Introduction
Tumorigenesis is characterized by enhanced
activities of glycolytic enzyme as well as distinct changes in the
glycolytic isoenzyme. Tumor cells must occupy a high rate of
nutrients and maintain a balance between the use of nutrients for
energy production and for anabolic processes, whereas less nutrient
uptake is required in most adult tissues and a greater fraction of
available nutrients are used for energy production rather than
macromolecule synthesis. This difference between metabolism of
tumor cells and their normal counterparts was first pointed out by
Warburg (1). Glucose is used for
anabolic processes preferentially rather than oxidative
phosphorylation in tumor cells, and this metabolic switch may be
required to support cell growth. The pyruvate kinase isoenzyme M2
(PKM2) is one of the most important regulators of the balance
between glycolytic energy regeneration [e.g. adenosine triphosphate
(ATP) synthesis] and the synthesis of cell building blocks (e.g.
protein, lipid and nucleic acid synthesis) in tumor cells. There
are four pyruvate kinase isoenzymes (M1, M2, L and R) that are
known. PKM2 is the embryonic form which is progressively replaced
by PKM1 in brain and muscle, PKL in kidney and liver, and PKR in
erythrocytes during differentiation. When adult tissue cells in
quiescence re-enter the cell cycle, the tissue-specific isoenzymes
are replaced by PKM2 (2). In
most, if not all, tumor cells PKM2 is overexpressed. PKM2
expression in cancer cells results in aerobic glycolysis and is
suggested to bestow a selective growth advantage, and thus, a
promising target. Many oncogenes impart a common alteration in cell
metabolism, inhibition of the M2 isoform might be of broad
applicability (3–5). Knockdown of PKM2 and replacement of
PKM2 by PKM1 were shown to reduce the carcinogenicity of human
tumor cell lines to form tumors in nude mouse xenografts (6,7).
Microarray studies have shown that pyruvate kinase which regulates
the rate-limiting final step of glycolysis, is one of the most
upregulated gene sets in cancer (8).
Cyclosporin A (CsA) is a noncytotoxic
immunosuppressant that was first discovered in 1970s and it was
initially used for immunosuppression following organ and marrow
transplantation. Subsequently, it has been applied in virtually all
branches of medicine where autoimmune or inflammatory processes
play a role in the pathology. Recent studies have explained that
CsA is also associated with cancer therapy. CsA is a substrate and
inhibitor of P-glycoprotein (P-gp) (9). It has been used as one of the
first-generation multidrug resistance (MDR) modulators to reverse
MDR and improve chemotherapy (10). Chemotherapy combined with CsA
increases the plasma concentration of chemotherapy drugs and
decreases the clearance of P-gp substrates, such as digoxin and
etoposide (11,12). On the other hand, CsA is an
inhibitor, but not a substrate for breast cancer resistance protein
(BCRP) (13). In the present
study, we found that CsA inhibits breast cancer cell proliferation
by influencing the glycolysis through downregulating the pyruvate
kinase subtype M2 (PKM2).
Materials and methods
Cell culture and transfection
Three human breast carcinoma cell lines MCF-7,
MDA-MB-435 and MDA-MB-231 were obtained from ATCC and cultured in
RPMI-1640 medium supplemented with 10% FBS. Normal breast primary
cells were obtained from normal breast tissue biopsies from the
Second Affiliated Hospital (Zhejiang University School of Medicine,
China) after patients had given their informed consent and were
processed as described before (14). Tissues were mechanically
dissociated with scissors and incubated at 37°C with constant
shaking in HBSS containing 500 IU/ml collagenase. Digestion was
monitored under an inverted microscope. Suspension was collected
and centrifuged, cells were then plated in DMEM containing 10
μg/ml insulin, 5×10−6 M cortisol 50 IU/ml
penicillin, 50 μg/ml streptomycin, 2 ng/ml EGF and 10% FBS.
Cells were cultured in a 5% CO2 95% air humidified
incubator. Medium was changed every 2 days. Human PKM2 encoding
gene was amplified by RT-PCR from total-RNA extracted from MCF-7
cells and was ligated to pEGFP-N1 (Clontech Laboratories, USA)
plasmid by T4 DNA ligase after digestion by the restricted
endonucleases, EcoRI and BamHI. MCF-7 cells were
transfected with pEGFP-N1-PKM2 expression vector using
Lipofectamine 2000 (Invitrogen Life Tecnologies, USA) according to
manufacturer’s instructions for 24 h before CsA treatment.
Assay of cell growth
Cell growth was measured using 3-(4,
5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT)
conversion assay. Cells were seeded in a 96-multiwell plate at a
concentration of 5×103 cells/well. After overnight
incubation, the medium was replaced with a fresh medium containing
CsA (Merck KGaA, Germany) at various concentrations and the plates
were incubated for 48 h. MTT (0.5 mg/ml) was added during the last
4 h of incubation and absorbance was measured at 570 nm.
Cell cycle analysis
MCF-7 cells were incubated with CsA at various
concentrations for 24 h. For cell cycle analysis, cells were
harvested, rinsed with PBS and fixed in 70% ethanol overnight at
4°C. The fixed cells were centrifuged and resuspended in 500
μl of PBS containing 0.1 mg/ml RNase-A and then incubated at
37°C for 30 min. Cells were then washed with PBS and resuspended in
PBS containing 0.05 mg/ml propidium iodide at room temperature for
30 min. DNA content was determined by LSR flow cytometry (BD
Biosciences, USA).
Mitochondrial membrane potential (ΔΨm)
assessment
ΔΨm was measured as the fluorescence of JC-1, a
lipophilic and cationic dye, accumulated in the mitochondria in a
potential-dependent manner. Cells were harvested and incubated for
20 min at 37°C before FACS analysis.
Western blot analysis
Cells were extracted in lysis buffer (Cell Signaling
Technology, Inc., USA) and the lysates were separated on a 12.5%
SDS-PAGE. Proteins were then transferred to a PVDF membrane
(Millipore, USA) in transfer buffer and were detected as we have
previously described (15).
Immunodetection was accomplished using the following primary
antibodies: anti-PKM2 (clone #3198; Cell Signaling Technology,
Inc.) and anti-β-actin (Santa Cruz Biotechnology, Inc., USA).
