Introduction
Niacin has long been used in the treatment of high
cholesterol, coronary heart disease, the skin disease Pellagra and
age-related macular degeneration (AMD) (1,2).
Niacin and its derivative, niacinamide, have been widely applied in
skin care products, including moisturizers, anti-aging products,
and rosacea treatments (3). It
has been well-accepted that niacin has protective effects against
skin photodamage induced by ultraviolet (UV)-radiation. A niacin
derivative, myristyl nicotinate (MN), may enhance epidermal
differentiation and barrier function in skin against photodamage,
suggesting that niacin delivery may protect against UV skin damage
and may be used in treating skin barrier impairment (4).
Niacin has the potential to influence cellular
processes including DNA repair, genomic stability, the immune
system, stress responses, signaling, transcription, apoptosis,
metabolism, differentiation, chromatin structure and life span. In
addition to its well-known redox functions in energy metabolism,
niacin and its derivatives in the form of NAD and NADP, are
required for the synthesis of cyclic ADP-ribose and NAADP, which
are two major mediators of intracellular calcium signaling pathways
(5).
A previous study (6) showed that niacin-deficient HaCaT
cells with low NAD status develop a decreased growth rate due to an
increase in apoptotic cells and an arrest in the G2/M cell cycle
phase accompanied with accumulation of reactive oxygen species and
increased DNA damage. This model of niacin deficiency has also
allowed the identification of NAD-dependent signaling events
critical in early skin carcinogenesis. Niacin deficiency in humans
would cause sun sensitivity in the skin, indicative of deficiencies
in responding to UV damage.
Furthermore, the identification of the nicotinic
acid receptor in human skin keratinocytes provides a further link
to niacin’s role as a potential skin cancer prevention agent and
supports the role of the nicotinic acid receptor as a potential
target for skin cancer prevention agents (7). While the exact mechanism through
which niacin serves as a modulator agent for a critical resistance
factor to prevent from UV-induced skin damage is still unknown.
Cellular energy loss (8) and
AMPK, AKT and eNOS activation (9)
have been suggested as some of the niacin protective
mechanisms.
UV is divided into UVC (200–280 nm), UVB (280–320
nm) and UVA (320–400 nm). UVB is of environmental significance,
penetrating into the papillary area of the dermis and inducing DNA
damages to the residing dendritic cells (DC)(10–12), as well as keratinocytes. These
cells are perturbed both phenotypically and functionally,
undergoing apoptosis upon UVB-radiation. Apoptosis can be induced
in the region that suffers the greatest exposure and cells
surrounding this area would also be partially damaged (10,12).
Apoptosis is the process of programmed cell death,
which involves a series of morphological changes, including cell
detachment, cell shrinkage, mitochondria leakage, chromatin
condensation, and DNA fragmentation. This process is controlled by
the balance between pro-apoptotic and anti-apoptotic signaling
pathways (13). The PI3K/AKT and
JNK phosphorylation cascades act as two important regulators, and
will have an anti-apoptotic role in a variety of tissue culture
models against stresses such as UV irradiation, matrix detachment,
cell cycle disturbance, and DNA damages (14,15).
Previous studies in dendritic cells as well as
keratinocytes have demonstrated that the cellular response to UV is
composed of transactivation of cell surface growth factors, such as
EGFR, and their downstream signal transduction machinery such as
MAPK and PI3K/AKT (12,16–19). UV activates all three MAPK
families including JNK, p38 and ERK in human keratinocytes and skin
dendritic cells (12,20–25). While MAPK (JNK and p38) is
responsible for UV-induced cell apoptosis, other cellular signals
such as AKT serve as survival signals to fight against UV-induced
widespread cell death (17,19,26,27). Activated AKT relays its survival
signals in a number of ways including phosphorylation and
inactivation of the pro-apoptotic BH3 protein BAD (28), members of the Forkhead family of
transcription factors (29) and
Mdm2, the negative regulator of p53 (30). However, the possible role of
mTORC1 (mTOR complex 1), another important downstream target of
AKT, in UV-induced cell death is not fully studied.
It is possible that niacin affects the cell status
by protecting against UV-induced apoptosis and death. To determine
the mechanism of niacin in HaCaT cells under UV, we measured the
level of AKT and MAPK after UV irradiation. In this study, we
demonstrate that UV radiation induces both AKT and MAPK activation.
AKT and TSC2 are required for UV-induced mTOR/S6 activation (S6K
and 4E-BP1 phosphorylation). Niacin pretreatment protects against
UV-induced cell death and apoptosis in keratinocytes (HaCaT cells)
by enhancing AKT/mTOR and S6 activation. Niacin may have a
functional role in the pro-survival mechanism through activating
the AKT/mTOR/S6 signaling pathway.
Materials and methods
Materials
Niacin (nicotinic acid) and DMSO were obtained from
Sigma (St. Louis, MO, USA). Monoclonal mouse anti-β-actin, goat
anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP antibody were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The Annexin V-FITC kit was purchased from BD Biosciences (San
Jose, CA, USA). The JC-1 probe cell apoptosis detection kit was
provided by the Beyotime Institute of Biotechnology (Haimen,
China). The p-EGFR (Tyr1068) antibody, and the p-AKT (Ser473),
p-AKT (Thr308), p-S6K (Thr389), p-4E-BP1 (Ser65), p-S6 (S235/236),
p-mTOR (Ser2448), p-p38 (Thr180/Tyr182), p-JNK(Thr183/Tyr185),
p-ERK1/2 (Thr202/Tyr 204), and p-TSC2 (Ser 1462) antibodies, the
AKT inhibitor LY294002, the mTOR inhibitor rapamycin, the
Tuberin/TSC2 siRNA, and the p38 MAPK, JNK and the AKT1/2 antibodies
were all from Cell Signaling Technology (Beverly, MA, USA). The
EGFR inhibitor, PD153035 was purchased from Invitrogen (Carlsbad,
CA, USA).
Cell culture, transfection and UV
treatment
The spontaneously immortalized human keratinocytes
(HaCaT cell line) were cultured at 37°C in RPMI-1640 supplemented
with 10% fetal bovine serum and 100 U/ml of penicillin/streptomycin
as previously reported (31,32). The procedures for UV irradiation
were similar to those described previously. For UV irradiation
experiments, the cells were starved for 1 day and then washed twice
with phosphate-buffered saline (PBS) before being exposed to UV
irradiation using a germicidal lamp (emission wave, 290–320 nm;
radiation intensity, 1.35 mW/cm2) SS-04B Sigma High-Tech
Co., Ltd. (Shanghai, China). Doses of UV-irradiation were
calibrated with a UVX radiometer also from Sigma High-Tech Co.,
Ltd. For siRNA transfection, cells were plated at a density of
3×106 cells per 100-mm culture dish, incubated
overnight, and then transfected with the indicated expression
vectors, using Lipofectamine 2000 (Invitrogen, USA).
