Introduction
Colorectal cancer is one of the leading causes of
mortality and is a major public health concern worldwide (Guo). The
incidence of colorectal cancer is also increasing among the Chinese
population (Jin). Chemoprevention is the most promising strategy
since other therapies are not effective in controlling
gastrointestinal cancers (Andre). However, recent studies have
focused on the antitumor properties of natural products since these
materials typically have fewer side-effects and are more suitable
for long-term use compared with chemically synthesized medicines
(16). Emodin, a naturally
occurring compound that may be extracted from various Chinese
plants, including Rheum officinale and Polygonum
cuspidatum (1), is known for
its anticarcinogenic effects that have been demonstrated in a
variety of experimental cancer models (2). Pharmacological studies have shown
that emodin exhibits anticancer effects in a number of human
cancers (3). In addition, previous
studies have shown that emodin exerts antiproliferative and
apoptosis-inducing effects on cell lines derived from ovarian
(4) and lung (5) cancer and leukemia (6). However, at present there is little
information demonstrating the possible antiproliferative effect of
emodin on colorectal cancer. Therefore, in the present study, the
inhibitory effect of emodin on cell proliferation in a colorectal
cancer cell line was investigated, as well as the molecular
mechanisms involved.
Materials and methods
Cell culture and treatment
LOVO colorectal cancer cells were purchased from the
American Type Culture Collection (ATCC, Manassas, VA, USA). The
cells were cultured as monolayers in RPMI-1640 medium (Gibco-BRL,
Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum
(Gibco-BRL, São Paulo, Brazil) and 1% penicillin/streptomycin. The
cells were grown at 37°C in a humidified atmosphere containing 5%
CO2. Emodin was purchased from Sigma-Aldrich (St. Louis,
MO, USA) and was freshly diluted to concentrations of 0, 10, 20 and
40 μmol/l with dimethyl sulfoxide (DMSO). The final concentration
of DMSO was <0.1%. The cells were treated with different
concentrations of emodin for 24 h and then analyzed.
Cell viability assay
Cell viability was determined using Cell-Counting
kit-8 (CCK-8, Dojindo Laboratories, Tokyo, Japan). Cells were
seeded into a 96-well plate at a density of 5×105
cells/well and cultured for 24 h. Emodin was then added to the
wells with final concentrations of 0, 10, 20 and 40 μmol/l; DMSO
alone was used for the control group. After 24 h, CCK-8 solution
was added (10 μl to each well containing 100 μl medium). The plates
were incubated at 37°C for 4 h, and the absorbance at 450 nm was
then measured using a Benchmark Plus microplate reader (Bio-Rad,
Hercules, CA, USA). All experiments were performed in
triplicate.
Reverse transcription polymerase chain
reaction (RT-PCR)
Total RNA was isolated from cells using
TRIzol® reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA) and quantified using UV absorption at 260 and 280 nm.
RT-PCR was performed in accordance with the reverse-transcription
PCR (RT-PCR) reaction kit (GIBCO, Grand Island, NY, USA)
instructions. RT-PCR primers were designed as follows: B-cell
lymphoma-2 (Bcl-2) forward, 5′-CTTTTGCTGTGGGGT TTTGT-3′ and
reverse, 5′-GTCATTCTGGCCTCTCTTGC-3′; Bcl-2-associated X protein
(Bax) forward, 5′-GGAGCT GCAGAGGATGATTG-3′ and reverse,
5′-CCTCCCAGA AAAATGCCATA-3′. The RT-PCR process was performed as
follows: Denaturation for 1 min at 93°C, followed by 31, 33 and 32
cycles of denaturing for 30 sec at 94°C, annealing for 1 min at
57°C, 60°C and 57°C for GAPDH, Bcl-2 and Bax, respectively, and
extension for 2 min at 72°C. The amplified products were visualized
using 1.5% agarose gel electrophoresis, stained with ethidium
bromide and images were then captured under ultraviolet light.
Densitometric analysis of three different observations was
performed using Quantity One Software (Bio-Rad). The quantity of
each transcript was normalized against GAPDH.
Western blot analysis
The cells were harvested at the indicated times, and
lysed with lysis buffer [50 mM Tris Cl, (pH 7.8), 150 mM NaCl, 1%
NP40, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride]. The
total protein (50 μg per well) was separated by 10% SDS-PAGE gels,
and electrophoretically transferred onto nitrocellulose membranes.