Quantitative RT-PCR
Total-RNA was isolated using TRIzol reagent
(Invitrogen Life Technologies). cDNA was synthesized using ReverTra
Ace qPCR RT kit (Toyobo, Co., Ltd., Japan) according to
manufacturer’s instructions. qRT-PCR reactions were run on an
Applied Biosystems 7500 Real Time PCR System machine as we have
previously described (16). Gene
expression levels in all samples were examined using SYBR-Green
(Takara Bio, Inc., Japan) according to the manufacturer’s
protocols. The following primer pairs were used for qRT-PCR, which
were designed according to a previous study (17): human PKM1 F,
5′-GTGCGAGCCTCAAGTCACT-3′ and R, 5′-GCTGCTAAACACTTATAAGAAG-3′;
human PKM2: F, 5′-GCCTGGCGCCCATTACCA-3′ and R,
5′-CCCACTGCAGCACTTGAAG-3′; human GAPDH F,
5′-TGCCAAATATGATGACATCAAGAA-3′ and R, 5′-GGA GTGGGTGTCGCTGTTG-3′.
After normalization of the expression of mRNA by GAPDH, the
relative levels of transcripts in CsA vs. untreated cells were
calculated using the 2-ΔΔCt method (18).
ATP measurement
ATP concentration was measured by the luminometric
ATP assay and normalized as described (19). Cells were lysed in 1% NP-40, and
when the ATP in the lysates and luciferin/luciferase enzyme complex
combined a reaction, which produced light, occured. The relative
light units were detected by Multimode Detector (DTX 880; Beckman
Coulter, USA). Intracellular ATP concentration was expressed as the
percentage of untreated to controls.
Measurement of pyruvate kinase
activity
Pyruvate kinase activity was measured according to
the absorbance at 340 nm owing to oxidation of NADH (7). The optical density was detected by
Multimode Detector (DTX 880; Beckman Coulter). Kinetic assays for
activity determinations contained cell lysate (2 μg), 1
mol/l Tris-HCl pH 8.0 (0.05 ml), 1 mol/l KCl (0.05 ml), 0.1 mol/l
MgCl2 (0.05 ml), 30 mmol/l ADP (0.025 ml), 2 mmol/l NADH
(0.05 ml), LDH (12 units), 50 mmol/l PEP (0.05 ml) and adequate
water to a total volume of 0.5 ml. The pyruvate kinase activity
measured under the various CsA concentration indicated expression
as the percentage of untreated to controls.
Statistical analysis
All experiments were repeated at least 3 times. Data
are presented as the mean ± SD. Data analysis was performed using
SPSS for Windows version 16.0. Statistical significance was
determined by Student’s t-test, with values of P<0.05 considered
to be statistically significant.
Results
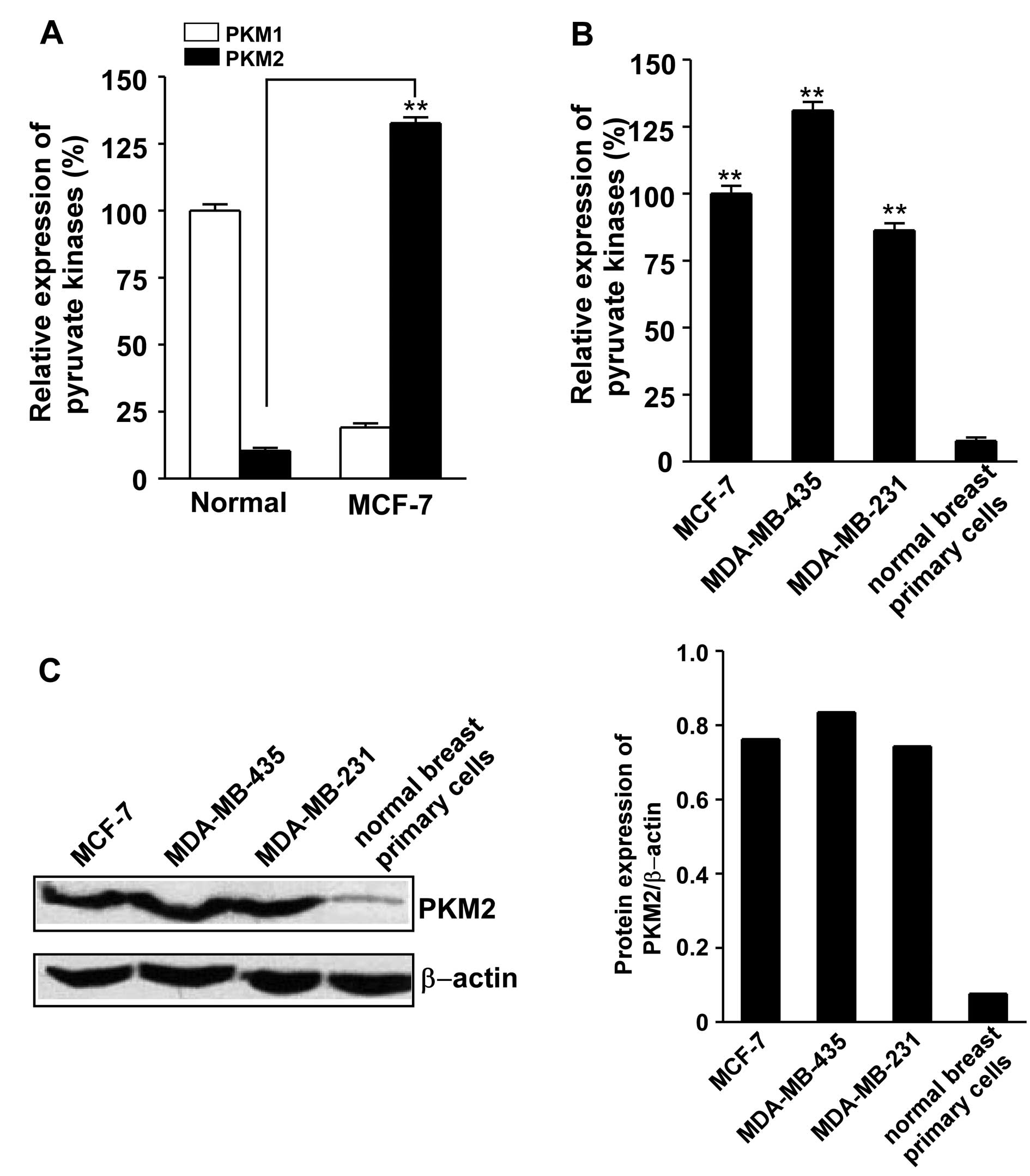
PKM2 is highly expressed in breast cancer
cell lines
As reported previously, PKM2 was highly expressed in
the 3 breast cancer cell lines MCF-7, MDA-MB-435 and MDA-MB-231.
