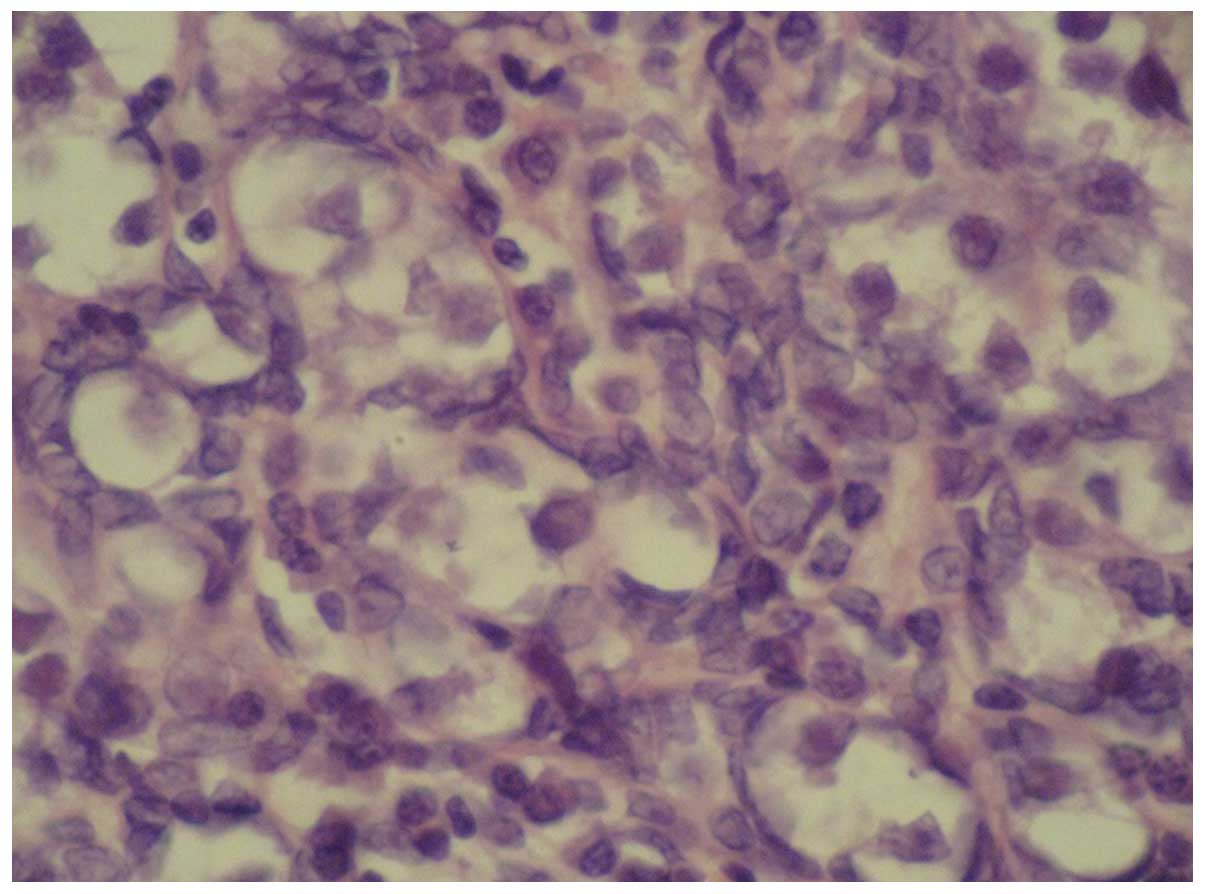
present case, the patient received bilateral gonadectomy. Fig. 1 shows the pathological features of
the ovarian gonadoblastoma: the supporting cells and granular cells
are arranged around the gonadoblastoma nests, and the mixed cells
are at the center of the nests. It is also noted that the
intercellular substance is calcified, and thus the section is
off-white or grayish yellow, like sandstone. Gonadoblastoma can be
either in a pure form or mixed with other germ cell tumors. In
50–60% of cases gonadoblastomas are associated with malignant germ
cell tumours, most presented as pure dysgerminom or less frequently
as immature teratoma, Yolk sac tumour (9). The mixed gonadoblastoma is
potentially malignant and has a poor prognosis. In the present
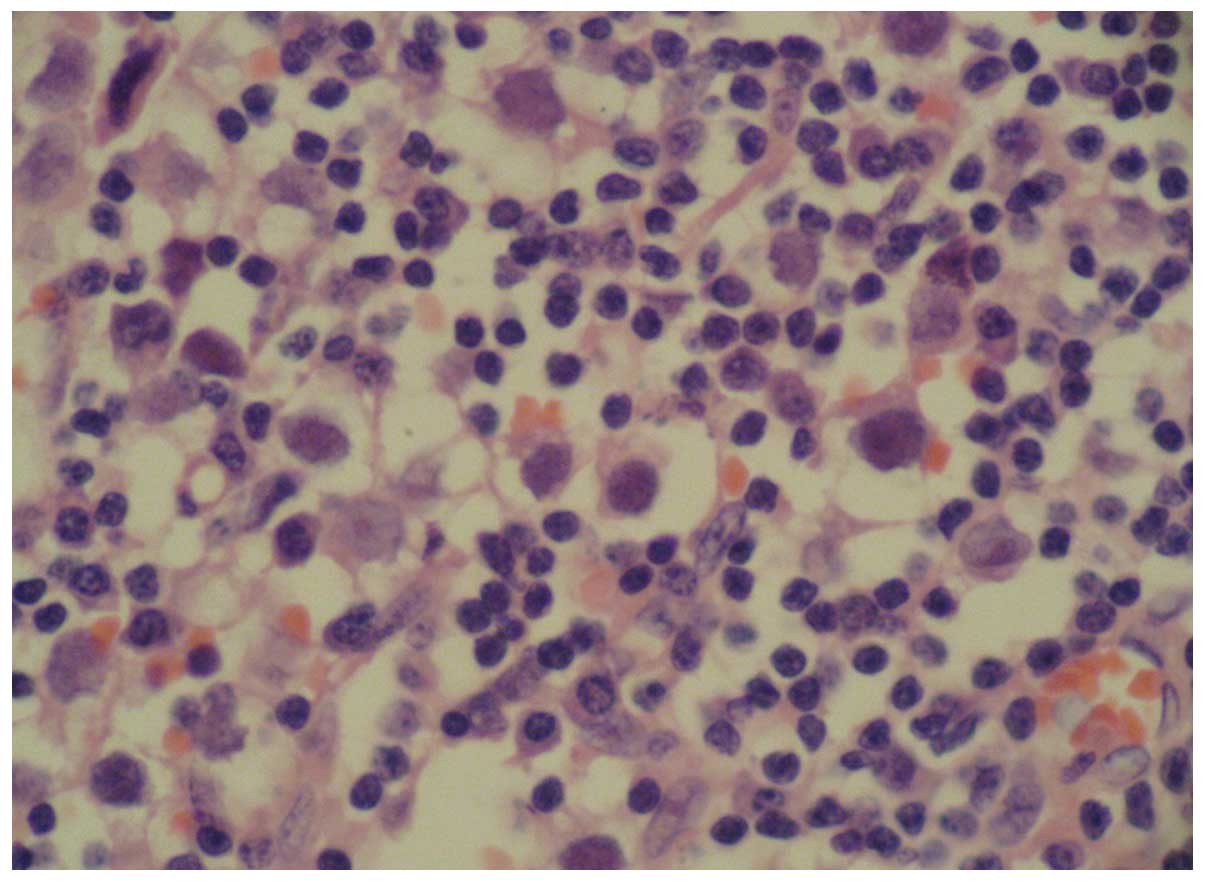
case, the patient was diagnosed as having stage-Ia gonadoblastoma
mixed with malignant dysgerminoma in the right ovary, and pure
benign gonadoblastoma in the left ovary, as shown in Fig. 2. Follow-up was performed at a fixed
interval. In this case, although the chromosomal sex of the SRS
patient was male, the patient had female external genitalia and had
been living as a female in the preceding years. Thus, estrogen
replacement therapy was administered periodically to promote the
development of secondary sexual characteristics and menstruation,
and at the same time, measures were taken to prevent osteoporosis.
It allowed the patient to have a more regular menstrual cycle, and
the four year follow-up showed no tumor recurrence.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81303108 and 81201871).
References
|
1
|
Hughes IA, Houk C, Ahmed SF and Lee PA:
Consensus statement on management of intersex disorders. Arch Dis
Child. 91:554–563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hersmus R, de Leeuw BH, Wolffenbuttel KP,