Following blocking for 1 h with 5% skimmed milk in Tris-buffered
saline (TBS) buffer (10 mM Tris and 150 mM NaCl), the membrane was
washed 3 times for 18 min each with TBST buffer (10 mM Tris, 150 mM
NaCl and 0.1% Tween-20). Immunoreactive bands were visualized using
horseradish peroxidase-conjugated secondary antibody (1:5,000
dilution; Beyotime Institute of Biotechnology, Haimen, Jiangsu,
China) and an enhanced chemiluminescene western blotting detection
kit (Amersham, Little Chalfont, Buckinghamshire, UK). The bands
were visualized using the enhanced chemilluminescence system, and
the band density was determined by Image J software (National
Institutes of Health, Bethestha, MD, USA. The antibodies were all
purchased from Beyotime Institute of Biotechnology.
Mitochondrial membrane potential
assay
Mitochondrial potential was analyzed using the
fluorescent potentiometric dye JC-1 as described previously
(7). Briefly, following 24 h of
treatment with 0–40 μmol/l emodin, cells were harvested, washed
twice with phosphate-buffered saline (PBS) and centrifuged for 8
min at 450 × g at room temperature. The cells were then suspended
with JC-1 (5 μg/ml) in serum-free RPMI-1640 and incubated for 15
min at 37°C. Following staining, the cells were collected at room
temperature and washed three times with pre-warmed PBS. The cell
pellet was then re-suspended in 1 ml PBS. Fluorescence emitted by
JC-1 was quantified using a fluorescence plate reader (Molecular
Devices, Sunnyvale, CA, USA) at 37°C. The fluorescence of the JC-1
monomer was measured at 485 nm excitation wavelength/530 nm
emission wavelength. The fluorescence of the JC-1 aggregate was
measured at 535 nm excitation wavelength/590 nm emission
wavelength. For each experiment, the ratios of JC-1 aggregate to
JC-1 monomer were normalized against untreated controls; the values
therefore represent a percentage of the mitochondrial function in
untreated cells.
Analysis of cytochrome c release
Cytochrome c release from the mitochondria
was evaluated by western blot analysis of cytosolic protein samples
(8,9). Cells were harvested following
treatment with emodin for 24 h and mixed with 100 μl cold lysis
buffer [50 mmol/l Tris-HCl (pH 7.4), 1 mmol/l NaF, 150 mmol/l NaCl,
1 mmol/l ethylene glycol tetraacetic acid, 1 mmol/l
phenylmethylsulfonyl fluoride (PMSF), 1% (v/v) Nonidet P-40 (NP-40)
and 10 μg/ml leupeptin], followed by centrifugation at 10,000 × g
for 30 min at 4°C. The supernatant was centrifuged two more times
under the same conditions to remove the nuclear debris. The
cytosolic extracts were then used for western blot analysis to
analyze the level of cytochrome c release. Western blot
analysis was subsequently performed as described above.
Statistical analysis
Data are expressed as the means ± standard deviation
of three assays. The statistical analysis was performed using a
one-way analysis of variance. A P-value of <0.05 was considered
to indicate a statistically significant difference. All statistical
analyses were performed using SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA).