The results showed that PKM2 mRNA was significantly overexpressed
in breast cancer line MCF-7 as compared to the normal breast
primary cells measured by qRT-PCR (Fig. 1A). PKM2 mRNA was also
overexpressed in MDA-MB-435 and MDA-MB-231 breast cancer cell lines
(Fig. 1B). The protein level of
PKM2 is upregulated in these breast cancer cell lines analyzed by
western blotting (Fig. 1C). These
results suggested that tumorigenesis is characterized by the
transformation from tissue-specific PKM1 to tumor-specific
PKM2.
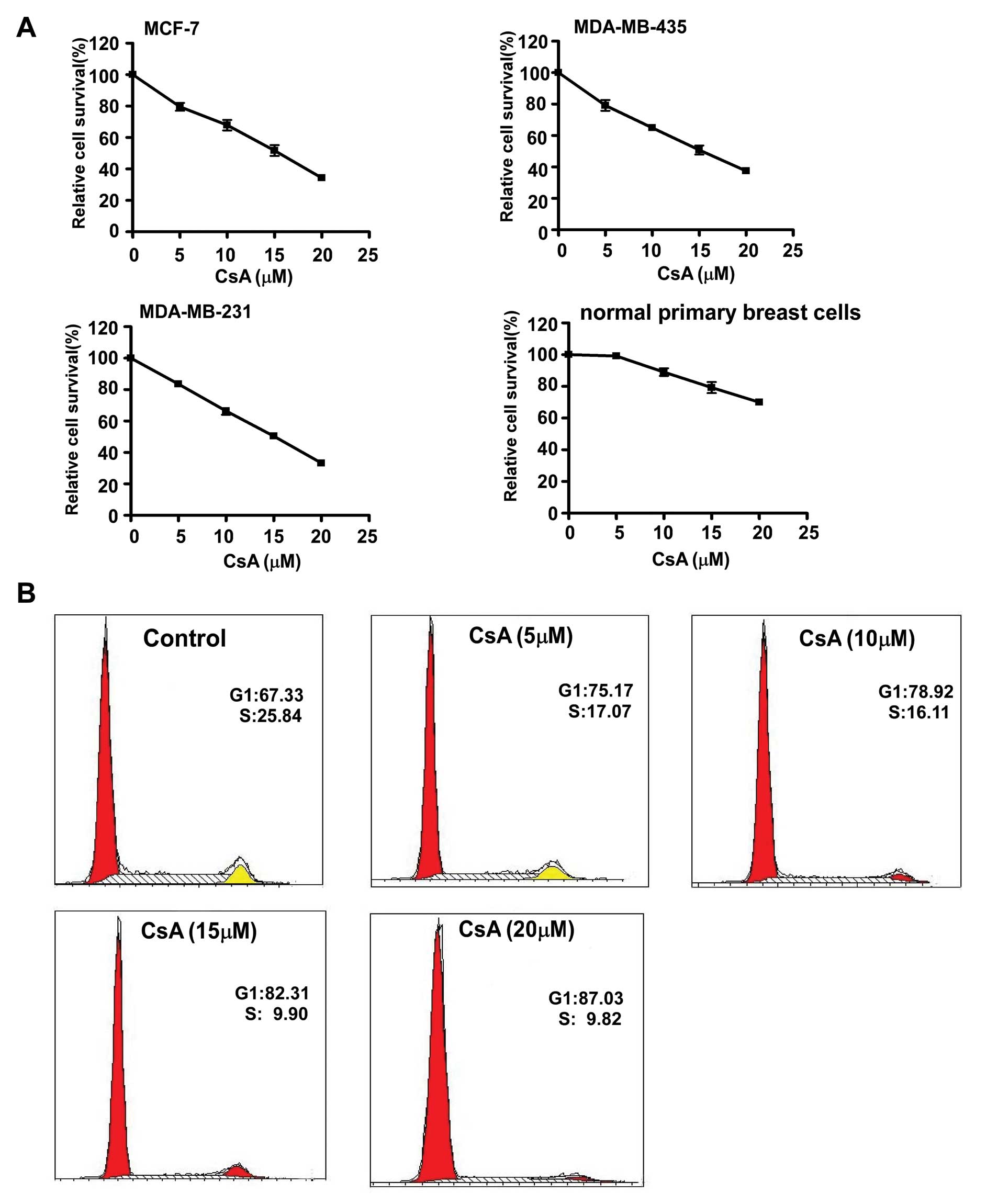
Effect of CsA on cell growth and
survival
Uncontrolled growth is an integral feature of all
malignant tumors. In order to investigate the effect of CsA on
breast cancer cell growth, MCF-7, MDA-MB-435, MDA-MB-231 and normal
breast primary cells were cultured in the presence of various
concentrations of CsA. The results showed that CsA significantly
inhibited the growth of cancer cell lines in a dose-dependent
manner, but the primary breast cells were much less sensitive to
different concentrations of CsA (Fig.
2A). Flow cytometry analysis revealed that CsA induced cell
cycle arrest in MCF-7 cells, as evidenced by the shift of cells
from the S to the G1 phase (Fig.
2B).