Cell viability assay (MTT assay)
Cell viability was measured by the
3-[4,5-dimethylthylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
(MTT) method (31). Briefly,
cells were seeded in 96-well plates at a density of
1×104 cells/well. Different seeding densities were
optimized at the beginning of the experiments. After incubation for
24 h, cells were exposed to fresh medium containing reagents at
37°C. After incubation for up to 24 h, 10 μl of MTT tetrazolium
salt (Sigma) dissolved in Hank’s balanced solution at a
concentration of 5 mg/ml was added to each well and incubated in a
CO2 incubator for 4 h. The DMSO (10 μl) was added to
dissolve formazan crystals and then lysed on a shaker for 15 min.
The absorbance value at 490 nm was measured with a microplate
spectrophotometer (SoftMax® Pro5, Molecular Devices,
USA).
Western blot analysis
As described previously (33), post treatment, cells were
collected and lysed in RIPA buffer (10 mM NaPO4, pH
7.4/300 mM NaCl/0.1% SDS/1% Nonidet P-40/1% deoxycholic acid/2 mM
EDTA) with protease inhibitors (Pierce, USA). Aliquots of 30–40 μg
of protein from each sample (treated as indicated in the legends)
were separated by 10% SDS-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto a polyvinylidenedifluoride (PVDF)
membrane (Millipore, Bedford, MA). After blocking with 5% BSA
(Beyotime) for 1 h, membranes were incubated with specific
antibodies overnight at 4°C followed by incubation with secondary
antibodies (HRP-conjugated anti-rabbit or anti-mouse IgG at the
appropriate dilutions) for 45 min to 1 h at room temperature.
Immunoblotted membranes were developed using the ECL western
blotting system (Thermo Scientific Biotechnology, USA).
Quantification of appropriate bands on western blots was performed
using the quantification software of the digital imaging system
(ChemiDoc™ XRS+, Bio-Rad, USA).
Confocal laser scanning microscope
(CLSM)
As previously described (33), all cells were fixed with 4%
formaldehyde in PBS for 30 min. Fixed cells were blocked with 10%
BSA in PBS for 30 min at room temperature. Anti-p-AKT and anti-p-S6
polyclonal antibodies were used at 1 μg/100 μl in buffer (0.5% BSA
in PBS) and incubated with the coverslip for 1 h. The antibodies
were detected using appropriate secondary antibodies (goat
anti-mouse or goat anti-rabbit; Beyotime, China) conjugated to Cy3
or FITC at 1:200. The processed slides were observed with a
Confocal Laser Scanning Microscope (TCS SP2, Leica, Germany).
Annexin V and propidium iodide (PI)
staining
A cell apoptosis detection kit was used for Annexin
V and PI staining. HaCaT cells were cultured in 35-mm dishes and
were exposed to different treatments. Cells were harvested after up
to 24 h incubation and washed in PBS twice. Cells were re-suspended
in the binding buffer, and then the fluorescein-conjugated Annexin
V and PI reagent were added to cell suspensions. After incubation
in the dark for 15 min the percentage of apoptotic cells and
necrotic cells were assessed by a flow cytometer (FACSCalibur, BD
Biosciences, USA).
Mitochondrial membrane potential
assay
The JC-1 probe was used to measure mitochondrial
depolarization. Briefly, cells were cultured in 6-well plates up to
~50% confluency, and after the indicated treatments cells were
incubated with 1 ml of JC-1 staining solution (5 μg/ml) at 37°C for
20 min and rinsed twice with PBS. Mitochondrial membrane potentials
were monitored by determining the relative amounts of dual
emissions from mitochondrial JC-1 monomers or aggregates using an
Olympus fluorescent microscope under Argon-ion 488 nm laser
excitation. Mitochondrial depolarization is indicated by an
increase in the green/red fluorescence intensity.
Statistical analysis
The values in the figures are expressed as the means
± standard deviation (SD). All experiments were repeated at least
three times. Experimental groups were compared by one-way ANOVA
analysis of SPSS 16.0. Mean differences were considered significant
(P<0.05) and highly significant (P<0.01).
Results
Niacin protects against UV-induced cell
death and apoptosis in keratinocytes (HaCaT cells)
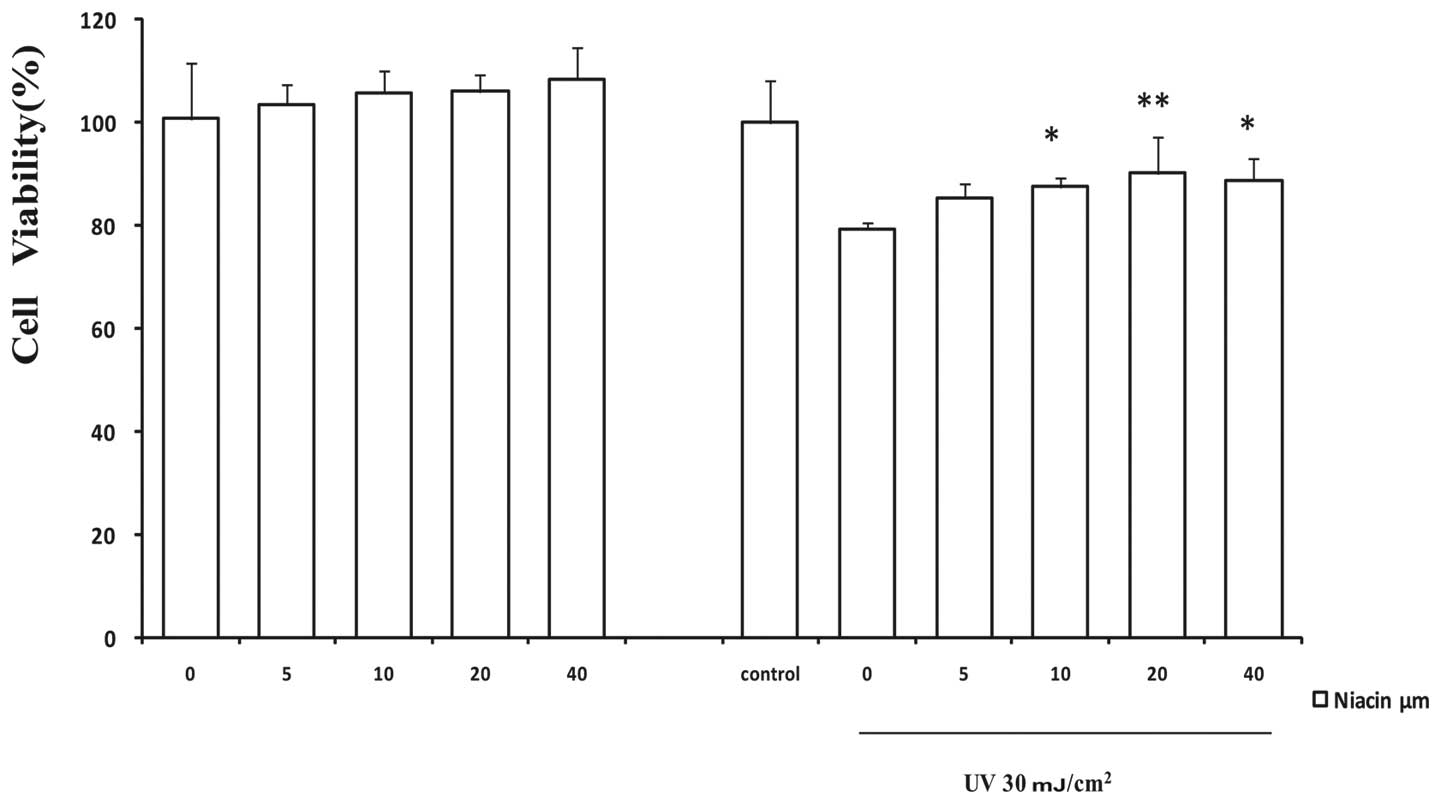
By using the cell viability MTT assay, first we
investigated the possible protective effects of niacin against
UV-induced cell death. As indicated in Fig. 1, cells were pretreated with a
dose-dependent course of niacin (0, 5, 10, 20 and 40 μM), after
with or without UV (30 mJ/cm2) as indicated in similar
studies. There was no significant dosage effect with up to 40 μM of
niacin in the control groups. As expected, UV treatment caused a
marked decrease in cell viability. The viable cells dropped to
79.35±1.21% (n=4) of the control within 24 h of 30
mJ/cm2 UV radiation. In the presence of niacin, the cell
viability was markedly recovered to 90.21±7.09% (P<0.01) at 20
μM of niacin in comparison to the UV-treated and niacin-free group.