Results
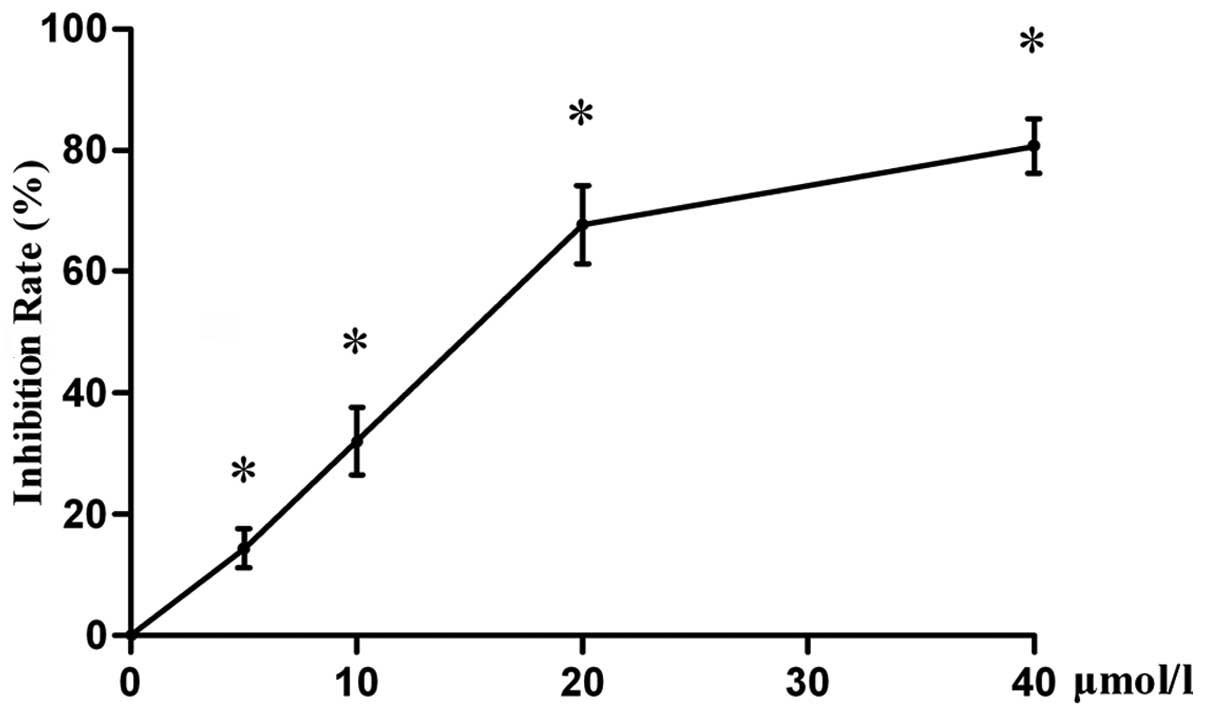
Effect of emodin on cell viability
In order to investigate the antiproliferative effect
of emodin on LOVO cells in vitro, cell proliferation was
measured using the CCK-8 assay. The results of the present study
(Fig. 1) demonstrated that emodin
inhibits cell proliferation in a concentration-dependent manner,
compared with that of vehicle-treated controls (P<0.05).
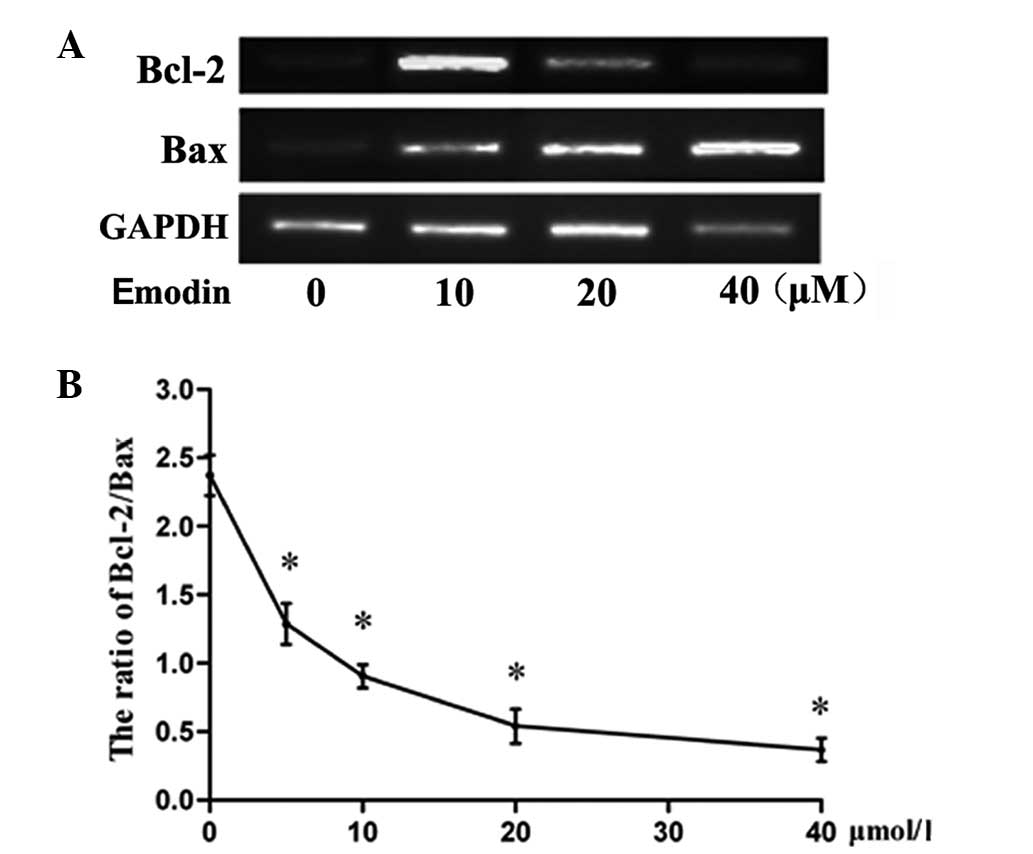
Effect of emodin on Bcl-2 and Bax
expression in LOVO cells
RT-PCR was performed to determined the Bcl-2 and Bax
expression levels of the cells. It was found that emodin
upregulated the expression of Bax in the treatment groups compared
with that in the control group (P<0.05), whereas it inhibited
the expression of Bcl-2 (Fig. 2).