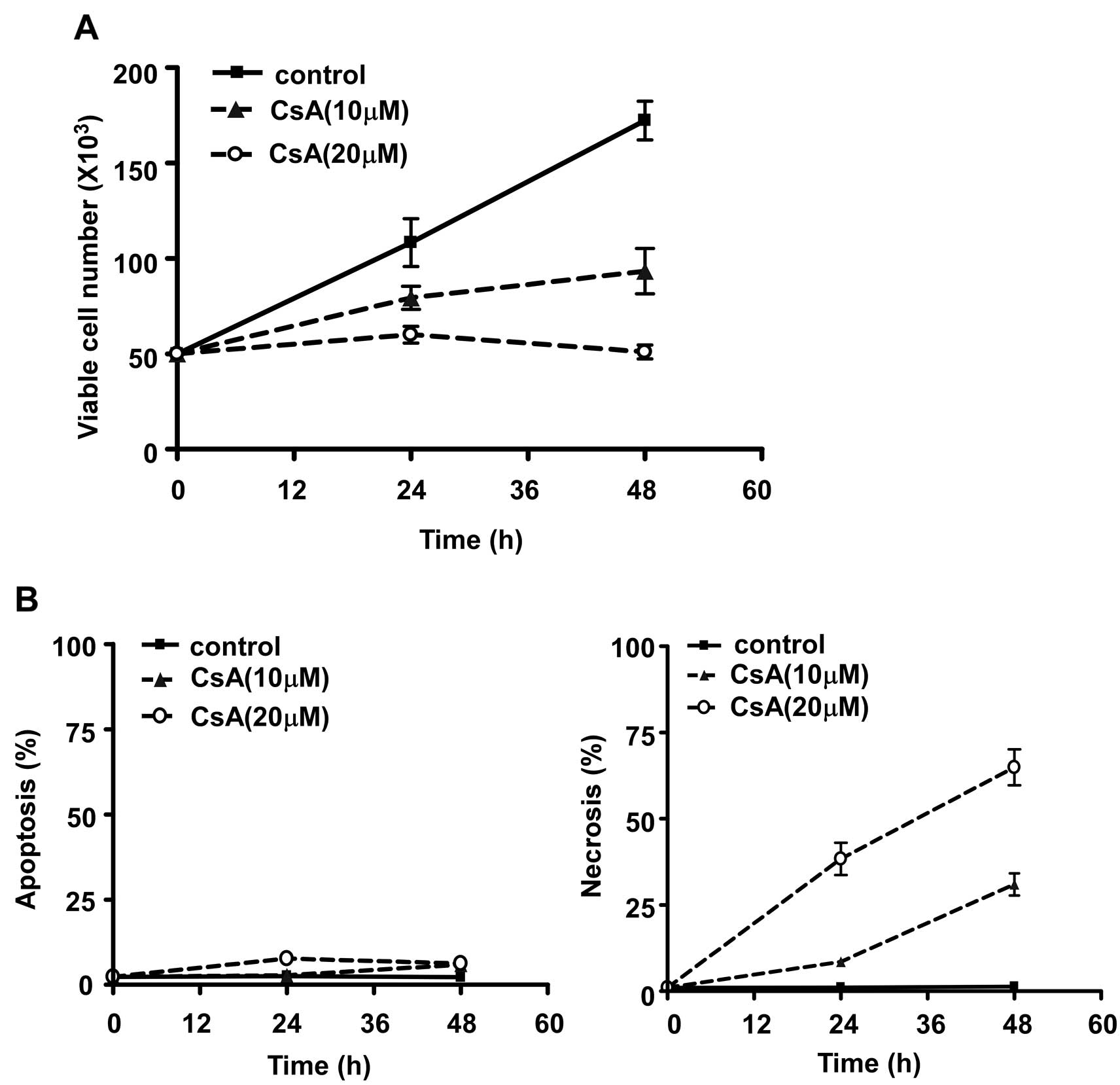
Furthermore, treatment with CsA significantly
decreased the number of viable MCF-7 cells in a time- and
dose-dependent manner (Fig. 3A).
To determine whether CsA inhibited cell growth by inducing
apoptosis of MCF-7 cells, we analyzed the percentage of apoptotic
cells gated as positive Annexin-V staining (20) and necrotic cells gated as PI
positive cell populations detected by the FACS assay. As shown in
Fig. 3B, CsA induced MCF-7 cell
death mainly via necrosis but not apoptosis and the cell death was
time-dependent.
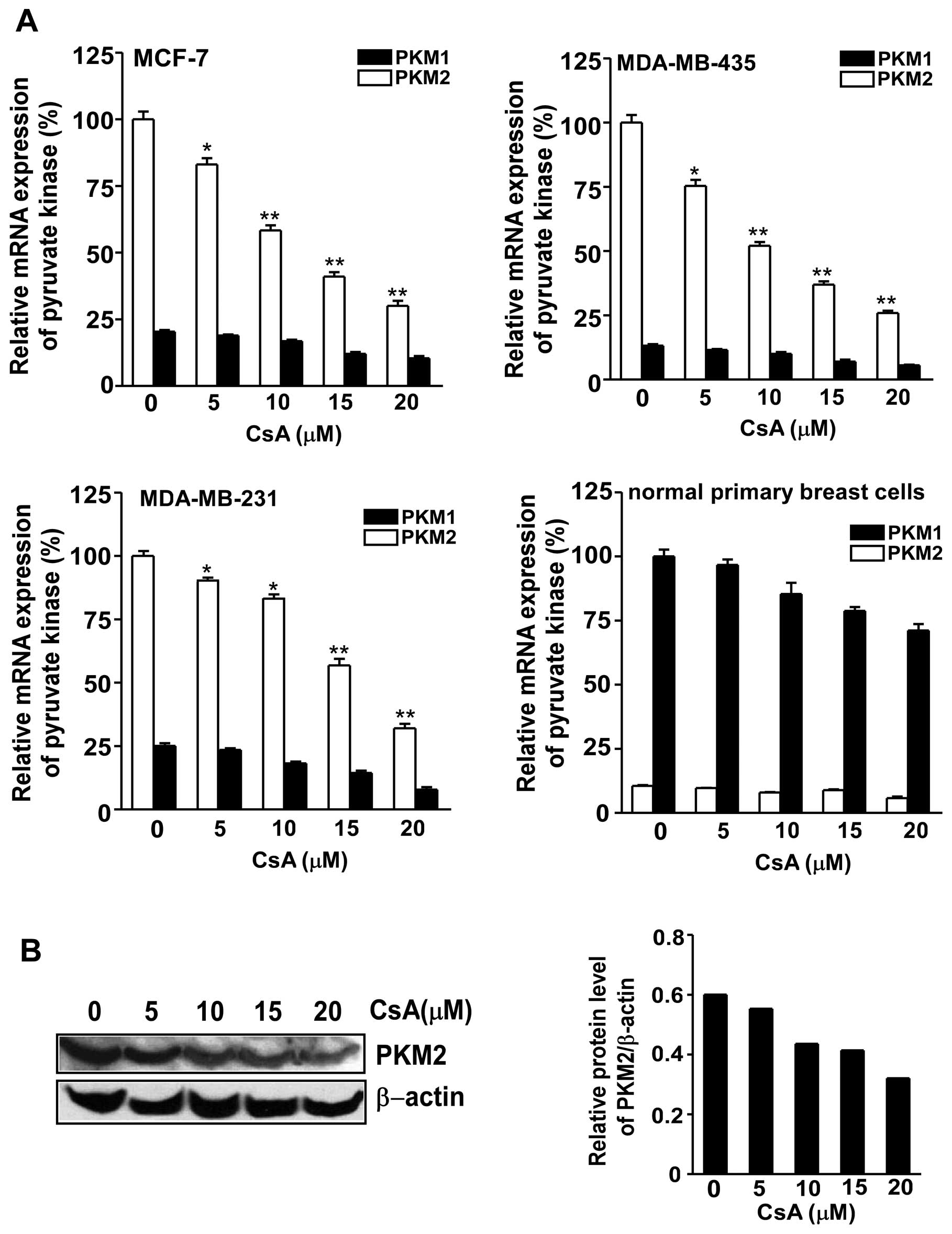
CsA inhibits the expression and the
activity of PKM2 in breast cancer cells
In order to test the inhibitory effect of CsA on the
pyruvate kinase M gene, qRT-PCR was performed to measure the
expression of PKM1/2 in MCF-7, MDA-MB-435, MDA-MB-231 and breast
primary cells. PKM2 mRNA expression was significantly inhibited in