Signiflcant improvement of cell viability occurred with a graded
increase in niacin concentrations from 5 to 20 μM, and showed a
declining trend at 40 μM with UV treatment. Next we assessed
whether niacin can inhibit UV-induced cell apoptosis. Data in
Fig. 2C clearly demonstrate that
20 μM with niacin pretreatment diminishes UV induced cell apoptosis
in HaCaT cells as evidenced by the Annexin V and PI staining assay
(Fig. 3). Normal cells stained
with the JC-1 probe emitted mitochondrial red fluorescence and
limited green fluorescence. Aggregated JC-1 has a high
mitochondrial membrane potential and emits red fluorescence but
will turn green after normal mitochondria is dispersed to the
monomeric form. Pretreatment with 20 μM niacin against UV
dramatically decreased the green/red fluorescence ratio (P<0.01)
in comparison with UV only, and the apoptotic cells dropped from
26.10±4.57% to 18.92±3.26% (P<0.01).
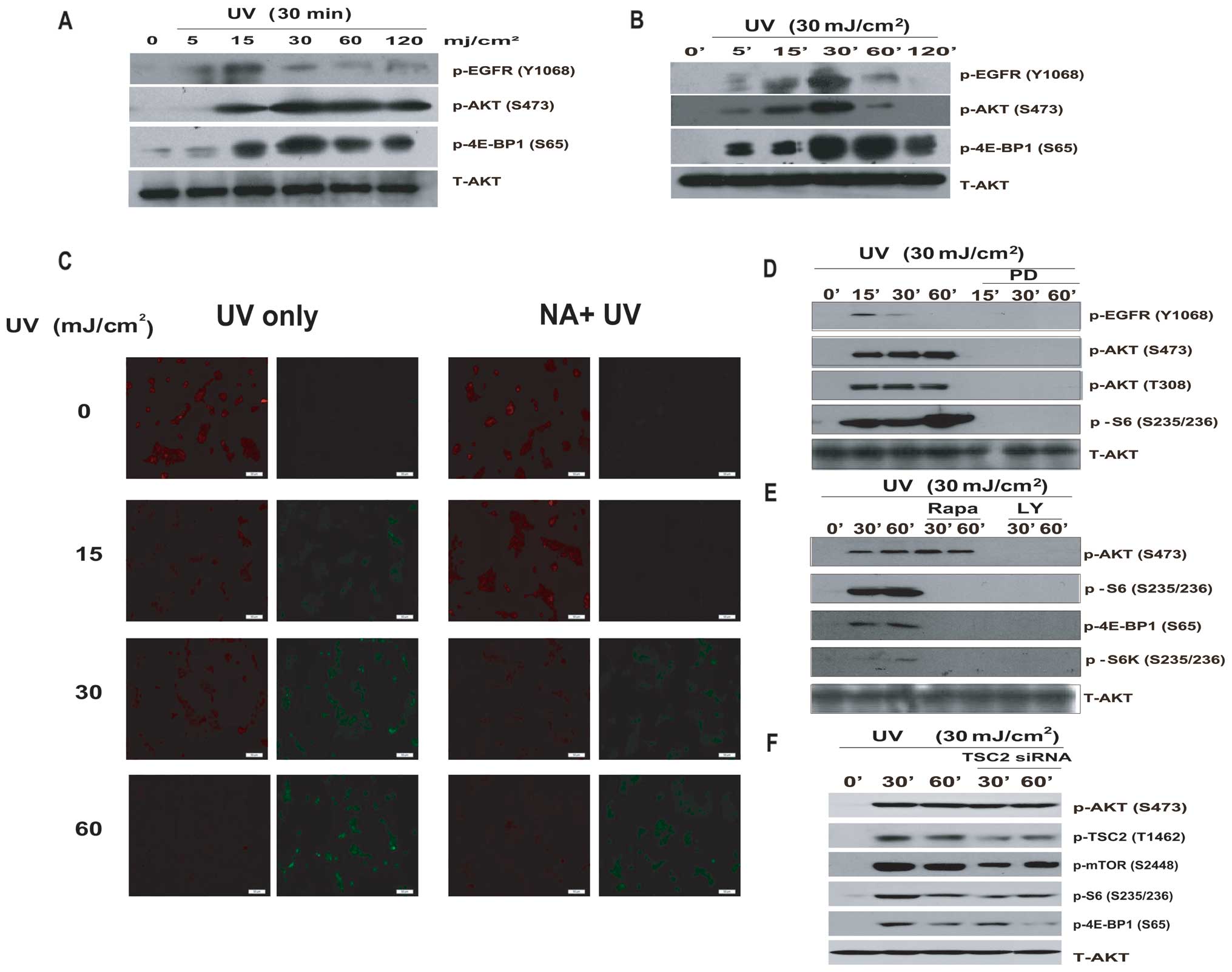 | Figure 2AKT, TSC2, mTOR and S6 are required
for UV-induced AKT cascade activation. (A and B) HaCaT cells were
exposed to different dosages of UV radiation (0, 5, 15, 30, 60, 120
mJ/cm2) for 30 min and for different durations of 30
mJ/cm2 UV respectively. p-EGFR (Tyr1068), p-AKT
(Ser473), p-4E-BP1 (Ser65) were detected by western blotting. (C)
HaCaT cells were pretreated with 20 μM niacin, followed by UV
radiation (0, 15, 30, 60 mJ/cm2), then stained with the
JC-1 probe and imaged by fluorescent microscope. The individual red
and green average fluorescence intensities are expressed as the
ratio of green to red fluorescence. An increase on the fluorescence
green/red ratio indicates a shift increase in mitochondrial
depolarization as an early apoptosis label. HaCaT cells were
pre-treated with (D) the EGFR inhibitor, PD153035 (PD, 1 μM); (E)
with the PI3K/AKT inhibitor LY294002 (LY, 10 μM) and mTOR inhibitor
rapamycin (Rapa, 100 nM), for 1 h, with UV (30 mJ/cm2)
radiation and cultured for 15, 30 and 60 min. (F) Cells were
transfected with Tuberin/TSC2 siRNAII (100 nM) for 48 h prior to UV
radiation (30 mJ/cm2), for 30 or 60 min. p-EGFR
(Tyr1068), p-AKT (Ser473), p-AKT (Thr308), p-TSC2 (Thr1462), p-mTOR
(Ser2448), p-S6K (Thr389), p-S6 (Ser235/236), p-4E-BP1 (Ser65) and
total AKT antibody were used by western blotting. All experiments
were repeated at least three times and similar results were
obtained. |