Furthermore, significant differences in the Bax/Bcl-2 ratio between
the treatment groups and the control cells were observed (Fig. 2).
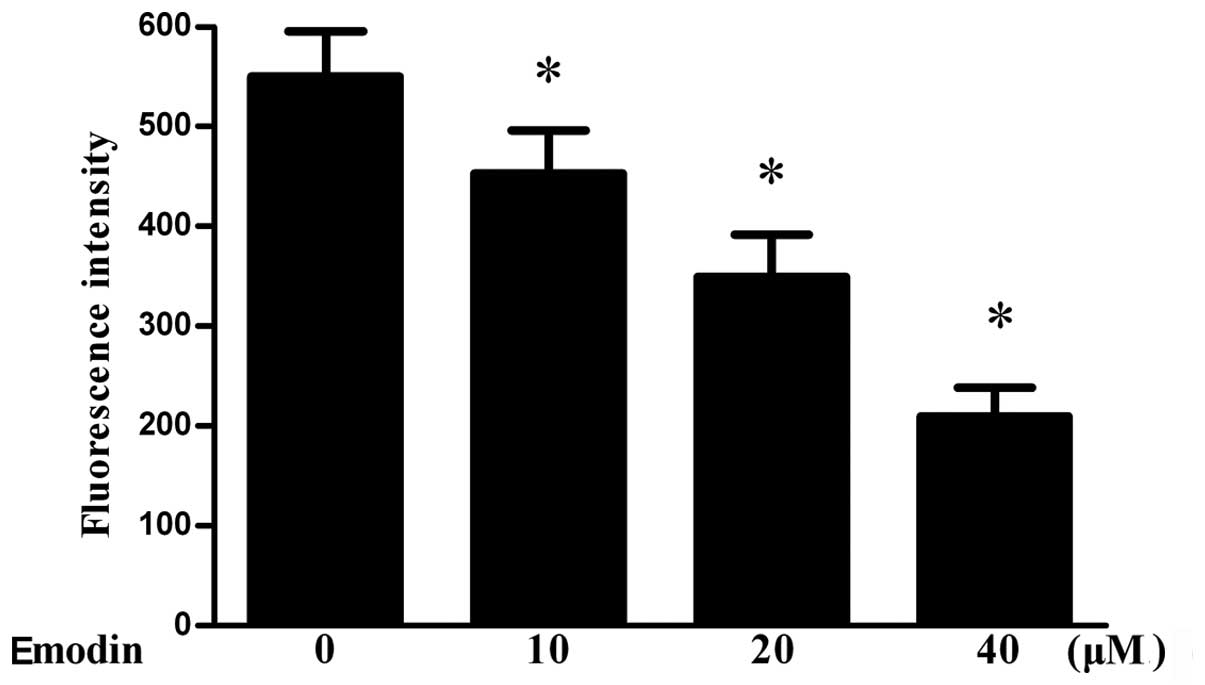
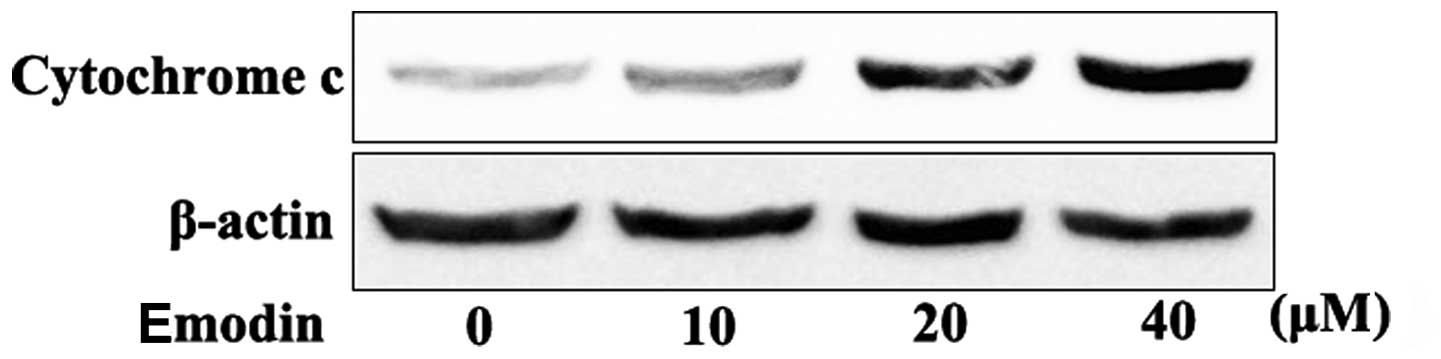
Effect of emodin on mitochondrial
membrane potential and release of cytochrome c
In order to determine if treatment with emodin
decreases the mitochondrial membrane potential, cells were treated
with different concentrations of emodin for 24 h prior to
measurement of the mitochondrial membrane potential. The results
indicated that the mitochondrial membrane potential was
significantly decreased by emodin (Fig. 3). In addition, it was found that
the release of cytochrome c was significantly increased in
cells incubated with 10–40 μmol/l emodin (Fig. 4).
Discussion
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is a
naturally occurring anthraquinone present in rhubarb and numerous
other plants. As well as being used as a laxative, emodin has a
number of other biological effects, including antibacterial,
antiviral, anti-inflammatory and anticancer effects (10). However, at present, there is little
evidence of the possible effects of emodin on proliferation in
colorectal cancer. Therefore, in the present study, the molecular
mechanism involved in the inhibitory effect of emodin on cell
proliferation was investigated.
The inhibitory effect of emodin on LOVO colorectal
cell proliferation was investigated, as well as the possible
underlying mechanisms. It was observed that emodin inhibited LOVO
cell proliferation, which is in accordance with a previous study
(11).
Bcl-2 family proteins function through different
pathways in the regulation of cell apoptosis (12). The Bcl-2 family primarily affects
the mitochondrial pathways (13).
Bcl-2 and its homologs prevent mitochondrial membrane disruption