the 3 cancer cell lines following exposure to CsA (Fig. 4A). The protein level of PKM2
expression in MCF cells was also downregulated after the treatment
with various concentrations of CsA (Fig. 4B).
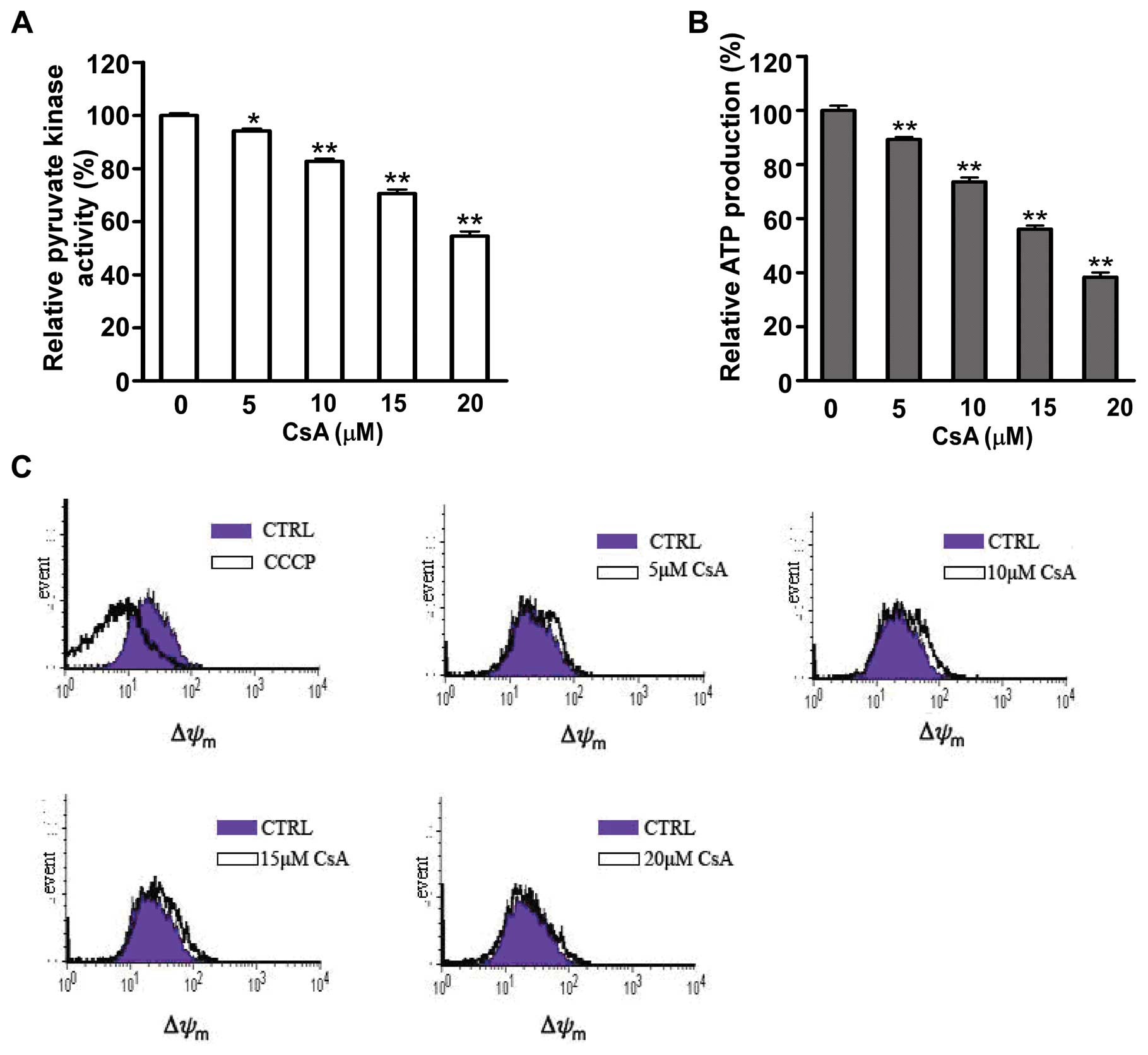
Furthermore, the enzyme activity of PKM2, which
catalyzes the dephosphorylation of phosphoenolpyruvate (PEP) to
pyruvate was measured by reduction of NADH. As shown in Fig. 5A, CsA exposure caused a 5–40%
inhibition of PKM2 activity in MCF-7 cells compared to untreated
cells with a dose-dependent manner. Pyruvate contributes to net
adenosine triphosphate (ATP) production within the glycolytic
pathway. The ATP production was detected in MCF cells after CsA
exposure. It was showed that decrease of PKM2 activity was
accompanied with reduction of ATP production (Fig. 5B). However, there was no
difference in mitochondria function between untreated MCF-7 cells
and cells treated with CsA (Fig.