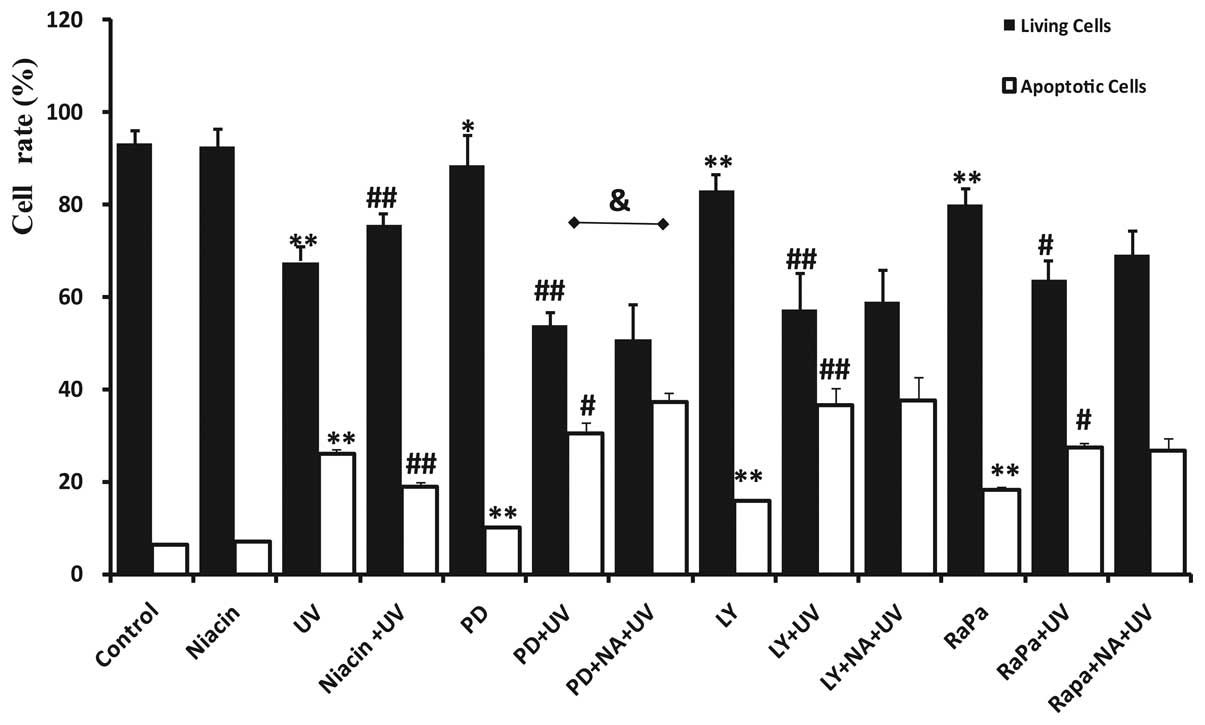 | Figure 3Niacin protects HaCaT cells from
UV-induced apoptosis cooperated with different AKT cascade
inhibitors. HaCaTs were pre-treated with 20 μM niacin (NA) for 30
min, and then incubated with different inhibitors for 30 min until
30 mJ/cm2 of UV radiation. PD, LY, RaPa are
abbreviations for PD153035, LY294002, and rapamycin respectively.
Twenty-four hours post-treatment the number and percentage of
apoptotic and necrotic cells were quantifled by FACS after cells
were washed and stained with Annexin V and PI. Apoptotic cells were
determined by counting the percentage of early apoptotic cells
Annexin V(+), PI(−) cells plus the percentage of late apoptotic
Annexin V(+), PI(+) cells. Normal living cells are presented as
Annexin V(−), PI(−), respectively. The values in the figure are
shown as mean ± SD of at least three separate experiments.
*P<0.05, **P<0.01 vs. the control
group; #P<0.05, ##P<0.01 vs. the UV
only group; and &P<0.05 when comparing the PD+UV
and PD+NA+UV group. |
AKT, TSC2 and mTOR are required for
UV-induced AKT cascade activation
We next examined whether the AKT cascade is
activated upon UV radiation. The results showed that UV radiation
activates the EGFR/AKT/mTOR pathway in a time- and dose-dependent
manner. UV radiation (30 mJ/cm2) activates AKT at the
highest level. The peaks of EGFR/AKT/mTOR activation take place
between 30 and 60 min at the UV dose of 30 mJ/cm2
(Fig. 2A and B). Previous studies
have demonstrated the key role of AKT in mTOR activation in
response to growth factors (32).
To investigate the role of AKT in UV-induced mTOR/S6 activation,
AKT cascade inhibitors were used. The results show that UV does not
induce AKT/mTOR/S6 activation in cells treated with the EGFR
inhibitor, PD153035, or the AKT inhibitor, LY294002 (Fig. 2D and E), suggesting that both EGFR
and AKT are required for UV-induced mTOR/S6 activation. Previous
studies have also shown that the regulation of S6K and 4E-BP1 by
AKT occurs primarily through the phosphorylation and inactivation
of TSC2 at Tyr1462 (34), leading
to increased phosphorylation of mTOR and downstream S6K and 4E-BP1.
We next examined the role of TSC2 in S6K activation upon UV
radiation. As shown in Fig. 2F,
UV-induced mTOR/S6 activation is modulated by TSC2. Transfection
with TSC2 siRNA specifically silenced TSC2 by approximately 60%,
and the downstream mTOR and S6 signals were partially inhibited
(Fig. 2F).