and the release of cytochrome c and other pro-apoptotic
factors, while Bax promotes these events. The expression of these
two opposing proteins may, in part, determine the fate of cells.
The ratio of Bcl-2/Bax is usually regarded as a criterion for
programmed cell death (14). The
results from the present study demonstrate that emodin upregulates
the expression of Bax, but inhibits the expression of Bcl-2. It was
also found that there were significant differences in the Bax/Bcl-2
ratio between the treatment and the control groups. These data are
consistent with emodin-inhibited cancer cell growth being
associated with the balance of Bcl-2/Bax.
In addition, the mitochondrial pathway is a major
apoptosis signaling pathway. Numerous studies have shown that the
mitochondrial membrane potential stimulates the mitochondrial
membrane to open, resulting in the release of cytochrome c
into the cytoplasm, activation of the caspase pathway and
degradation of important intracellular proteins, and consequently
the induction of apoptosis (15).
In the present study, it was found that the mitochondrial membrane
potential was significantly decreased by emodin in a concentration-
and time-dependent manner. In addition, the results from the
western blot analysis revealed that cytochrome c was
released from the mitochondria into the cytoplasm, suggesting that
the mitochondrial apoptosis pathway is involved in emodin-induced
cell line apoptosis. These results are in accordance with a
previous study by Wang et al (16), which demonstrated that emodin
inhibited HeLa proliferation by the induction of apoptosis through
the intrinsic mitochondrial pathway.
In conclusion, the results from the present study
suggest that emodin inhibits cancer cell growth via the regulation
of Bcl-2/Bax and that the mitochondrial apoptosis pathway may be
involved.