5C).
All above data suggested that decreased PKM2
activity led to reduction of ATP production, which may result in
cell death after CsA treatment in MCF-7 cells (21).
Inhibition of cell growth by CsA is
mainly dependent on PKM2 activity
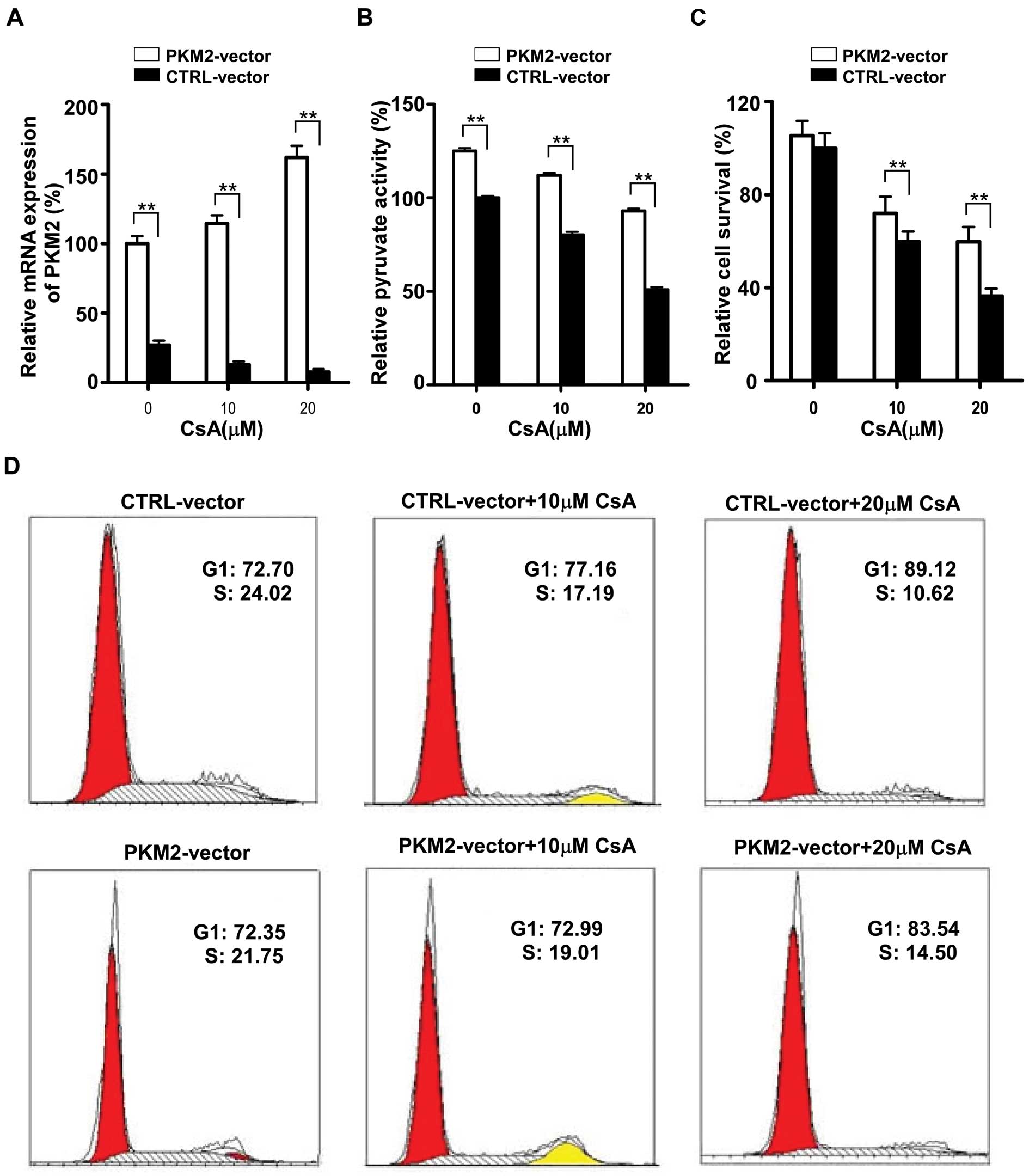
To determine whether the effect of CsA is PKM2
dependent, we next investigated the role of ectopic overexpression
of PKM2 to protect the MCF-7 cells from CsA-induced death.
Interestingly, CsA failed to inhibit the proliferation of MCF-7
cells which were transiently transfected with PKM2-pEGFP-N1
plasmid. As shown in Fig. 6A, a
3–5-fold increase in the expression of PKM2 was observed in MCF-7
cells after PKM2-pEGFP-N1 plasmid transfection, and CsA failed to
downregulate the expression of PKM2 in MCF-7 cells with PKM2
plasmid transfection compared to the mock control (MCF-7 cells
transfected with control pEGFP-N1 plasmid). Surprisingly, PKM2
expression was increased in transfected MCF-7 cells treated with
CsA when compared to the transfected cells without CsA treatment.
It may be due to the fact that the cells which did not gain the
plasmid were inhibited by CsA, resulting in a high proportion of
plasmid-gained cells.
The activity of PKM2 was also determined in
plasmidtransfected cells after CsA exposure. It was showed that the
activity of PKM2 was increased after transfection and that CsA
failed to downregulate the enzyme activity (Fig. 6B). To further demonstrate that the
ectopic overexpression of PKM2 induced resistance to CsA, MTT assay
and cell cycle analysis were also used to determine the growth of
MCF-7 cells. It was shown that ectopic overexpression of PKM2
protected MCF-7 cells from the inhibition effect of CsA (Fig. 6C) and partially reversed the cell
cycle arrest induced by CsA (Fig.