Niacin, in contrast to AKT cascade
inhibitors, affects UV-induced apoptosis in HaCaT cells
As demonstrated, niacin protects against UV-induced
cell death and cell apoptosis. The AKT cascade activation is
induced by UV radiation. Taken together, we conclude that the AKT
cascade signal activation serves as a critical upstream signal
against UV, serving as a pro-survival signal against UV-induced
cell apoptosis. To determine whether the AKT signaling pathway is
involved in the pro-survival and anti-apoptotic effects of niacin,
cells were treated with AKT cascade kinase inhibitors
independently, such as the EGFR inhibitor, PD153035, the AKT
inhibitor, LY294002 and the mTOR inhibitor, rapamycin. As
previously shown, treatment with PD153035, LY294002 and rapamycin
resulted in the increase of UV-induced apoptosis. Living cells were
decreased successively from 93.21±2.74% to 80.08±3.20% while the
percentages of apoptotic cells were successively increased from
6.35±0.32% in the control to 18.39±0.55% (P<0.01) in the
rapamycin group. Independent treatment with LY294002 and rapamycin
seemed to affect cells efficiently. The contribution of the AKT
cascade signal activation after UV was further detected by
pretreating cells with inhibitors before UV exposure. Treatment
with LY294002 and rapamycin were found to have less influence on
the UV-induced loss of cell viability in the cells in comparison
with PD98059. However, LY294002 has a more critical role in the UV
effect. The UV-induced apoptosis ratio was 36.71±3.67% compared to
others. Last, we treated niacin-treated cells with inhibitors prior
to UV. This led to an insignificant decrease of UV-induced cell
apoptosis (P>0.05) except for PD98059 which increaed apoptotic
cells (P<0.05).
Niacin protects against UV-induced
apoptosis via enhancement of AKT and mTORC/S6 activation
We have shown that niacin pretreatment protects
against UV-induced cell death and apoptosis in keratinocytes (HaCaT
cells) (Figs. 1 and 2). AKT and the downstream signal mTOR/S6
are important for the protective effect of niacin, so next we
examined whether niacin is able to induce AKT/mTOR/S6 activation in
HaCaT cells. The results showed that niacin strongly induces AKT
and the downstream mTOR/S6 activation at 30 min (Fig. 4A). Furthermore, niacin induces
little activation of MAPK (ERK and JNK) compared to the UV group
which strongly activates MAPK (p38) (Fig. 4C). We also demonstrated that
UV-induced AKT/mTOR/S6 activation serves as a pro-survival signal
(Fig. 2). Next we examined the
possible role of niacin on UV induced AKT/mTOR/S6 activation. As
shown in Fig. 4B, niacin
pretreatment largely enhances UV-induced AKT/mTOR/S6 activation in
HaCaT cells. The AKT inhibitor, LY294002 largely neutralizes the
protective effects of niacin (Fig.
4E) and shows a low mitochondrial membrane potential,
indicating that AKT activation is necessary for niacin’s protective
effects against UV-induced cell apoptosis.
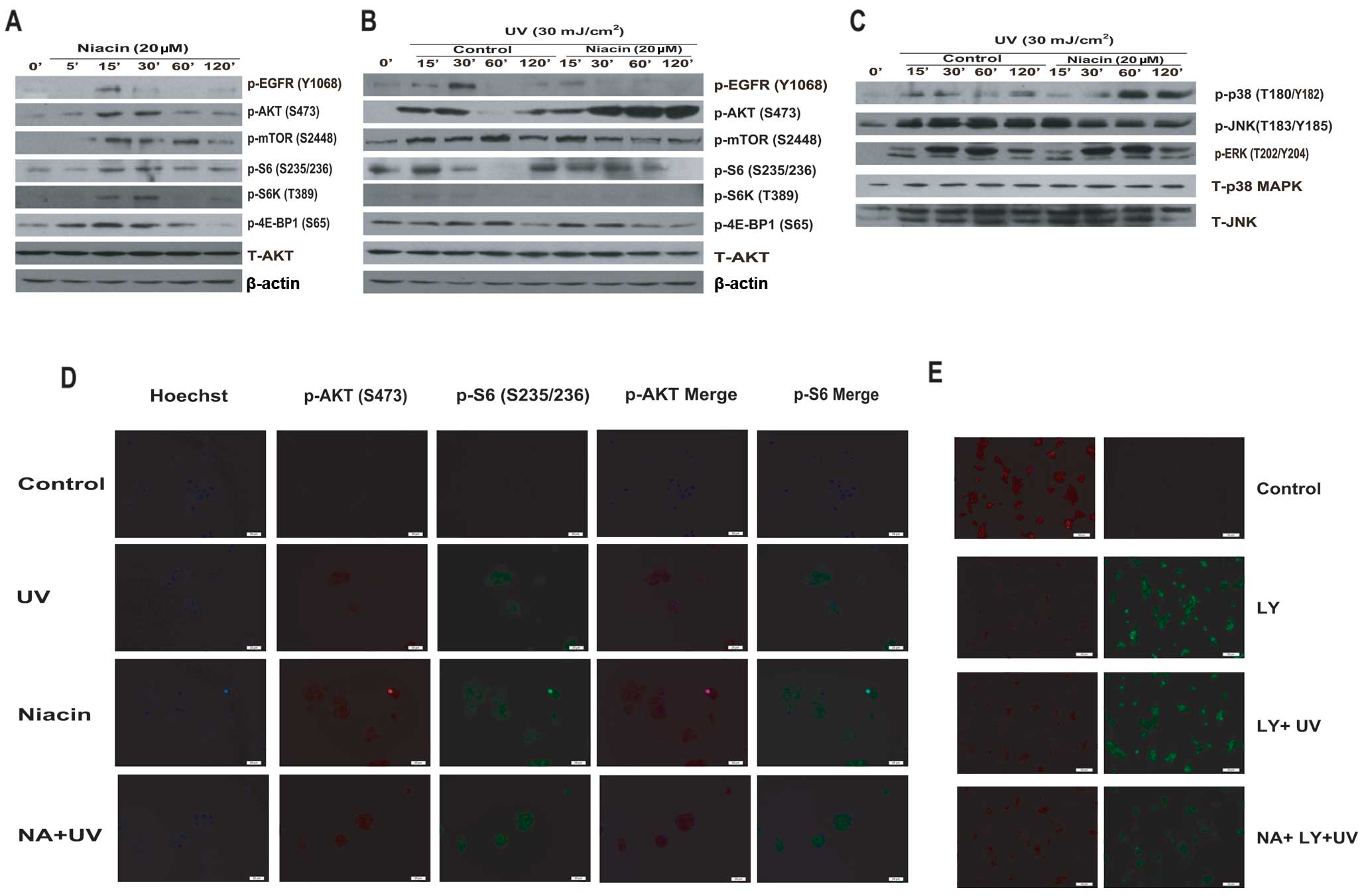 | Figure 4Niacin protects against UV-induced
apoptosis via enhancement of AKT/mTOR/S6 activation. (A) HaCaT
cells were treated with niacin (NA, 20 μM) and cultured for 5, 15,
30, 60 and 120 min. p-EGFR (Tyr1068), p-AKT (Ser473), p-mTOR
(Ser2448), p-S6K (Thr389), p-S6 (Ser235/236), p-4E-BP1 (Ser65),
total AKT and β-actin antibody were used for western blotting. (B
and C) HaCaT cells were pretreated with or without niacin (20 μM)
for 1 h, followed by UV (30 mJ/cm2) radiation and
cultured for the indicated time, p-EGFR (Tyr1068), p-AKT (Ser473),
p-mTOR (Ser2448), p-S6K (Thr389), p-S6 (Ser235/236), p-4E-BP1
(Ser65), p-p38 (Thr180/Tyr182), p-JNK (Thr183/Tyr185), p-ERK1/2
(Thr202/Tyr204), p38 MAPK, JNK total AKT and β-actin were detected
by western blotting. (D) HaCaT cells were pretreated with niacin
(20 μM) for 1 h, followed by UV radiation (30 mJ/cm2)
and cultured for 1 h, p-AKT (Ser473) and p-S6 (Ser235/236) were
detected by confocal microscopy. (E) HaCaT cells were pre-treated
with niacin and LY294002 separately or in combination, after UV (30
mJ/cm2) radiation; mitochondrial depolarization was
observed the same way in Fig.