References
|
1
|
Ma YS, Weng SW, Lin MW, et al: Antitumor
effects of emodin on LS1034 human colon cancer cells in vitro and
in vivo: roles of apoptotic cell death and LS1034 tumor xenografts
model. Food Chem Toxicol. 50:1271–1278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jing X, Ueki N, Cheng J, Imanishi H and
Hada T: Induction of apoptosis in hepatocellular carcinoma cell
lines by emodin. Jpn J Cancer Res. 93:874–882. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu A, Chen H, Wei W, et al:
Antiproliferative and antimetastatic effects of emodin on human
pancreatic cancer. Oncol Rep. 26:81–89. 2011.PubMed/NCBI
|
|
4
|
Li J, Liu P, Mao H, Wanga A and Zhang X:
Emodin sensitizes paclitaxel-resistant human ovarian cancer cells
to paclitaxel-induced apoptosis in vitro. Oncol Rep.
21:1605–1610. 2009.PubMed/NCBI
|
|
5
|
Ko JC, Su YJ, Lin ST, et al: Suppression
of ERCC1 and Rad51 expression through ERK1/2 inactivation is
essential in emodin-mediated cytotoxicity in human non-small cell
lung cancer cells. Biochem Pharmacol. 79:655–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang CG, Yang JQ, Liu BZ, et al:
Anti-tumor activity of emodin against human chronic myelocytic
leukemia K562 cell lines in vitro and in vivo. Eur J Pharmacol.
627:33–41. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shang T, Joseph J, Hillard CJ and
Kalyanaraman B: Death-associated protein kinase as a sensor of
mitochondrial membrane potential: role of lysosome in mitochondrial
toxin-induced cell death. J Biol Chem. 280:34644–34653. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bae JH, Park JW and Kwon TK: Ruthenium
red, inhibitor of mitochondrial Ca2+ uniporter, inhibits
curcumin-induced apoptosis via the prevention of intracellular
Ca2+ depletion and cytochrome c release. Biochem
Biophys Res Commun. 303:1073–1079. 2003.PubMed/NCBI
|
|
9
|
Grishko V, Rachek L, Musiyenko S, Ledoux
SP and Wilson GL: Involvement of mtDNA damage in free fatty
acid-induced apoptosis. Free Radic Biol Med. 38:755–762. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu JD, Liu S, Wang W, et al: Emodin
induces chloride secretion in rat distal colon through activation
of mast cells and enteric neurons. Br J Pharmacol. 165:197–207.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen H, Wei W, Guo Y, et al: Enhanced
effect of gemcitabine by emodin against pancreatic cancer in vivo
via cytochrome C-regulated apoptosis. Oncol Rep. 25:1253–1261.
2011.PubMed/NCBI
|
|
12
|
Reed JC: Bcl-2 family proteins. Oncogene.
17:3225–3236. 1998. View Article : Google Scholar
|
|
13
|
Wang JB, Qi LL, Zheng SD and Wu TX:
Curcumin induces apoptosis through the mitochondria-mediated
apoptotic pathway in HT-29 cells. J Zhejiang Univ Sci B. 10:93–102.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pettersson F, Dalgleish AG, Bissonnette RP
and Colston KW: Retinoids cause apoptosis in pancreatic cancer
cells via activation of RAR-gamma and altered expression of
Bcl-2/Bax. Br J Cancer. 87:555–561. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
16
|
Wang YX, Yu H, Zhang YY, et al: Emodin
induces apoptosis of human cervical cancer HeLa cells via intrinsic
mitochondrial and extrinsic death receptor pathway. Cancer Cell
Int. 13:712013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo LD, Chen XJ, Hu YH, Yu ZJ, Wang D and
Liu JZ: Curcumin inhibits proliferation and induces apoptosis of
human colorectal cancer cells by activating the mitochondria
apoptotic pathway. Phytother Res. 27:422–430. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin P, Wu ZT, Li SR, Li SJ, Wang JH, Wang
ZH, Lu JG, Cui XJ, Han Y, Rao J and Sheng JQ: Colorectal cancer
screening with fecal occult blood test: A 22-year cohort study.
Oncol Lett. 6:576–582. 2013.PubMed/NCBI
|
|
19
|
Andre T, Boni C and Mounedji-Boudiaf L:
Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for
colon cancer. N Engl J Med. 350:2343–2351. 2004. View Article : Google Scholar
|