6D).
Discussion
The metabolic state of tumor cells is different from
that of normal counterparts and a high glycolytic capacity is
crucial for cancer cell survival and proliferation (22). For this purpose, a switch of
isoenzyme pattern and activity is initiated in metabolic
phenotypes. Pyruvate kinase is a key enzyme of glycolysis.
Different isoforms of this enzyme exist (pyruvate kinases L, R, M1,
M2) and are tissue-specifically expressed in various organisms.
PKM1 and PKM2 are different splicing products of the same gene
(23). They differ only in 22
amino acid residues in an exon of 45 amino acids (24). The M1 isoform is expressed in most
adult tissues while the M2 isoform is strongly overexpressed in
most, if not all, cancers (2,25).
Recent studies have showed that, in human cancer cells, epidermal
growth factor receptor (EGFR) activation induces translocation of
PKM2, but not PKM1, into the nucleus where K433 of PKM2 binds to
c-Src-phosphorylated Y333 of β-catenin. This interaction is
required for both proteins to be recruited to the CCND1 promoter,
leading to HDAC3 removal from the promoter, histone H3 acetylation
and cyclin D1 expression. PKM2-dependent β-catenin transactivation
is instrumental in EGFR-promoted tumor cell proliferation and brain
tumor development (5). Many
studies have indicated that PKM2 might be a promising target for
cancer intervention (3,4,25).
Here we showed that the 3 breast cancer cell lines exhibited
substantially high PKM2 expression as compared to the normal
primary breast cells, suggesting that tumor M2-PK provided a good
discrimination of benign disease from a malignant one, including
breast cancers (26,27).
We found that CsA suppressed the cell growth, cell
cycle progression and G1/S phase transition of breast cancer cell
lines. Recent studies showed that CsA inhibited the expression of
some oncogenes. CsA could suppress cancer cell proliferation
probably through regulating the expression levels of c-Myc, p21 and
PCNA via inhibition of CaN/NFAT activity (28,29). Our data demonstrated for the first
time that CsA downregulated the cancer-specific PKM2 which promotes
the progression of the tumor. The enzyme activity of PKM2 was also
inhibited in MCF-7 cells treated with CsA. As pyruvate kinase
catalyzes the last step in the glycolytic sequence and is
responsible for net ATP production in this pathway, we measured the
production of ATP in MCF-7 treated with CsA and found a significant
reduction. This suggests that CsA caused inhibition of PKM2
expression, resulting in decrease of the intracellular ATP in tumor
cells and then leading to a deceleration of cell proliferation and
even induction of cell death. In order to confirm this view, we
measured the function of mitochondria showing that the decrease in
the ATP production is not dependent on mitochondria. Moreover,
transient transfection with PKM2 expression vector into MCF-7 cells
partially reversed the inhibition effect of CsA. Thus, we conclude
that the most likely target of CsA action as inhibitor of breast
cell growth is the tumor-specific PKM2.
As shown in our results, the sensitivity of the
normal tissue cells was much lower than the tumor cell lines when
treated with CsA. We supposed that the capacity of CsA to decrease
the expression of PKM1 and PKM2 was different and M2-PK was more
sensitive. There are some differences of function between M1-PK and
M2-PK, a well known one is that the nonallosteric M1 isoenzyme
exists only as a tetramer, which is a constitutively active enzyme
while M2-PK is an allosteric enzyme that can exist in two different
conformations (a tetrameric form which is active and a dimeric form
which is inactive) (17,30). A previous study has demonstrated
that the tetramer:dimer ratio of M2-PK is directly controlled by
different signal metabolites and oncoproteins (2). CsA decreased the expression and
activity of pyruvate kinase M2 and thus reduced the production of
ATP in cancer cells rather than that in normal tissues.
CsA is a kind of classic immunosuppressant, so
general administration of CsA to patients with breast cancers may
impair anti-tumor immune responses of the host and be detrimental.
However, local administration of CsA may be beneficial because this
could inhibit breast cancer proliferation without inhibiting
general immunity. Moreover, this application might impair the
suppressive function of
CD4+CD25+Foxp3+ regulatory T cells
(Tregs) which infiltrated into tumor tissues. Tregs are known to
suppress anti-tumor immune responses (31,32) and their differentiation and
suppressive function are guided by the interaction of transcription
factor Foxp3 with NFAT (33),
which is also inhibited by CsA.
In conclusion, we demonstrated that CsA was able to
effectively inhibit PKM2 overexpression in breast cancer cells,
which is relative to cancer cell proliferation. Although it remains
to be further investigated whether CsA treatment would be
beneficial in breast cancer treatment, the mechanism suggested here
might be useful to develop efficient approaches to treat breast
cancers.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (81072405),
the ‘Program for New Century Excellent Talents in University’ from
the Ministry of Education of China (NCET08-0486) and the Zhejiang
Provincial Natural Science Foundation of China (R2100528). It was
also sponsored by the Zhejiang Provincial Program for the
cultivation of high-level innovative health talents.
References
|
1.
|
O WarburgOn the origin of cancer
cellsScience123309314195610.1126/science.123.3191.30913298683
|
|
2.
|
S MazurekCB BoschekF HugoPyruvate kinase
type M2 and its role in tumor growth and spreadingSemin Cancer
Biol15300308200510.1016/j.semcancer.2005.04.00915908230
|
|
3.
|
D AnastasiouG PoulogiannisJM
AsaraInhibition of pyruvate kinase M2 by reactive oxygen species
contributes to cellular antioxidant
responsesScience33412781283201110.1126/science.121148522052977
|
|
4.
|
MS GoldbergPA SharpPyruvate kinase
M2-specific siRNA induces apoptosis and tumor regressionJ Exp
MedJan232012(Epub ahead of print)10.1084/jem.20111487
|
|
5.
|
W YangY XiaH JiNuclear PKM2 regulates
beta-catenin transactivation upon EGFR
activationNature480118122201110.1038/nature1059822056988
|
|
6.