2C. |
Discussion
Despite previous research showing that niacin
reduces the immunosuppressive effects of UV both in vivo and
in vitro (35), at least
in part by providing energy repletion to UV-irradiated cells for it
can normalize subsets of apoptosis and cellular energy loss effects
(8,36), the precise molecular mechanisms
underlying the cytoprotective and anti-apoptotic effects remain
largely unknown. Here, we showed that treatment with niacin led to
efficient suppression of UV-induced cell death and apoptosis in
keratinocytes (Figs. 1 and
2). We also found that the
treatment of niacin could recover the changed mitochondrial
membrane potential. These data suggest that niacin can protect
against UV-induced mitochondrial dysfunction (Fig. 2). The fact that niacin protects
the cells from apoptotic cell damage appears to be applicable to
the protection from UV irradiation of keratinocytes, which are the
primary cell type in the epidermis and play a key role in the
body’s initial line of defense.
To understand the molecular mechanism of the
anti-apoptotic effect of niacin in UV-treated keratinocytes, first
we analyzed several signaling pathways related to UV-induced
apoptosis in HaCaT cells. We found that the short time-dependent
regulation of the AKT cascade and MAPK feedback activation is
involved in HaCaT cells after UV exposure. AKT is well known to be
differentially activated depending on the type of extracellular
stimuli. As suggested in previous studies, MAPK/JNK/p38 activation
may be essential for UV-induced apoptosis, but the activation of
AKT, mTOR, S6 can be the pro-surviving signals against UV-induced
apoptosis. Some plant flavonoids with anti-apoptotic properties
activate the MAPK/AKT/NF-κB pathway (37–39). We found that niacin pretreatment
largely enhances UV-induced AKT/mTOR/S6 activation in HaCaT cells
(Fig. 4). Therefore, the PI3K/AKT
cascade is considered as the canonical pathway involved in the
protection of UV-induced apoptosis followed by EGFR kinase
activation. As indicated in the Fig.
3 apoptosis assay, EGFR could relatively strengthen the damage
of UV; AKT and mTOR inhibitors could largely neutralize the
protective effects of niacin against UV, suggesting that AKT
signaling activation is necessary for niacin’s protective effects
against UV-induced cell death and apoptosis.
In addition to stimulating cell growth, mTOR also
promotes cell survival. eIF4E serves as an important downstream
effectors of mTOR in the control of cell survival through the
4E-BP1 (40–46). The translation and expression of
mRNAs encoding a few major anti-apoptotic proteins including (XIAP,
c-IAP1, Bcl-XL and BCl-2) are mTORC1 and cap-dependent (47). S6K1 is another mTOR target that
plays a role in the apoptosis resistance of cancer cells (48–50). Previous studies (51) together with our current work
demonstrate that mTOR activation serves as a pro-survival
UV-induced cell apoptosis signal (Figs. 2 and 4). AKT, by phosphorylation and
inhibition of TSC2, serves as the upstream signal for UV-induced
mTORC1 activation (Fig. 2).
Importantly, UV-induced TSC2 and S6K phosphorylation were almost
abolished by pharmacological inhibitors of AKT (Fig. 2). Taken together, we conclude that
AKT by phosphorylating and inhibiting TSC2, acts as an upstream
signal for UV-induced mTOR activation, and the latter serves as a
pro-survival signal against UV-induced cell death and
apoptosis.
Although the PI3K/AKT and its downstream substrate
mTOR (S6K and 4E-BP1 phosphorylation, rapamycin sensitive) pathway
are well-established, the identity of the kinase responsible for
phosphorylating AKT at Ser473 remains elusive until in recent
years, when it was revealed to be mTORC2 (46,52–54). mTORC2 is a complex of mLST8,
rictor (rapamycin-insensitive companion of mTOR), and mSin1 with
mTOR (46,52–56). Aside from AKT hydrophobic motif
phosphorylation, mTORC2 has also been implicated in the
phosphorylation of the AGC kinase, PKCα (46,57), the regulation of actin
cytoskeleton reorganization (46)
and development of prostate cancer in PTEN deficient mice (58). Ablation of mTORC2 activity impairs
the phosphorylation of only a subset of AKT targets including
FoxO1/3a, while leaving other AKT targets such as TSC2 and glycogen
synthase kinase 3 (GSK-3) as well as the mTORC1 effectors S6K and
4E-BP1 unaffected (46,57,59).
In conclusion, we found for the first time that
niacin pretreatment protects against UV-induced cell death and
apoptosis by enhancing the pro-survival pathways including AKT,
mTOR and S6 in skin keratinocytes (HaCaT cells). Oral and external
niacin and nicotinamide are both safe and inexpensive and appear to
be promising chemopreventive supplements for reducing the
mutagenic, immunosuppressive and cell damage effects of sunlight
(60). To our knowledge, this is
also the first study providing a molecular mechanism to support
that niacin can be utilized as a skin photodamage protective
agent.
Acknowledgements
This research was supported by a grant from the
National Natural Science Foundation of China (30872280 to Dr Aie
Xu).