et al: New insights into type II germ cell tumor pathogenesis based
on studies of patients with various forms of disorders of sex
development (DSD). Mol Cell Endocrinol. 291:1–10. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu GF, Jin RL and Jia HP: Diagnosis of the
missense mutation of SRY gene in a patient with 46XY female sex
reverse. Medical & Pharmaceutical Journal of Chinese People’s
Liberation Army. 23:4–6. 2011.(In Chinese).
|
|
4
|
Bao MS, Wu XB, Chen SH, et al: Analysis of
SRY gene in patients with sex reversal syndrome. The Journal of
Practical Medicine. 23:348–350. 2007.(In Chinese).
|
|
5
|
Sukumaran A, Desmangles JC, Gartner LA and
Buchlis J: Duplication of dosage sensitive sex reversal area in a
46, XY patient with normal sex determining region of Y causing
complete sex reversal. J Pediatr Endocrinol Metab. 26:775–779.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Domenice S, Corrêa RV, Costa EM, et al:
Mutations in the SRY, DAX1, SF1 and WNT4 genes in Brazilian
sex-reversed patients. Braz J Med Biol Res. 37:145–150. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hersmus R, Stoop H, Turbitt E, et al: SRY
mutation analysis by next generation (deep) sequencing in a cohort
of chromosomal Disorders of Sex Development (DSD) patients with a
mosaic karyotype. BMC Med Genet. 13:1082012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cools M, Drop SL, Wolffenbuttel KP,
Oosterhuis JW and Looijenga LH: Germ cell tumors in the intersex
gonad: old paths, new directions, moving frontiers. Endocr Rev.
27:468–484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanagal DV, Prasad K, Rajesh A, et al:
Ovarian gonadoblastoma with dysgerminoma in a young girl with 46,
XX Karyotype: A Case Report. J Clin Diagn Res. 9:2021–2022.
2013.PubMed/NCBI
|