|
HR ChristofkMG Vander HeidenMH HarrisThe
M2 splice isoform of pyruvate kinase is important for cancer
metabolism and tumour
growthNature452230233200810.1038/nature0673418337823
|
|
7.
|
HR ChristofkMG Vander HeidenN WuPyruvate
kinase M2 is a phosphotyrosine-binding
proteinNature452181186200810.1038/nature0666718337815
|
|
8.
|
B AltenbergKO GreulichGenes of glycolysis
are ubiquitously overexpressed in 24 cancer
classesGenomics8410141020200410.1016/j.ygeno.2004.08.01015533718
|
|
9.
|
T SaekiK UedaY TanigawaraHuman
P-glycoprotein transports cyclosporin A and FK506J Biol
Chem2686077608019937681059
|
|
10.
|
B TanD Piwnica-WormsL RatnerMultidrug
resistance transporters and modulationCurr Opin
Oncol12450458200010.1097/00001622-200009000-0001110975553
|
|
11.
|
G EnglundP HallbergP ArturssonAssociation
between the number of coadministered P-glycoprotein inhibitors and
serum digoxin levels in patients on therapeutic drug monitoringBMC
Med28200410.1186/1741-7015-2-815061868
|
|
12.
|
J Carcel-TrullolsF Torres-MolinaA
AraicoEffect of cyclosporine A on the tissue distribution and
pharmacokinetics of etoposideCancer Chemother
Pharmacol54153160200415114410
|
|
13.
|
CQ XiaN LiuGT MiwaInteractions of
cyclosporin A with breast cancer resistance proteinDrug Metab
Dispos35576582200717220244
|
|
14.
|
P BerthonG PancinoP de
CremouxCharacterization of normal breast epithelial cells in
primary cultures: differentiation and growth factor receptors
studiesIn Vitro Cell Dev Biol28A7167241992
|
|
15.
|
QQ WangH LiT OliverIntegrin beta 1
regulates phagosome maturation in macrophages through Rac
expressionJ Immunol18024192428200818250451
|
|
16.
|
Y LiuQ ChenY SongMicroRNA-98 negatively
regulates IL-10 production and endotoxin tolerance in macrophages
after LPS stimulationFEBS
Lett58519631968201110.1016/j.febslet.2011.05.02921609717
|
|
17.
|
JD DombrauckasBD SantarsieroAD
MesecarStructural basis for tumor pyruvate kinase M2 allosteric
regulation and catalysisBiochemistry4494179429200515996096
|
|
18.
|
KJ LivakTD SchmittgenAnalysis of relative
gene expression data using real-time quantitative PCR and the
2(-Delta Delta C(T)) methodMethods25402408200111846609
|
|
19.
|
EL LarsonAE AielloC
Gomez-DuarteBioluminescence ATP monitoring as a surrogate marker
for microbial load on hands and surfaces in the homeFood
Microbiol20735739200310.1016/S0740-0020(03)00041-8
|
|
20.
|
A IshaqueM Al-RubeaiUse of intracellular
pH and annexin-V flow cytometric assays to monitor apoptosis and
its suppression by bcl-2 over-expression in hybridoma cell cultureJ
Immunol Methods221435719989894897
|
|
21.
|
M LeistB SingleAF CastoldiIntracellular
adenosine triphosphate (ATP) concentration: a switch in the
decision between apoptosis and necrosisJ Exp
Med18514811486199710.1084/jem.185.8.14819126928
|
|
22.
|
K GarberEnergy deregulation: licensing
tumors to
growScience31211581159200610.1126/science.312.5777.115816728625
|
|
23.
|
K TaniMC YoshidaH SatohHuman M2-type
pyruvate kinase: cDNA cloning, chromosomal assignment and
expression in hepatomaGene7350951619882854097
|
|
24.
|
K YamadaT NoguchiRegulation of pyruvate
kinase M gene expressionBiochem Biophys Res
Commun256257262199910.1006/bbrc.1999.022810079172
|
|
25.
|
R LiJ LiuH XueDiagnostic value of fecal
tumor M2-pyruvate kinase for CRC screening: A systematic review and
meta-analysisInt J CancerJan192012(Epub ahead of
print)10.1002/ijc.27442.
|
|
26.
|
J SchneiderHG VelcovskyH MorrComparison of
the tumor markers tumor M2-PK, CEA, CYFRA 21-1, NSE and SCC in the
diagnosis of lung cancerAnticancer Res2050535058200011326667
|
|
27.
|
J SchneiderG SchulzeComparison of tumor
M2-pyruvate kinase (tumor M2-PK), carcinoembryonic antigen (CEA),
carbohydrate antigens CA 19-9 and CA 72-4 in the diagnosis of
gastrointestinal cancerAnticancer Res2350895093200314981971
|
|
28.
|
M BuchholzA SchatzM WagnerOverexpression
of c-myc in pancreatic cancer caused by ectopic activation of
NFATc1 and the Ca2+/calcineurin signaling pathwayEMBO
J2537143724200616874304
|
|
29.
|
T MasuoS OkamuraY ZhangCyclosporine A
inhibits colorectal cancer proliferation probably by regulating
expression levels of c-Myc, p21(WAF1/CIP1) and proliferating cell
nuclear antigenCancer
Lett2856672200910.1016/j.canlet.2009.05.001
|
|
30.
|
JO WoollRH FriesenMA WhiteStructural and
functional linkages between subunit interfaces in mammalian
pyruvate kinaseJ Mol
Biol312525540200110.1006/jmbi.2001.497811563914
|
|
31.
|
A GallimoreA GodkinRegulatory T cells and
tumour immunity - observations in mice and
menImmunology123157163200810.1111/j.1365-2567.2007.02748.x18067556
|
|
32.
|
TJ CurielG CoukosL ZouSpecific recruitment
of regulatory T cells in ovarian carcinoma fosters immune privilege
and predicts reduced survivalNat
Med10942949200410.1038/nm109315322536
|
|
33.
|
Y WuM BordeV HeissmeyerFOXP3 controls
regulatory T cell function through cooperation with
NFATCell126375387200610.1016/j.cell.2006.05.04216873067
|