References
|
1
|
M StuderM BrielB LeimenstollTR GlassHC
BucherEffect of different antilipidemic agents and diets on
mortality: a systematic reviewArch Intern
Med165725730200510.1001/archinte.165.7.72515824290
|
|
2
|
TI MetelitsinaJE GrunwaldJC DuPontGS
YingEffect of niacin on the choroidal circulation of patients with
age related macular degenerationBr J
Ophthalmol8815681572200410.1136/bjo.2004.04660715548814
|
|
3
|
DL BissettJE OblongCA BergeNiacinamide: A
B vitamin that improves aging facial skin appearanceDermatol
Surg31860865200510.1111/j.1524-4725.2005.3173216029679
|
|
4
|
EL JacobsonH KimM KimA topical lipophilic
niacin derivative increases NAD, epidermal differentiation and
barrier function in photodamaged skinExp
Dermatol16490499200710.1111/j.1600-0625.2007.00553.x17518989
|
|
5
|
JB KirklandNiacin and carcinogenesisNutr
Cancer46110118200310.1207/S15327914NC4602_0214690785
|
|
6
|
CA BenaventeEL JacobsonNiacin restriction
upregulates NADPH oxidase and reactive oxygen species (ROS) in
human keratinocytesFree Radic Biol
Med44527537200810.1016/j.freeradbiomed.2007.10.00617997992
|
|
7
|
CA BenaventeMK JacobsonEL JacobsonNAD in
skin: therapeutic approaches for niacinCurr Pharm
Des152938200910.2174/13816120978718576019149600
|
|
8
|
J ParkGM HallidayD SurjanaDL
DamianNicotinamide prevents ultraviolet radiation-induced cellular
energy lossPhotochem
Photobiol86942948201010.1111/j.1751-1097.2010.00746.x20492562
|
|
9
|
SV PenumathsaM ThirunavukkarasuSM
SamuelNiacin bound chromium treatment induces myocardial Glut-4
translocation and caveolar interaction via Akt, AMPK and eNOS
phosphorylation in streptozotocin induced diabetic rats after
ischemia-reperfusion injuryBiochim Biophys
Acta17923948200910.1016/j.bbadis.2008.10.018
|
|
10
|
L MeunierUltraviolet light and dendritic
cellsEur J Dermatol9269275199910356402
|
|
11
|
W KolgenH BothH van WeeldenEpidermal
langerhans cell depletion after artificial ultraviolet B
irradiation of human skin in vivo: apoptosis versus migrationJ
Invest
Dermatol118812817200210.1046/j.1523-1747.2002.01742.x11982758
|
|
12
|
S NakagawaT OhtaniM Mizuaship38
Mitogen-Activated protein kinase mediates dual role of ultraviolet
B radiation in induction of maturation and apoptosis of
monocyte-derived dendritic cellsJ Invest
Dermatol123361370200410.1111/j.0022-202X.2004.23238.x15245437
|
|
13
|
BS McGowanEF CiccimaroTO ChanAM FeldmanThe
balance between pro-apoptotic and anti-apoptotic pathways in the
failing myocardiumCardiovasc
Toxicol3191206200310.1385/CT:3:3:19114555786
|
|
14
|
SR DattaA BrunetME GreenbergCellular
survival: a play in three AktsGenes
Dev1329052927199910.1101/gad.13.22.290510579998
|
|
15
|
RE BurkeInhibition of mitogen-activated
protein kinase and stimulation of Akt kinase signaling pathways:
two approaches with therapeutic potential in the treatment of
neurodegenerative diseasePharmacol
Ther114261277200710.1016/j.pharmthera.2007.02.002
|
|
16
|
L ZhuangB WangGA ShinderGM ShivjiTW MakDN
SauderTNF receptor p55 plays a pivotal role in murine keratinocyte
apoptosis induced by ultraviolet B irradiationJ
Immunol1621440144719999973400
|
|
17
|
Y XuJJ VoorheesGJ FisherEpidermal growth
factor receptor is a critical mediator of ultraviolet B
irradiation-induced signal transduction in immortalized human
keratinocyte HaCaT cellsAm J
Pathol169823830200610.2353/ajpath.2006.050449
|
|
18
|
C SachsenmaierA Radler-PohlR ZinckA
NordheimP HerrlichHJ RahmsdorfInvolvement of growth factor
receptors in the mammalian UVC
responseCell78963972199410.1016/0092-8674(94)90272-07923365
|
|
19
|
YS WanZQ WangY ShaoJJ VoorheesGJ
FisherUltraviolet irradiation activates PI 3-kinase/AKT survival
pathway via EGF receptors in human skin in vivoInt J
Oncol18461466200111179472
|
|
20
|
R PfundtI van Vlijmen-WillemsM BergersM
WingensW CloinJ SchalkwijkIn situ demonstration of phosphorylated
c-jun and p38 MAP kinase in epidermal keratinocytes following
ultraviolet B irradiation of human skinJ
Pathol193248255200110.1002/1096-9896(2000)9999:9999%3C::AID-PATH780%3E3.0.CO;2-Y11180173
|
|
21
|
W ChenQ TangMS GonzalesGT BowdenRole of
p38 MAP kinases and ERK in mediating ultraviolet-B induced
cyclooxygenase-2 gene expression in human
keratinocytesOncogene2039213926200110.1038/sj.onc.120453011439356
|
|
22
|
AM BodeZ DongMitogen-activated protein
kinase activation in UV-induced signal transductionSci
STKE2003167200312554854
|
|
23
|
M PapoutsakiF MorettiM LanzaA
p38-dependent pathway regulates DeltaNp63 DNA binding to
p53-dependent promoters in UV-induced apoptosis of
keratinocytesOncogene2469706975200510.1038/sj.onc.120883516007154
|
|
24
|
N ChouinardK ValerieM RouabhiaJ
HuotUVB-mediated activation of p38 mitogen-activated protein kinase
enhances resistance of normal human keratinocytes to apoptosis by
stabilizing cytoplasmic p53Biochem
J365133145200210.1042/BJ2002007212071847
|
|
25
|
QS ZhangDA MaddockJP ChenCytokine-induced
p38 activation feedback regulates the prolonged activation of AKT
cell survival pathway initiated by reactive oxygen species in
response to UV irradiation in human keratinocytesInt J
Oncol19105710612001
|
|
26
|
S WangWS El-DeiryTRAIL and apoptosis
induction by TNF-family death
receptorsOncogene2286288633200310.1038/sj.onc.120723214634624
|
|
27
|
Y LiZ BiB YanY WanUVB radiation induces
expression of HIF-1α and VEGF through the EGFR/PI3K/DEC1 pathwayInt
J Mol Med187137192006
|
|
28
|
SR DattaH DudekX TaoAkt phosphorylation of
BAD couples survival signals to the cell-intrinsic death
machineryCell91231241199710.1016/S0092-8674(00)80405-59346240
|
|
29
|
A BrunetA BonniMJ ZigmondAkt promotes cell
survival by phosphorylating and inhibiting a Forkhead transcription
factorCell96857868199910.1016/S0092-8674(00)80595-410102273
|
|
30
|
LD MayoDB DonnerA phosphatidylinositol
3-kinase/Akt pathway promotes translocation of Mdm2 from the
cytoplasm to the nucleusProc Natl Acad Sci
USA981159811603200110.1073/pnas.18118119811504915
|
|
31
|
C CaoS HealeyA AmaralATP-sensitive
potassium channel: a novel target for protection against UV-induced
human skin cell damageJ Cell
Physiol212252263200710.1002/jcp.2102617301957
|
|
32
|
C CaoY SunS HealeyEGFR-mediated expression
of aquaporin-3 is involved in human skin fibroblast
migrationBiochem J400225234200610.1042/BJ2006081616848764
|
|
33
|
C GuanF LinM ZhouThe role of VIT1/FBXO11
in the regulation of apoptosis and tyrosinase export from
endoplasmic reticulum in cultured melanocytesInt J Mol
Med265765201020514423
|
|
34
|
JM DickensonS ReederB ReesS AlexanderD
KendallFunctional expression of adenosine A2A and A3 receptors in
the mouse dendritic cell line XS-106Eur J
Pharmacol4744351200310.1016/S0014-2999(03)02041-712909194
|
|
35
|
G SivapirabuE YiasemidesGM HallidayJ
ParkDL DamianTopical nicotinamide modulates cellular energy
metabolism and provides broad-spectrum protection against
ultraviolet radiation-induced immunosuppression in humansBr J
Dermatol16113571364200910.1111/j.1365-2133.2009.09244.x
|
|
36
|
DL DamianPhotoprotective effects of
nicotinamidePhotochem Photobiol
Sci9578585201010.1039/b9pp00146h20354654
|
|
37
|
ER LeeJH KimHY ChoiK JeonSG
ChoCytoprotective effect of eriodictyol in UV-irradiated
keratinocytes via phosphatase-dependent modulation of both the p38
MAPK and Akt signaling pathwaysCell Physiol
Biochem27513524201110.1159/00032997321691069
|
|
38
|
AnggakusumaYantiJK HwangEffects of
macelignan isolated from Myristica fragrans Houtt. On
UVB-induced matrix metalloproteinase-9 and cyclooxygenase-2 in
HaCaT cellsJ Dermatol Sci571141222010
|
|
39
|
A SvobodovaA ZdarilovaJ VostalovaLonicera
caerulea and Vaccinium myrtillus fruit polyphenols protect HaCaT
keratinocytes against UVB-induced phototoxic stress and DNA damageJ
Dermatol Sci56196204200910.1016/j.jdermsci.2009.08.004
|
|
40
|
AL EdingerCB ThompsonAkt maintains cell
size and survival by increasing mTOR-dependent nutrient uptakeMol
Biol Cell1322762288200210.1091/mbc.01-12-058412134068
|
|
41
|
SK MungamuriX YangAD ThorK
SomasundaramSurvival signaling by Notch1: mammalian target of
rapamycin (mTOR)-dependent inhibition of p53Cancer
Res6647154724200610.1158/0008-5472.CAN-05-383016651424
|
|
42
|
VA PolunovskyAC GingrasN
SonenbergTranslational control of the antiapoptotic function of
RasJ Biol Chem2752477624780200010.1074/jbc.M00193820010811643
|
|
43
|
S LiN SonenbergAC GingrasTranslational
control of cell fate: availability of phosphorylation sites on
translational repressor 4E-BP1 governs its proapoptotic potencyMol
Cell Biol2228532861200210.1128/MCB.22.8.2853-2861.200211909977
|
|
44
|
LA LiottaPS SteegWG
Stetler-StevensonCancer metastasis and angiogenesis: an imbalance
of positive and negative
regulationCell64327336199110.1016/0092-8674(91)90642-C1703045
|
|
45
|
CG ProudThe eukaryotic initiation factor
4E-binding proteins and apoptosisCell Death
Differ12541546200510.1038/sj.cdd.440158815818413
|
|
46
|
V FacchinettiW OuyangH WeiThe mammalian
target of rapamycin complex 2 controls folding and stability of Akt
and protein kinase CEMBO
J2719321943200810.1038/emboj.2008.12018566586
|
|
47
|
CG ProudRole of mTOR signalling in the
control of translation initiation and elongation by nutrientsCurr
Top Microbiol Immunol279215244200414560960
|
|
48
|
HQ WangT QuanT HeTF FrankeJJ VoorheesGJ
FisherEpidermal growth factor receptor-dependent,
NF-kappaB-independent activation of the phosphatidylinositol
3-kinase/Akt pathway inhibits ultraviolet irradiation-induced
caspases-3, -8, and -9 in human keratinocytesJ Biol
Chem2784573745745200310.1074/jbc.M300574200
|
|
49
|
Y MamaneE PetroulakisO LeBacquerN
SonenbergmTOR, translation initiation and
cancerOncogene2564166422200610.1038/sj.onc.120988817041626
|
|
50
|
A PannerCD JamesMS BergerRO PiepermTOR
controls FLIPS translation and TRAIL sensitivity in glioblastoma
multiforme cellsMol Cell
Biol2588098823200510.1128/MCB.25.20.8809-8823.200516199861
|
|
51
|
C CaoS LuA SowaPriming with EGFR tyrosine
kinase inhibitor and EGF sensitizes ovarian cancer cells to respond
to chemotherapeutical drugsCancer
Lett266249262200810.1016/j.canlet.2008.02.06218400375
|
|
52
|
DD SarbassovDA GuertinSM AliDM
SabatiniPhosphorylation and regulation of Akt/PKB by the
rictor-mTOR
complexScience30710981101200510.1126/science.110614815718470
|
|
53
|
RC HreskoM MuecklermTOR. RICTOR is the
Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytesJ Biol
Chem2804040640416200510.1074/jbc.M50836120016221682
|
|
54
|
C ShiotaJT WooJ LindnerKD SheltonMA
MagnusonMultiallelic disruption of the rictor gene in mice reveals
that mTOR complex 2 is essential for fetal growth and viabilityDev
Cell11583589200610.1016/j.devcel.2006.08.01316962829
|
|
55
|
MA FriasCC ThoreenJD JaffemSin1 is
necessary for Akt/PKB phosphorylation, and its isoforms define
three distinct mTORC2sCurr
Biol1618651870200610.1016/j.cub.2006.08.00116919458
|
|
56
|
J MasriA BernathJ MartinmTORC2 activity is
elevated in gliomas and promotes growth and cell motility via
overexpression of rictorCancer
Res671171211720200710.1158/0008-5472.CAN-07-222318089801
|
|
57
|
DA GuertinDM StevensCC ThoreenAblation in
mice of the mTORC components raptor, rictor, or mLST8 reveals that
mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not
S6K1Dev Cell11859871200610.1016/j.devcel.2006.10.00717141160
|
|
58
|
DA GuertinDM StevensM SaitohmTOR complex 2
is required for the development of prostate cancer induced by Pten
loss in miceCancer
Cell15148159200910.1016/j.ccr.2008.12.01719185849
|
|
59
|
PT BhaskarN HayThe two TORCs and AktDev
Cell12487502200710.1016/j.devcel.2007.03.02017419990
|
|
60
|
E YiasemidesG SivapirabuGM HallidayJ
ParkDL DamianOral nicotinamide protects against ultraviolet
radiation-induced immunosuppression in
humansCarcinogenesis30101105200910.1093/carcin/bgn24819028705
|